
Abstract
Introduction
Segmental spinal dysgenesis, a rare developmental malformation, usually manifests during pregnancy or at birth. The resulting gross spinal instability necessitates spinal stabilization, which is inherently challenging in neonates.
Methods
We report four cases of segmental dysgenesis: three in the thoracolumbar region and one at the cervicothoracic junction. The latter was maintained in a custom orthosis that restricted all craniospinal motion while allowing routine care. Two neonates underwent surgical stabilization. The fourth patient will remain in a brace until 12–14 months old when fusion is planned.
Results
Fusion with rib autografts failed in the two neonates. One patient has been followed for 13 years and is paraplegic. The second patient was lost to follow up. The patient with the cervicothoracic dysgenesis maintained normal neurologic function until his death at 8 months of cardiac failure. The fourth patient is 12 months old and has been maintained in a thoracolumbar orthosis with stable neurologic function.
Conclusion
Several factors contribute to the challenge of creating a stable fusion in neonates. Incomplete ossification of the vertebral bodies and poor results with allograft materials restrict fusion options. Neurologic deficits often prevent ambulation and decrease the axial-loading forces that enhance fusion. To allow children to grow and develop, we advocate rigid spinal immobilization for 12–18 months before spinal fusion (preferably, rib or fibular autograft). Given the already narrow spinal canal, the use of instrumentation is controversial. We advocate the use of instrumentation in infants only when a sound construct cannot be obtained with the graft alone.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Segmental spinal dysgenesis is a rare developmental abnormality that usually affects the lumbar and thoracolumbar spine. The condition is often discovered at or before birth when the anomalies are detected on prenatal screening ultrasonography. Patients have a variety of neurological deficits, ranging from normal function to paraplegia. A component of spinal stenosis is thought to contribute to the neurological deficits and may be the initial indication for surgical intervention. The classification of vertebral body anomalies is controversial and has been reported under many different names in the literature. We present four cases treated at our institution and review the imaging characteristics and diagnostic criteria for segmental dysgenesis, the proposed embryology in comparison to other disorders, and our management strategy.
Case studies
Case 1
This newborn baby boy was referred to the neurosurgery service because lower extremity and spinal deformities were noted at birth. On examination, the infant had pes equinovarus. Painful stimulus above the level of the deformity elicited flexion at the hips and ankles, and extension at the knees. Anal wink was present but urodynamic testing was not performed. Imaging showed high lumbar segmental dysgenesis at L1–L2 and a myelocystocele. In addition to the segmental dysgenesis with normal lumbar vertebrae distal to the L1–L2 defect, he also had partial sacral agenesis. Myelography revealed no nerve root sleeves at the affected level. Surgical repair was undertaken when the boy was 6 weeks old via an anterior approach that allowed both myelocystocele repair and spinal fusion. Abnormal fibrous tissue was removed from the L1 to L3 vertebral body region. A rib strut graft supplemented with additional pieces of bone was constructed to span the defect. Pseudarthrosis developed over the following year and was associated with increased kyphosis and progression to complete paraplegia at L1. The patient was lost to follow-up after the 1-year visit.
Case 2
This newborn girl was referred to the neurosurgery service because a spinal anomaly was noted at birth. Examination revealed a palpable bony defect at the thoracolumbar junction and purposeful movement of the hips during flexion and extension. There was minimal withdrawal to pain in the lower extremities with good rectal tone. Formal urologic testing was not performed. Plain X-rays and 3D computed tomography (CT) scan showed 13 thoracic vertebrae and three lumbar vertebrae, with anomalies at T13 and L2 and absence of the L1 vertebra. The conus was not clearly identified. The spinal cord tapered severely at the thoracolumbar junction, widened again caudal to L2, and was tethered to the sacrum.
At 3.5 weeks old she underwent anterior corpectomy and decompression at the affected level. Intraoperatively, the spinal cord was rotated 45° to the right as it coursed behind the anomalous bodies. The nerve roots corresponding to T13 and L2 coursed cephalad from their exit site to the foramina. Absence of the L1 root was noted bilaterally. After the fibrous tissue was removed at L1, a graft was constructed with two pieces of rib. Complications included a nerve root avulsion at the L2 level.
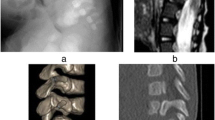
Lower extremity function appeared unchanged after surgery. Postoperative CT showed that one of the grafts had shifted. One day after surgery, the patient therefore underwent surgical exploration. At reoperation, no cord compression was found, and a stay suture was placed around the grafts. Although fusion eventually developed at the surgical site, some subsidence and subluxation occurred. Lower extremity motor function did not improve. She had a neurogenic bladder, but painful stimulation throughout the lower extremities elicited a cry and wince. At age 6, she developed back pain, which was managed conservatively for 2 years. Imaging revealed a “block vertebrae” anteriorly at the prior fusion site and severe bony stenosis at the superior end of her fusion. At the age of 8, she underwent laminectomy for decompression at L2 through L4 (Fig. 1a and b). She has frequent bladder infections, but otherwise she functions independently as a paraplegic at age 15.

a Sagittal and b axial T2-weighted MRIs of patient at age 11 show fused bony anomalies in the lumbar spine. The spinal cord is still visible caudal to the defect
Case 3
This boy was born with C7-T1 dysgenesis (Fig. 2a and b) and congenital heart disease. His neurologic examination was normal with the exception of a clenched fist on the left. He was managed in a custom craniocervicothoracic orthosis until he died at 8 months from heart disease (Fig. 3a and b). His neurologic examination, however, had remained stable.

a Anteroposterior and b lateral 3D computed tomographic reconstructions of segmental spinal dysgenesis involving C7 and T1

a Anterior and b posterior views of custom cranial cervicothoracic orthosis
Case 4
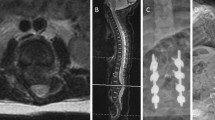
This boy was born with T12-L1 dysgenesis, a horseshoe kidney, an undescended testicle, an inguinal hernia, bilateral hip dislocations, and pes equinovarus. Physical examination revealed a lump in the lumbar region. He exhibited no spontaneous motion of the lower extremities, but a painful stimulus elicited a verbal response. CT revealed segmental absence of the T12 and L1 vertebrae, with preservation of the vertebral formation in the distal lumbar spine (Fig. 4a and b). Magnetic resonance imaging (MRI) showed the presence of a spinal cord distal to the affected level (Fig. 4c). He was managed in a custom thoracolumbosacral orthosis with good alignment. At 19 months, his lower extremities were contracted in flexion with no significant response to pain. His mother reported only fair compliance with the brace. MRI showed interval collapse with kyphosis at the thoracolumbar junction (Fig. 5). We plan to proceed with a posterior fusion when he is about 3 years old or once imaging reveals bone quality, in which posterior instrumentation will be safe and efficacious. Once he has reached the age of 6 years, we will augment the posterior fusion with anterior arthrodesis.

a Lateral and b anteroposterior 3D computed tomography reconstructions show vertebral defects. c Sagittal T2-weighted magnetic resonance image shows defect with patent canal and neural tissue caudal to defect

Sagittal MRI of patient at 19 months shows interval collapse and kyphosis at the thoracolumbar level
Discussion
As cited by Scott et al. [15] Hohl first reported the concept of spinal agenesis in 1852. Many early reports involved sacral agenesis and anomalies of the caudal spinal column. As strict definitions emerge, most of these cases are now considered separate entities. In 1988, Scott et al. [15] proposed the use of the term “segmental spinal dysgenesis” for a subset of patients with segmental defects. Faciszewski et al. [6] further defined the condition to include focal spinal stenosis at the affected level; lack of neurocentral junctions; absence of pedicles, spinous processes, and transverse processes; and no osseous abnormalities caudad to the involved level. They also described an osseous ring around the spinal canal, which was open posteriorly in 3 of 17 patients. The abnormal bone in the region of the affected vertebral bodies tends to be displaced posteriorly. This malalignment contributes to both the spinal stenosis and kyphosis associated with this syndrome. Absence of nerve roots has been reported in many series [6, 7, 15] and probably also contributes to the neurologic dysfunction.
A variety of associated malformations has also been reported: pes equinovarus deformities, Klippel–Feil anomaly, renal abnormalities, situs inversus, and tetralogy of Fallot. Three of our four patients had additional anomalies, including a myelocystocele and congenital heart disease. These associated findings are not surprising and may correlate temporally with disruption in the development of the spinal structures.
Most reported cases of segmental spinal dysgenesis have involved the lumbar or thoracic spine [2, 6, 7, 9, 15, 17]. We present a case in which the defect occurred at C7-T1. It is unclear whether the same embryological mechanisms can account for this case. Desai et al. [4] and Scott et al. [15] each reported one case involving the lower thoracic levels, and these are the highest levels of involvement found in the literature. Rastogi et al. [14] reported a patient with segmental spinal dysgenesis and a dorsal dermal sinus at T10, an interesting case of dysraphic and dysgenetic abnormalities coinciding.
Embryology
The precise embryological derangement has yet to be determined. It appears to involve a failure of the caudal notochord and cell mass, specifically, a chorda-mesodermal derangement during gastrulation as described by Tortori-Donati et al. [17]. Martinez-Frias [12], who has published extensively on segmentation anomalies and developmental field defects, has proposed investigation at the molecular level to improve characterization of these events. Vitamin A deficiency can result in a variety of developmental anomalies similar to segmental spinal dysgenesis. Maden et al. [11] studied vitamin A-deficient quail embryos and found apoptosis among the neural crest cells and failure of neurites to grow from the neural tube. Van Allen et al. [18] proposed an alternative five-site closure mechanism for the neural tube. In this model, the upper lumbar spine results from a watershed area between the first and fifth closure sites and may predispose to anomalies. This theory remains to be proved embryologically.
Unlike in congenital vertebral dislocation, which is not associated with spina bifida or dysraphism, there are several reports of patients with occult dysraphism and segmental spinal dysgenesis [1, 15, 17]. These reports indicate that the defect can occur before the end of the fourth week when the neural tube is closed. Dias et al. [5] cite this difference to account for the preserved alignment in congenital vertebral dislocation. The vertebral body and posterior elements develop from three pairs of chondrification centers [5]. A defect in formation of the sclerotome from which these centers are derived may result in agenesis of the corresponding portion of the vertebral body. As in sacral agenesis, maternal diabetes may be a predisposing factor, as are pharmacologic toxins like lithium [15].
Imaging
MRI, the gold standard for imaging spinal anomalies, is the most sensitive modality for imaging soft tissue defects such as dysraphism, cord tethering, and syringomyelia [13]. It is ideal for visualizing cerebrospinal fluid and neural structures that pass through the affected level. Tortori-Donati et al. [17] found that the patient’s clinical status correlated with the amount of neural tissue visualized on MRI at the level of the lesion. CT with 3D reconstruction is essential for visualizing the bony defects and aids in planning the reconstruction. Myelography has almost been replaced by MRI, although it is useful for evaluating spinal stenosis and the presence of the nerve root sleeves. Abnormalities of nerve root sheaths are also associated with other congenital defects such as the absence of the pedicle in posterior arch defects. Myelography in such patients shows a common dural sheath for the nerve roots at and below the level of the absent pedicle [3].
Treatment
Given the rarity of segmental spinal dysgenesis, options for treatment are as controversial as the underlying embryological mechanisms. Some authors advocate surgical stabilization with fusion at the time of diagnosis; others use a staged strategy of anterior and posterior fusion after a period of bracing. This decision is particularly relevant to patients who present at birth with this segmental defect. It is also important for patients who have only minimal or no neurologic deficits, which can be difficult to determine in the neonate. Any treatment strategy must be individualized based on specific anatomical and clinical factors. Any patient whose neurological function deteriorates should be reevaluated immediately with the potential for changing the management strategy.
The two issues to be addressed when treating these children are spinal instability and neural compression. The lack of normal bony elements and the abnormal alignment of defective bone frequently lead to overt spinal instability. The lower thoracic and lumbar spine must sustain axial loading in the upright position. Consequently, many patients will require an anterior approach. Ribs and fibular grafts provide optimal support but are slower to become incorporated than iliac crest and cancellous bone. The latter is best suited for posterolateral fusion where there is less of an axial load. The addition of decompression to the fusion procedure must be determined by the patient’s neurologic status and radiologic evidence of compression. Of 17 patients in the series by Faciszewski et al. [6], 10 required decompression, although only 2 patients improved neurologically. The drawback of decompression is that it further destabilizes the spine.
Previous publications by Flynn et al. [7] and Faciszewski et al. [6] both recommended that arthrodesis be performed as soon as the diagnosis is made. They also recognized that achieving solid fusion in this population is difficult. The six patients reported by Flynn et al. [7] required 15 procedures to achieve a stable fusion. The immaturity of the osseous bed also challenges fusion. Faciszewski et al. [6] stated that bracing should have no role in the management of segmental spinal dysgenesis. However, the 17 patients in that series required 45 fusion procedures to obtain stability, and only 2 patients recovered neurologic function. We believe that the morbidity associated with undergoing multiple attempts at fusion must be weighed against the risk of neurologic decline in a properly fitted brace. We do not believe that “waiting for neurologic decline” is the same as bracing to enable bony maturity. Given the outcomes of previously published reports, we cannot advocate neonatal fusion in the absence of neurologic decline.
In one of the larger series of very young children with congenital spinal disorders, Kim et al. [10] treated 26 patients with congenital kyphosis. They compared two groups: one under the age of 3 years and one over the age of 3 years. They compared a posterior-only approach with a combined anterior and posterior approach. For both age groups, they concluded that the rate of pseudarthrosis was low if instrumentation was used, and that kyphosis may correct gradually over time after surgery in the younger group. The instrumentation varied from taping or wiring posterior elements to Harrington and Texas Scottish Rite Hospital constructs. The lack of spinal stenosis and rarity of neurologic deficits associated with congenital kyphosis may allow instrumentation to be used in more cases than in segmental spinal dysgenesis.
The use of intraoperative electrophysiologic monitoring is becoming more common, especially since it became possible to monitor motor-evoked potentials (MEPs). Although this modality may be useful in an older population, variability is significant in neonates and young infants, improving at 10 to 14 months. Adult patterns emerge after 3 years of age [8], but anecdotal evidence supports the monitoring of MEPs at 18 months. Variability is greater in the younger patients, with latencies decreasing and amplitude increasing over time [16].
Conclusion
This series illustrates the complexity of segmental spinal dysgenesis and the divergent outcomes possible with treatment. In light of the poor results of surgery in infants, we believe that bracing is the optimal initial management strategy for very young children with this disease. The main goal of spinal stabilization in this population may be simply to allow the patient to sit without support. Minimizing the number of procedures required to achieve a solid fusion is in the patient’s best interest. We believe that bracing enables the bones to mature to a point where fusion rates improve. However, any patient who deteriorates neurologically while braced may be treated sooner. Once arthrodesis has been performed, it is important to follow the child radiographically into maturity to evaluate for pseudarthrosis, progression of kyphosis, and spinal cord tethering.
References
Buyse G, Van Calenbergh F, Choux M, Demaerel P, Sciot R, Verpoorten C (2003) Segmental spinal cord hypoplasia and meningocele with preservation of medullary function: case report. Surg Neurol 59:505–507
Cherny WB, Rekate HL (1993) Segmental spinal dysgenesis in the newborn (abstract). Child’s Nerv Syst 9:360
Cox HE, Bennett WF (1984) Computed tomography of absent cervical pedicle. J Comput Assist Tomogr 8:537–539
Desai K, Nadkarni T, Bhayani R, Goel A (2003) Congenital thoracic cord segmental amyelia: a rare manifestation of segmental spinal dysgenesis. Pediatr Neurosurg 38:102–106
Dias MS, Li V, Landi M, Schwend R, Grabb P (1998) The embryogenesis of congenital vertebral dislocation: early embryonic buckling? Pediatr Neurosurg 29:281–289
Faciszewski T, Winter RB, Lonstein JE, Sane S, Erickson D (1995) Segmental spinal dysgenesis. A disorder different from spinal agenesis. J Bone Jt Surg Am 77:530–537
Flynn JM, Otsuka NY, Emans JB, Hall JE, Hresko MT (1997) Segmental spinal dysgenesis: early neurologic deterioration and treatment. J Pediatr Orthop 17:100–104
Geneva IE, Krasteva MB, Kostianev SS (2002) Age-related changes of the somatosensory evoked potentials in healthy children. Folia Med (Plovdiv) 44:13–18
Hughes LO, McCarthy RE, Glasier CM (1998) Segmental spinal dysgenesis: a report of three cases. J Pediatr Orthop 18:227–232
Kim YJ, Otsuka NY, Flynn JM, Hall JE, Emans JB, Hresko MT (2001) Surgical treatment of congenital kyphosis. Spine 26:2251–2257
Maden M, Gale E, Zile M (1998) The role of vitamin A in the development of the central nervous system. J Nutr 128:471S–475S
Martinez-Frias ML (2004) Segmentation anomalies of the vertebras and ribs: one expression of the primary developmental field. Am J Med Genet A 128:127–131
Philips MF, Dormans J, Drummond D, Schut L, Sutton LN (1997) Progressive congenital kyphosis: report of five cases and review of the literature. Pediatr Neurosurg 26:130–143
Rastogi H, Behari S, Phadke RV, Gupta RK, Kumar S, Mittal P (1996) Spinal segmental maldevelopment with a dermal sinus. Neuroradiology 38:658–660
Scott RM, Wolpert SM, Bartoshesky LE, Zimbler S, Karlin L (1988) Segmental spinal dysgenesis. Neurosurgery 22:739–744
Szelenyi A, Bueno de Camargo A, Deletis V (2003) Neurophysiological evaluation of the corticospinal tract by D-wave recordings in young children. Child’s Nerv Syst 19:30–34
Tortori-Donati P, Fondelli MP, Rossi A, Raybaud CA, Cama A, Capra V (1999) Segmental spinal dysgenesis: neuroradiologic findings with clinical and embryologic correlation. AJNR Am J Neuroradiol 20:445–456
Van Allen MI, Kalousek DK, Chernoff GF, Juriloff D, Harris M, McGillivray BC, Yong SL, Langlois S, MacLeod PM, Chitayat D et al (1993) Evidence for multi-site closure of the neural tube in humans. Am J Med Genet 47:723–743
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bristol, R.E., Theodore, N. & Rekate, H.L. Segmental spinal dysgenesis: report of four cases and proposed management strategy. Childs Nerv Syst 23, 359–364 (2007). https://doi.org/10.1007/s00381-006-0228-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-006-0228-y