
Abstract
In this paper, we present a strategy for imaging measurements of absolute concentration values of gas-phase SiO in the combustion synthesis of silica, generated from the reaction of hexamethyldisiloxane (HMDSO) precursor in a lean (ϕ = 0.6) hydrogen/oxygen/argon flame. The method is based on laser-induced fluorescence (LIF) exciting the Q(42) rotational transition within the A1Π − X1Σ (1, 0) electronic band system of SiO at 231 nm. Corrections for temperature-dependent population of the related ground state are based on multi-line SiO–LIF thermometry utilizing transitions within the A1Π − X1Σ (0, 0) electronic band around 234 nm. Corrections for local collisional quenching are based on measured effective fluorescence lifetimes from the temporal signal decay using a short camera gate stepped with respect to the laser pulse. This fluorescence lifetime measurement was confirmed with additional measurements using a fast photomultiplier. The resulting semi-quantitative LIF signal was photometrically calibrated using Rayleigh scattering from known gas samples at various pressures and laser energies as well as with nitric oxide LIF. The obtained absolute SiO concentration values in the HMDSO-doped flames will serve as a stringent test case for recently developed flame kinetic mechanisms for this class of gas-borne silicon dioxide nanoparticle synthesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Silica (SiO2) nanoparticles and coatings are conveniently synthesized in gas-phase premixed flames with a variable amount of a silicon-carrying precursor (e.g., hexamethyldisiloxane, HMDSO) [1,2,3]. To fully understand the chemical route from precursor decomposition to particle formation, experimental data on temperature and concentration fields of intermediate species such as SiO are of great importance for comparison to, or even validation of, chemical kinetics mechanisms simulating such processes [4, 5]. In the case of flame-synthesis of silica (SiO2) nanoparticles, the gas-phase radical intermediate SiO plays a pivotal role in the formation of silica [6], and due to its simple diatomic character facilitates use of laser diagnostics for its concentration measurement [5, 7,8,9]. Ease of measurement and kinetics modeling also results from low-pressure conditions and corresponding expanded reaction zones—allowing for more accurately resolved concentration profiles [10]. Measuring absolute concentration values is of interest, since they provide more stringent targets for model validation. The appropriate diagnostics technique to measure absolute SiO concentration in low-pressure silica-synthesis flames via a combined laser-induced fluorescence (LIF) method is the focus of this study.
Early measurements of SiO applied LIF to plasma discharges, chemical vapor deposition (CVD), evaporation processes or flames [11,12,13,14], with the focus on spectroscopic characterization of SiO. Nevertheless, attempts to measure spatially resolved concentrations of SiO have been performed [8, 9, 15] using LIF, albeit on a relative scale. Glumac [5] applied LIF to a low-pressure (2.6 kPa) premixed H2/O2 stagnation flame (ϕ = 2) doped with hexamethyldisilazane (HMDS). Relative concentrations were derived from applying quenching corrections to their measured LIF signal. Similarly, LIF was applied to low-pressure premixed flames to study the reaction scheme of other Si-based precursors hexamethyldisiloxane (HMDSO) and tetramethylsilane (TMS) under fuel-lean conditions, whereby relative concentration profiles of SiO were also measured [8, 9]. In the same environment, SiO LIF has also been used for determining local temperature [7] probing the relative population of several rovibrational ground states.
The utility of relative concentrations derives from the spatial distribution measured from LIF after temperature and quenching corrections have been applied, therefore guiding development of kinetics models [8, 9]. Hitherto, most LIF-based SiO measurements delivered only relative concentrations. Zachariah and Burgess [7], however, demonstrated early on pointwise absolute measurements using a PMT for LIF signal detection. The detection system was calibrated to yield absolute concentrations of SiO via simultaneous line-of-sight absorption measurements. Such a strategy is facilitated under sufficiently absorbing conditions (thus sufficiently high volumetric SiO concentrations), which in their work was achieved in flames under atmospheric pressure [7]. Under low-pressure conditions, such as those studied here, the absorption is too weak for accurate absorption measurements. Alternatively, Motret et al. [16] determined absolute SiO concentrations in dielectric barrier discharges in Ar/SiH4/N2O mixtures based on optical emission spectroscopy (OES) and calibrated detectors. Although such a scheme obviates the need for a laser, OES suffers from uncertainties originating from self-absorption and non-equilibrated populations, therefore requiring much care with spectral interpretation and also lacks in spatial resolution.
Laser-induced fluorescence measurements allow for local measurements and with the use of a camera as a detector, LIF achieves 2D spatially resolved imaging capabilities. The challenge of LIF, however, lies in the approach to interpret signal in terms of absolute concentrations or species number densities. When using LIF, and after selecting an interference-free transition and accounting for thermal population and collisional quenching (thus, fluorescence quantum yield), intensity calibration relative to a known standard is necessary towards absolute measurements.
Most importantly for quantitative LIF, proper line selection is crucial to avoid problems associated with spectral interference from either the target species in question, herein SiO, or from other species simultaneously present in the combustion gases. In our work, we investigate potential interference within the LIF process by measuring excitation-emission spectra covering the entire range potentially relevant for laser excitation and LIF signal detection. Additional to laser-induced interference, contribution from spontaneous emission from SiO at the flame conditions and other species (including thermal radiation from hot particles) in the flame environment is also accounted for during recording and can also be reduced by short detector-gate times. A line that also offers strong intensity is beneficial towards higher signal-to-noise in the final absolute concentration result and also allows easy discrimination against spontaneous emission from the flame environment.
Selection of a line that offers low temperature sensitivity in the expected temperature range also helps—but over an extended temperature range, the local temperature still needs to be measured for proper correction of the local variation in ground-state population of the probed state. In this work, we use a wavelength-scanning imaging approach for measuring the temperature images via multi-line thermometry based on SiO LIF [17].
Various approaches have been suggested for avoiding the influence of collisional quenching in LIF measurements. Saturation LIF [18, 19] is intrinsically quenching-independent, but care is needed due to simultaneous occurrence of saturated and non-saturated contributions from an uneven spatial and temporal laser beam profile. Also, for full saturation, laser fluences are required that might interact with local chemistry and thus change the system while being studied, e.g., by photolysis of water. Using laser-induced predissociative fluorescence (LIPF), the upper excited state of the monitored species is led to rapid dissociation (on a ps time scale) due to potential-energy curve crossing with a repulsive state. The resulting short excited-state lifetime prevents energy transfer through collisions [20, 21]. This technique has limited applicability (met for, e.g., OH [22, 23] and O2) and is not applicable to SiO.
For LIF in the non-saturated linear regime, information about local quenching is required. This is also important for semi-quantitative measurements when the fluorescence lifetime changes over the observed area or in between various operating conditions. Here, knowledge about relative variations in quenching is sufficient. If the fluorescence quantum yield is not part of the calibration strategy (such as via the calibration with LIF measurements under reference conditions or with comparative measurements against absorption data), absolute information about the fluorescence quantum yields is required. In this work, we determine 2D maps of the fluorescence lifetime (thus collisional quenching) by time-stepping the detection of SiO LIF relative to the excitation laser.
For quantitative interpretation of the resulting (corrected) LIF signal intensities, various options exist, most of them however are not applicable to an unstable intermediate such as SiO which exists at low concentrations in the probe volume only. For LIF of stable molecules, measurements under well-controlled conditions can be used as a reference for calibration, such as gas mixtures in the case of application of organic tracer molecules [24] or flames where the species of interest is added in small concentrations under conditions where it does not significantly interact with the local reactive environment such as NO added to lean flames [25].
Under specific conditions, self-calibrating systems have been reported. For example, when the absorber concentration or pressure is high enough, absorption will manifest itself in the LIF signal along the beam path. This approach has been used for calibrating Fe–LIF measurements with excitation of two bands with significantly different absorption cross-sections [26]. Also, the back-and-forth technique introduced by Versluis et al. [27] relies on this principle and works well with strong absorption, whereby two counter-propagating laser beams excite a target species in a mutually shared plane. This technique has proven to be useful with OH and NO under atmospheric pressure [27, 28]. In both cases, it is important, that the species generating LIF signal is the only one significantly contributing to laser attenuation. In the case of low absorption, e.g., low-pressure conditions, LIF calibrated by cavity ring down (CRD) absorption can also be used [29, 30] owing to its high sensitivity. Such potential utility of this approach is offset by special care needed to couple the TEM00 mode of the laser beam into the cavity; once achieved, the spatial resolution determined by the beam waist is typically limited to ~ 1 mm. Furthermore, use of a cylindrical beam necessitates stitching successive images together to achieve an equivalent 2D fully resolved image. Reaction chambers designed for nanoparticle synthesis flames also experience window fouling, and would pose significant challenges to CRD measurements where high-quality cavity-mirrors are extremely sensitive to the slightest degradation in reflectivity.
The problems associated with the above-mentioned calibration methods with unstable intermediates under low-pressure conditions can be overcome by using photometric calibration for the laser/detector system used. This, however, then requires a full quantitative assessment of the LIF process, including absorption cross-sections, ground-state populations and fluorescence quantum yields. If the photometric calibration is performed against calibrated light sources, the influence of variation of the optical path and filters must also be considered. This can be avoided if a method is used that applies as many of the components of the final detection system as possible. Potential reference schemes are by spontaneous Raman or Rayleigh scattering, which have been used, e.g., for quantifying acetone and 3-pentanone fluorescence intensities in heated cells [31] or for calibrating CH– and C2–LIF measurements [32,33,34,35]. Care must be taken to correctly consider the polarization-dependence of the Rayleigh signal (in contrast to the isotropic character of the LIF signal).
Alternatively, LIF of a well-characterized species that can be added to the environment of interest (e.g., NO) can also be used to perform the same task. In this study, we use Rayleigh scattering as well as NO LIF for calibrating our detection setup—pressurizing the chamber with N2 for Rayleigh or a predetermined NO/Ar mixture for NO LIF from a well-characterized rovibronic line [36, 37] at ambient temperature. Both methods have the advantage that the same laser beam is used as for the SiO–LIF measurements, solving for open questions of the probe volume that would otherwise need to be addressed by a photometric calibration with a calibration lamp. The NO–LIF approach provides the additional advantage that with slight detuning of the laser to an NO resonance, the filter setup used for discriminating SiO LIF against elastic scattering can be maintained. Also, unlike Rayleigh scattering, NO LIF provides the same polarization characteristics as SiO LIF thus reducing one additional error source. On the other hand, Rayleigh scattering prevents the need to quantify collisional quenching of the reference NO–LIF signal. Here, we compare both approaches.
Together with SiO–LIF imaging measurements, concentration distributions of the intermediate SiO can be quantified absolutely in our low-pressure (i.e., 3 kPa) stationary silica-synthesis flames. Furthermore, all such measurements can be performed using a single camera setup for the detector calibration, SiO LIF, in addition to the supporting temperature and effective lifetime measurements through a combined wavelength and detector delay-time tuning approach that yield combined temperature, fluorescence quantum yield and, hence, absolute SiO concentration images.
2 Data analysis strategy
In this work, we excite the target SiO molecule in the linear regime using laser radiation. For a two level system, and given the emission is integrated over all time after the laser pulse of energy \(~E\), the spectrally integrated LIF signal \(~{I_{{\text{LIF}}}}\) is [38]:
where the number density of SiO can be related to the mole fraction, \(~x\), via the ideal gas law: \(~{n_{{\text{SiO}}}}=px/kT\). Temperature \(T\) governs the Boltzmann fraction\(~{f_{\text{B}}}\), as well as the overlap integral \(~\Gamma\) and the fluorescence quantum yield \(~{\tau _{{\text{eff}}}}/{\tau _0}\). Constants associated with excitation include the Einstein absorption coefficient \(~B_{{12}}^{J}\) and the laser linewidth \(~\Delta {\nu _{\text{L}}}\); and for detection, the product \(~\Omega \varepsilon \eta l\) consists—from left to right—of the detection solid angle, the transmission efficiency, the wavelength dependent detection sensitivity of the camera's detector and the length of the probe volume, respectively. The above relation is valid for small laser pulse energies \(~E\), whereby the LIF signal is linear with energy such that a normalized signal can be defined \(~{S_{\text{F}}}={I_{{\text{LIF}}}}/E\). With this, the following sections outline how the SiO mole fraction can be inferred from experiments and data analysis invoking the above linear LIF relation.
2.1 Relative concentration determination
The expression for relative concentration is given by (2), comprising terms for the energy-normalized laser-induced fluorescence (LIF) signal \({S_{\text{F}}}\), temperature \(T\), and fluorescence lifetime \(~{\tau _{{\text{eff}}}}\).
Such a measure of concentration can be forced to be relative to any predefined basis. Each term is explained in turn below.
2.1.1 Fluorescence measurement
The often-used relation above (Eq. 2) is based on measuring the fluorescence \({S_{\text{F}}}\) corresponding to exciting the peak of an isolated line in the excitation spectrum of SiO after being normalized by the laser-pulse energy. The overlap factor \(\Gamma\) must also be considered, to account for the finite linewidth of the laser relative to the absorption linewidth [33, 39]. This factor is dimensionless and is the product of the full width at half maximum (FWHM) of the laser profile \(\Delta {\nu _{{\text{laser}}}}\) and the overlap integral \(\int_{{ - \infty }}^{\infty } {{g_{{\text{laser}}}}~{g_{{\text{line}}}}~{\text{d}}\nu } ~\); the latter is evaluated at the peak position of the convolved product of the area-normalized laser and frequency-dependent absorption line profiles, \(~{g_{{\text{laser}}}}(\nu )\) and \({g_{{\text{line}}}}(\nu )\), respectively. The ratio \({S_{\text{F}}}/\Gamma\) in Eq. (2) can be evaluated directly via integration of the line profile \(S(\bar {\nu })\) in the excitation spectrum, given by Eq. (3):
In this work, we perform spectral scans across the Q(42) line of the A1Π − X1Σ (1, 0) electronic band system to evaluate the integral, where in situ spectral scans are viable under our steady-state conditions when using a laser with a low repetition rate of 10 Hz. However, it is also feasible to extend our diagnostics capability towards instantaneous measurement for unsteady systems under observation. This can be done in principle by pre-evaluating \(\Gamma\) through the flame, and then measuring only the peak fluorescence \(~{S_{\text{F}}}\) on a single laser-shot basis. Although this strategy offers the capability for instantaneous measurements, it is liable to incur greater uncertainty from imprecise spectral positioning of the peak position of the line. Furthermore, at higher pressures collisional broadening becomes important whereupon \(\Gamma\) starts to depend on local composition, and therefore can no longer be simply correlated with temperature only—as would be the case in the low-pressure Doppler-broadened regime.
2.1.2 Temperature correction
For interpreting the LIF signal intensity, temperature information is essential to account for both the temperature term in the ideal gas law and the population of the lower level of the rovibronic transition. Within a flame, where conditions are typically thermally equilibrated, the Boltzmann distribution can be used to calculate the fraction of SiO molecules \({f_{\text{B}}}\) with the desired initial state for excitation. In this work, spatially resolved 2D temperature imaging was performed using multi-line SiO–LIF thermometry that pixel-wise evaluates a sequence of imaging measurements while scanning the laser wavelength across multiple SiO–LIF transitions with different ground-state energies [17]. The resulting temperature images enable to correct for the proportion of SiO molecules contributing to the fluorescence signal, through the fB/T term in Eq. (2).
2.1.3 Collisional quenching correction
Owing to variations of temperature and chemical composition within a flame, collisional quenching is spatially non-uniform. The amount of quenching can be spatially quantified by measuring the effective fluorescence lifetime throughout a flame. The lifetime \({\tau _{{\text{eff}}}}\) is inversely related to the quenching rate \(~Q\), given by Eq. (4), where the Einstein \({A_{jk}}\) coefficients are summed over all de-excitation pathways:
Under low-pressure flame conditions, the rate of collisions is reduced leading to lower rates of quenching such that \(Q~\) is of the same order of magnitude as \(\sum\nolimits_{k} {{A_{jk}}}\). This facilitates the direct quantification of quenching through measuring the lifetime \({\tau _{{\text{eff}}}}\) of the fluorescing target species via time-resolved LIF, where lifetimes are correspondingly longer and can be measured more accurately.
Conventionally, a fast photomultiplier tube (PMT) is employed to measure the decaying fluorescence signal after excitation with a laser pulse. Firstly, an instrument function is measured by recording the temporal profile of the signal obtained from elastically scattered light originating from the laser pulse. This represents the temporal duration of the pulse (i.e., FWHM = 7 ns) and the time response of the detection system (PMT rise-time: 0.78 ns). Secondly, the time-resolved fluorescence of exciting SiO in the flame is recorded using the PMT. By employing a mono-exponential decay curve and convolving with the instrument function, a synthetic fluorescence transient can be fitted allowing lifetimes to be evaluated within the flame. In this study, we instead use a fast-gated (5 ns minimum width) ICCD camera to perform lifetime measurements in imaging mode. This way, and together with the 30 mm high laser sheet, time-resolved images of the decaying fluorescence were resolved in 2 ns increments. This was done by scanning the delay between the laser pulse and recording the LIF signal. In a similar way to the PMT approach, pixel-wise decay curves were acquired and then fitted to extract lifetimes, \(~{\tau _{{\text{eff}}}}\).
2.2 Calibration
The expression for absolute mole fraction in Eq. (5) is similar in form to (2) except for the additional determination of: the absorption coefficient, \(B_{{12}}^{J}\) (m3/J/s2), and natural lifetime, \({\tau _0}\) (s), for line Q(42) of SiO; system pressure, \(p\) (Pa), and the detection calibration term \(C\):
It should be noted that the fundamental spectroscopic constants \(B_{{12}}^{J}\) and \({\tau _0}\) can normally be measured easily in the case of sufficiently isolated lines in the excitation spectrum. However, in our study the SiO radical is an intermediate which is only formed in situ, and hence absorption cells with a priori known amounts of SiO cannot be used to determine \(~B_{{12}}^{J}\). Parameters \(B_{{12}}^{J}\) and \({\tau _0}\) evaluated from theory, as well as from other experimental attempts, harbor uncertainties which will be the subject of discussion later in the paper.
Detection channel calibration, as expressed through \(~C\), can conventionally be realized via Rayleigh or Raman scattering or well-characterized fluorescence, such as NO LIF. Such a scheme is straightforward and useful provided signal strength is sufficiently high and that (in case of using Rayleigh signal) unwanted elastic scattering from surfaces can be minimized. The term \(C\) corresponding to Rayleigh calibration is expressed in Eq. (6), with \(\partial \sigma /\partial \Omega\) denoting the scattering cross-section of N2 calculated from [40] (20.6 × 10−27 cm2/sr at 231 nm) at a viewing angle orthogonal to the incident laser beam of wavenumber \(\bar {\nu }\) and spectral width \(~\Delta {\nu _{{\text{laser}}}}\). The calibration is performed by filling the reactor chamber with N2 gas of temperature \(~T\) (typically 295 K) to a series of known pressures. For each pressure, a Rayleigh image is recorded resulting in pixel-wise evaluation of \(\partial {S_{\text{s}}}/\partial p\) as a function of HAB from the stack of images.
Alternatively, NO LIF at ambient conditions can also be exploited for calibration purposes. Equation 7 comprises terms analogous to the SiO case, but now for line S21(17.5) in the A2Σ+ − X2Π (0, 0) band near 225 nm of NO. In this case, a mixture of NO in Ar (2000 ppm) was filled to \(p=\) 3 kPa in the chamber at room temperature (295 K). In this calibration scheme, an optical filter is fixed between the reactor and camera—designed to suppress light of scattered incident wavelength for both the SiO–LIF measurement and NO–LIF calibration. Owing to slightly different emission spectra between SiO and NO, the latter passes 18% more light through our filter with all else being equal; this is expressed in our filter ratio (i.e., \(~F\) = 1.18) in Eq. (7):
Considering its ease of use and relatively strong signal, NO LIF is an attractive alternative and is facilitated by its comprehensive spectroscopic characterization in the literature [41, 42] with good knowledge of \(~{f_{\text{B}}}\), \({\tau _0}\) and \(B_{{12}}^{J}\) for line S21(17.5). Furthermore, given that NO and SiO possess similar molecular structure their emission spectra are very similar. Exciting the S21(17.5) line of NO at 225 nm, and the Q(42) line of SiO at 231 nm exhibits a good degree of overlap of emission bands in the range 230–290 nm [11, 43]. Such overlap enhances the accuracy of the detection channel calibration owing to the same spectral region of the camera’s response curve and detection channel transmission being employed.
3 Methodology
3.1 Experimental setup
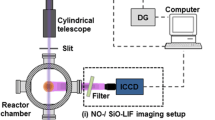
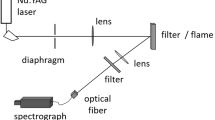
Particulars of the setup employed in this study have already been described in Refs. [8, 9]. Briefly, the setup shown in Fig. 1 comprises the reactor, laser system, and detection modules. A vertically translatable burner is mounted on a water-cooled (303 K) flange, which is housed in a cylindrical chamber of diameter 10 cm. The burner consists of a sintered-bronze matrix of 36 mm diameter, which can stabilize upward-burning premixed flat flames. The surface temperature of the matrix is monitored with a thermocouple attached to the burner rim. The SiO2 synthesis flame is stationary without temporal variations and typically operated at a chamber pressure of 3 kPa with flow rates of the feed gases summarized in Table 1.

Schematic of the arrangement for: (i) NO- and SiO–LIF imaging with an ICCD camera; (ii) SiO fluorescence lifetime measurements using a PMT, and (iii) spectrally resolved measurements of SiO LIF using a spectrometer. FCU frequency conversion unit, FG delay generator, ICCD intensified charged-coupled device, PMT photomultiplier tube
The vapor-phase precursor HMDSO (hexamethyldisiloxane) was introduced to the feed gases via the use of a bubbler, which is independently characterized in terms of evaporation rate and thus HMDSO concentration in the feed gas by mass-loss measurements. NO could be added to the flame gases for corresponding NO–LIF measurements. Optical access to the flame is possible via two rectangular (50 × 25 mm2) quartz windows for transmission of the laser sheet, while fluorescence can be observed perpendicular to the laser sheet through a 90-mm diameter quartz window.
SiO and NO were excited with the light generated by a narrowband tunable dye laser system in combination with frequency conversion. A dye laser (Sirah, Cobra Stretch, 0.24 cm−1 FWHM linewidth) was operated with Coumarin 47 and pumped by the tripled output (355 nm) of a flashlamp-pumped Nd:YAG laser (Spectra Physics, Lab Series, 10 Hz repetition rate), producing tunable radiation between 435 and 485 nm. UV radiation for exciting SiO and NO between 218 and 243 nm was generated by frequency-doubling in the thermally stabilized BBO crystal of the frequency conversion unit.
The laser beam was formed into a vertical light sheet with a 30 × 1 mm2 cross-section by a micro-lens array beam homogenizer (Limo) and a focusing lens before being steered with a 90° quartz prism into the burner chamber. LIF measurements were restricted to 0–30 mm height above the burner plate (HAB). Pulse-to-pulse intensity fluctuations were recorded by an energy monitor (LaVision) and stored with each image for subsequent intensity correction. LIF images were collected perpendicular to the light-sheet plane and focused using a UV-lens (B. Halle, f = 100 mm, f# = 2) onto an intensified CCD camera (LaVision, Imager Intense, 1375 × 1039 pixel sensor with a high-speed intensifier LaVision, HS-IRO) yielding a projected pixel size of 40 µm/pixel and a gate width set to 500 ns. Elastically scattered light was suppressed by custom-made longpass filters to transmit from beyond 225 to 235 nm for NO A − X (0, 0) and SiO A − X (0, 0) LIF measurements, respectively. Typical laser fluences of up to 0.1 mJ/cm2 were used, corresponding to laser-pulse energies of ~ 25 µJ/pulse at the measurement volume, resulting in linear LIF behavior.
3.2 SiO–LIF line selection
The choice of SiO line to excite was determined by both practical and theoretical considerations. In this work, we chose A ← X (1, 0) of SiO, whose bandhead lies at 43,486 cm−1 (229.96 nm) with a wavelength region readily accessible using our dye laser. The (1, 0) band delivers strong fluorescence owing to excitation originating from the lowest vibrational level (i.e., v″ = 0) and due to a high Frank–Condon factor; both aspects leading to stronger absorption of laser radiation. Although strong absorption is advantageous for improving LIF signal-to-noise ratios on the one hand, too much absorption can lead to distortion in the final inferred concentration profiles using the photometric calibration method for the laser/detector system in this work. For our axisymmetric low-pressure flame with a small path length, it was confirmed that the laser-beam attenuation was negligible which was evident from the symmetry of the SiO–LIF images in our experiments. However, in general, absorption effects need to be assessed based on measurement conditions thus guiding the appropriate choice of wavelength region. In the following, we examine the possibility of spectral interference from other species in the flame relevant to the (1, 0) band, as well as selecting a more specific spectral region that yields good precision in SiO mole-fraction determination.
For selective measurements of SiO, interference from LIF of OH, hot O2 [44], or other intermediate species in the flame must be ruled-out. This is achieved by mapping-out the LIF signal via an excitation–emission chart, as illustrated in Fig. 2. After excitation of three example lines in A − X (1, 0) in the 230.9–231.1 nm range, emission is seen from A − X (1, 0) in addition to the red-shifted (1, 1), (1, 3), (1, 4), (1, 5), (1, 6), and (1, 7) bands (λ = 231–292 nm). It should be noted that the (1, 2) emission band is missing due to a forbidden transition. Figure 2 also indicates, at least for the measurement corresponding to HAB = 10 mm and HMDSO concentration of 50 ppm, that spectral interference is not evident owing to lack of irregular features in the chart for these conditions. If interference were present, features other than from SiO would appear in the emission spectrum shown on the horizontal-axis in Fig. 2.

Excitation–emission chart for the A − X (1, 0) band of the flame at 3 kPa with 50 ppm HMDSO. Each horizontal band in the chart represents the spectrally resolved emission. The arrow in the excitation spectrum indicates the position where the emission spectrum was extracted. The arrow in the emission spectrum indicates the emission where the excitation spectrum was extracted
The excitation spectrum in Fig. 2 highlights the chosen line Q(42) amidst neighboring lines—representing good mutual separation at this excitation wavelength within the (1, 0) band. Figure 3a shows the Q(42) transition in the A1Π–X1Σ+ (1, 0) SiO band at 231 nm in more detail—used in this study for assessing the concentration levels (i.e., mole fraction) of SiO within the synthesis flame. The (1, 0) band offers the strongest signals for SiO, whose excitation wavelengths are easily accessible using common dye laser systems. Excitation of a rovibronic line from the Q-branch further enhances the resulting LIF intensity. It should be noted that an excitation scan, whilst not essential in our scheme, is a preferred way of measuring SiO LIF owing to reduced uncertainty in finding the peak position as well as higher effective signal-to-noise in the evaluated signal. In this fashion, LIF images were captured with the camera and averaged over 50 laser pulses, then repeated again for an incremental change in the laser wavelength over the spectral range shown in Fig. 3a. Excitation spectra were then extracted for each pixel from the stack of LIF images. Figure 3a shows an example excitation spectrum fitted with a Gaussian profile corresponding to our low-pressure conditions of 3 kPa for an axial pixel position of 10 mm HAB in the flame. Despite a good fit to the experimental spectrum, there is evidence of a small degree of interference on the right wing of the line profile. Such interference is to be expected to originate from overlapping bands of SiO with (1, 0) used in this study.

a Gaussian fit to isolated rovibronic line Q(42) at 230.99 nm in the A1Π ← X1Σ+(1, 0) system of SiO to infer concentration. b Fit of SiO–LIF excitation spectrum using LIFSim near 235 nm in the A1Π ← X1Σ+ (0, 0) system of SiO to infer temperature
Notwithstanding signal strength and a good degree of spectral isolation, line Q(42) was selected also on its temperature insensitivity when aiming to measure mole fractions—resulting in greater precision of concentration measurements after temperature correction. It is noteworthy that selecting for low temperature sensitivity depends on the ultimate quantity to be measured. In the case of concentration measurements, either mole fraction (x/ppm) or number density (n/m−3) can be used. Each quantity requires the appropriate line to be selected for temperature insensitivity; for n, Boltzmann fraction fB should be invariant with temperature T, whereas for x, the choice is fB/T. Such dependence is evident from Eq. (2), derived from the linear LIF signal (i.e., Eq. (1)). Figure 4 shows the temperature dependence of the two Boltzmann-related variables for line Q(42), manifesting that our line choice is appropriate for mole-fraction measurement (red curve) given the temperature range in our flames. Flame temperature was measured using SiO-thermometry, as described in detail in Ref. [17], using a set of well-separated lines near 235 nm shown in Fig. 3b. LIFSim was applied to perform local spectral fitting to derive an image of temperature, which was then used to perform the above-mentioned temperature correction to the LIF signal.

Boltzmann dependences of the selected rovibronic line Q(42) with temperature for SiO. The blue solid line corresponds to the Boltzmann fraction, fB; the pink dashed line for temperature-normalized Boltzmann fraction, fB/T. For comparison, the respective dependences for the line P(37) are also shown
4 Results and discussion
The following sections discuss each of the correction procedures applied to the raw SiO–LIF signal towards final derivation of a 2D spatially resolved image of absolute SiO mole fraction. Such corrections range from photometric calibration, having the greatest impact owing to inherent light sheet normalization, to fluorescence lifetime considerations.
4.1 Photometric calibration
NO LIF excited at S21(17.5) was used to perform the photometric calibration, with an example image recorded by the camera shown in Fig. 5a—averaged over 50 laser shots. The image was taken using a gate width of 500 ns, which is sufficient to capture the entire LIF signal after the laser pulse (FWHM = 7 ns). The photometric calibration was performed at conditions of ambient temperature of 295 K, chamber pressure of 3 kPa, and with a gentle flow of 2000 ppm NO/Ar mixture through the reactor. The centerline shown in Fig. 5a represents where a column of binned pixels was used to extract a vertical cross-section as shown in Fig. 5b. Each superpixel corresponds to a width of 2.24 mm and height 0.32 mm.

a Averaged image of NO–LIF/Rayleigh signal at ambient conditions for photometric calibration, showing centerline over which a column of binned pixels was used. b Centerline profiles of NO LIF, as well as from N2 Rayleigh scattering, based on the reciprocal of the calibration term and common units. c Plot of Rayleigh signal as a function of N2 pressure in the reactor chamber
The intensity structure of the laser sheet resulting from the use of the beam homogenizer is evident from both the image and centerline profile seen in Fig. 5a, b, respectively. Because of the homogeneous distribution of NO, these calibration measurements are also ideal for measuring the light sheet inhomogeneity and are used for flat-field correction of the SiO–LIF measurements. Along the centerline, the NO–LIF signal intensity is mostly uniform between the two horizontal bright edges, with this inner region being used for the measurements in this study. In these measurements, NO LIF is in the linear regime of excitation, which was verified by repeating the measurement with various laser intensities. The observed intensities in Fig. 5 also depend upon the detection efficiencies of each pixel in the imaging system—noting that this is the primary purpose of the photometric calibration. The calibration in this study is therefore composed of contributions from both laser-sheet intensity structure and detection efficiency, which are both embedded within the signal term \({S_{\text{F}}}\) of the photometric calibration term \(C\) in Eq. (7). When the calibration term is multiplied in Eq. (5), with the aim of yielding absolute mole fraction of SiO, the two above-mentioned contributions cancel due to the same laser sheet and imaging system being used for both calibration and SiO LIF measurements.
In a similar fashion, Rayleigh scattering measurements based on pure N2 were also investigated for the purpose of comparing side-by-side the two approaches towards photometric calibration. Using Rayleigh scattering also results in a qualitatively similar image as shown in Fig. 5a. For such calibration, the change of scattering signal as a function of N2 pressure in the chamber (i.e., \(\partial {S_{\text{S}}}/\partial p\)) is required as part of the calibration term \(C\) in Eq. (6). Figure 5c shows an example plot of how scattering signal varies with N2 pressure for a given superpixel on the centerline. Owing to Rayleigh signals being typically three orders of magnitude weaker than those of LIF, it should be noted that more points are therefore required to more accurately infer the gradient \(~\partial {S_{\text{S}}}/\partial p\). It was found in this work that a minimum of ~ 5 points was required using Rayleigh to reproduce the same shape of profile as for NO LIF seen in Fig. 5b. Similar to the NO–LIF calibration, the localized signal depends directly on the laser intensity, which determines the gradient of the plot in Fig. 5c. However, the gradient is also governed by the degree of polarization of the incident laser beam. Since a Rayleigh scattering signal can only be detected with the electric field component of the incoming laser beam having a non-zero projection onto the imaging plane, the intensity of the Rayleigh signal and hence the gradient \(\partial {S_{\text{S}}}/\partial p\) depends on how much of the incident light is vertically polarized. In our setup, 100% horizontal polarization would result in zero signal. Figure 5b also shows the corresponding centerline profile for Rayleigh scattering based on a common scale (i.e., the reciprocal of the calibration term for each of the calibration approaches). By comparing both scales, the profile derived from the Rayleigh method is 94% in magnitude to that of the NO–LIF method. Assuming accurate NO–LIF calibration, the mismatch between profiles in Fig. 5b is most likely attributable to a degree of depolarization of the incident laser beam caused by the use of optics (e.g., homogenizer, lens, prism, etc.).
4.2 SiO–LIF profiles
The abundance of SiO in our flames is apparent from the vertical extent of the LIF signal seen in Fig. 6a. Such an SiO–LIF image corresponds to the case of 2050 ppm HMDSO loading to the base flame (ϕ = 0.6) averaged over 50 laser pulses, exhibiting a peak signal-to-noise ratio of ~ 20 within the unbinned image. There is an obvious gap immediately above the burner plate showing little SiO LIF, being consistent with the finite time needed for sufficient decomposition and oxidation of HMDSO towards SiO generation [9].

a Averaged image of SiO–LIF on laser sheet illumination for HMDSO loading of 2050 ppm, b centerline profiles of both SiO LIF and light-sheet normalized LIF
A high degree of symmetry is also evident in the raw SiO–LIF image in Fig. 6a in spite of a slight divergence of the light sheet in the direction of propagation shown in Fig. 5a, which would contribute to the reduction of the local laser fluence. Furthermore, symmetry is also maintained in spite of small, but finite, absorption of the laser radiation from the SiO along the light path under these conditions (i.e., 3 kPa, 2050 ppm HMDSO, ϕ = 0.6), hence supporting our rationale for adopting the photometric calibration approach for determining absolute SiO concentrations.
Figure 6b shows the result of normalizing the raw SiO LIF by the NO–LIF photometric calibration image, thus removing the laser sheet structure from the profile. After this first stage of correction, the image of normalized LIF unveils the existence of a prominent band of relatively high SiO concentration near to the burner surface—it is interesting to note that this structure correlates to the visible chemiluminescence seen in the naked flame. Centerline profiles of raw and normalized LIF are plotted in Fig. 6b both showing a rise from zero value at the burner surface. The raw LIF profile rises and falls via two peaks, and it should be noted that accurate measurement requires sufficient spatial resolution. The obvious advantage of using a camera to measure the LIF profile is the ability to faithfully capture the rapid swings in the profile that would not otherwise be easily reproduced via the use of a PMT and shifting the burner in coarse increments.
4.3 Temperature correction
Commonly, NO–LIF based thermometry is invoked to spatially resolve temperature in flames—suitable especially under fuel-lean conditions. However, flames doped with silicon-based precursors, such as HMDSO in our case, release SiO as an intermediate which spectrally interferes with NO—thus rendering NO thermometry impractical. Alternatively, the SiO native to such HMDSO-doped flames can be usefully employed as an interference-free temperature marker [17]. Here, we used LIFSim to generate a 2D distribution of temperature—as displayed in Fig. 7a—for the flame (ϕ = 0.6) doped with 2050 ppm HMDSO resulting from fitting of SiO excitation spectra, exemplified by Fig. 3b. Figure 7a shows a clear thermal distribution typical of premixed flat flames, surrounded by a region of low signal-to-noise. It would be expected for this outer region that there are diminished levels of SiO further downstream where SiO is later consumed towards final production of SiO2. On the other hand, in the region immediately adjacent to the burner plate, low signal-to-noise in Fig. 7a is due to insufficient presence of SiO during its nascent production. In spite of the above-mentioned advantages of SiO-thermometry, it is clear that the use of SiO as a temperature marker is limited to the regions where it is present in sufficient quantity.

a Temperature field of the doped flame (2050 ppm HMDSO) derived from SiO–LIF thermometry. The black dashed line indicates the centerline, whose profiles for temperature (blue plot) and Boltzmann ratio (violet plot) are shown in b. The temperature profile of the undoped base flame (red plot) measured using NO–LIF thermometry is also shown for comparison
The black dashed centerline in Fig. 7a represents where a column of binned pixels was used to take vertical profiles from the temperature field—as displayed in Fig. 7b. Each superpixel corresponds to a width of 2.24 mm and height 0.32 mm, as outlined earlier. The temperature profile of the undoped base flame was measured using standard NO–LIF thermometry [25] as shown by the red plot in Fig. 7b. Such a profile is consistent with previous measurements on this premixed lean flame [8, 9]—corresponding to an adiabatic temperature of 2380 K. This base-flame profile can be compared to that generated from the SiO-thermometry approach used for our doped flames; Fig. 7b also displays an example temperature profile for the 2050 ppm HMDSO case. In our experiments, the profile for the doped case shows a marked earlier rise in temperature compared to the base flame, until it reaches a similar peak temperature to that of the base flame of ~ 1250 K at around 13 mm HAB. The earlier rise of flame temperature is attributable to the extra hydrocarbon content added to the flame from the HMDSO dopant, which results in an increase in effective equivalence ratio from 0.6 to 0.7 in the case of 2050 ppm HMDSO. Figure 7b shows how the Boltzmann ratio fB/T, as discussed in Sect. 3.2, varies across the doped flame. Despite the appreciable temperature rise from 800 to 1250 K, the Boltzmann ratio remains relatively invariant, which is useful for minimizing the temperature dependence for the SiO mole fraction determination.
4.4 SiO–LIF lifetime correction
The final correction required for quantification of SiO in our flames takes account of the collisional quenching of the excited molecule. Figure 8 shows three example images of SiO LIF taken in 6 ns increments after the laser pulse using the shortest camera gate available (i.e., 5 ns). The temporal dynamics of the LIF process, temporally fitted for an individual pixel, is illustrated below via a fluorescence decay curve in Fig. 8.

Illustration of how fitting was performed to extract reliable lifetimes. Solid lines denote PMT measured signals for a single point in the flame. Black squares show times after the laser pulse where measurements were taken with the camera; three examples of instantaneous SiO LIF captured with a 5-ns camera gate are shown in 6-ns increments
The blue curve in Fig. 8 portrays a notional rise and fall of SiO LIF during and after the laser pulse. The green curve shows a notional pulse–response function (PRF), which combines both the dynamics of the laser pulse along with the temporal-response characteristics of the detection system. The green curve is outlined by measuring elastic scattering from the chamber—averaged over 50 laser pulses—using the shortest gate width of the camera (5 ns) and then sequentially scanning over the profile of the PRF by tuning the delay between the laser pulse and camera acquisition. Figure 8 shows that the FWHM of the PRF is 12.5 ns—and given that the FWHM of the laser pulse has previously been measured as 7 ns using a PMT with a rapid rise time of 0.78 ns [17]—it can be inferred that there is a significant influence on the PRF width other than from just the laser pulse duration in the case of using a fast camera. To better understand the contributions to the PRF width, it can be envisaged that the PRF shown in Fig. 8 is the result of a convolution of the (1) laser pulse, (2) camera gate, and (3) jitter contribution. Approximating each function as Gaussian—and knowing the laser pulse and gate width to be 7 and 5 ns, respectively—the jitter results in a contribution of 9 ns. Such jitter originates from non-ideal instability between the laser output and the triggering of the camera.
Conventionally, a monoexponential step-function is convolved with the measured PRF—the result of which is fitted to the experimental decay curve to infer the fluorescence lifetime. However, when the PRF is significantly widened by the above-mentioned contributions for instance, the accuracy of the deconvolution process to extract the lifetime is compromised. Alternatively, a selected part of the decay curve can be fitted without invoking knowledge of the PRF—shown as “Region of fitting” in Fig. 8 immediately after the PRF has diminished. A monoexponential is simply directly fitted to this region of the decay curve—indicated by the red curve in Fig. 8. In this scheme, we first locate the time at the point we can no longer detect the laser pulse using our fast-camera system, and then from this point forward measure the fluorescence decay in small steps. In the lower half, Fig. 8 demonstrates such measurements using 2-ns intervals along this section of the curve. We adopted such a method of lifetime measurement because sampling all of the LIF transient (blue curve), in addition to the PRF (green curve), resulted in erratic lifetime profiles using our camera system. It is also worthwhile noting that in spite of the short lifetimes seen in SiO, the success of using our method for measuring such rapid decay demonstrates potential in less demanding quenching systems with longer lifetimes.
Figure 9 shows the evaluated lifetimes profiled along the centerline with height above burner. There is a weak monotonic dependence with height, despite a significant change in temperature from ~ 500 → 1200 K over the 25 mm range, as seen in Fig. 7b. In view of many influences on the lifetime, it appears from the form of the profile that lifetime is chiefly governed by temperature. This is expected as quenching cross-sections often decrease with temperature, thus promoting rises in lifetimes.

a Two-dimensional distribution of the evaluated fluorescence lifetime and b profile along the centerline for the base flame ϕ = 0.6
4.5 Absolute SiO concentrations
Figure 10 shows the final profile of SiO concentrations, after photometric calibration and correction for temperature and quenching effects. Taking the example case of 2050 ppm HMDSO loading, shown by the purple curve in Fig. 10, the section of the profile closer to the burner surface is scaled-up by normalization with the profile \({f_{\text{B}}}/T\) exhibited in Fig. 7b—in accordance with Eq. (5); similarly, such scaling also holds true for effective lifetime variation. This scaling is consistent with the relative increase in the peak around 3 mm HAB when the blue curve in Fig. 6b is compared to its purple counterpart in Fig. 10. Flames with two lower values of HMDSO loading are also shown (green and orange curves), in which the SiO levels scale roughly in proportion to the loading. Furthermore, the prominent initial peaks in the profiles for 530 and 1230 ppm HMDSO show a slight shift away from the burner surface. This can be explained by the fact that HMDSO precursor also acts as a fuel due to its hydrocarbon content, and therefore slightly increases the effective equivalence ratio of the flame—as evinced by the temperature increases seen in Fig. 7b.

a Example 2D image of SiO absolute mole fraction for our base flame (ϕ = 0.6) with 2050 ppm HMDSO. b Corresponding centerline profiles for all loadings of 530, 1230, and 2050 ppm HMDSO. Profiles are based on the spectroscopic parameters stated in the text
Once all the corrections have been applied, the mole fraction profiles in Fig. 10 can be rendered absolute in magnitude. The remaining parameters in Eq. (5) that govern the mole fraction include the Einstein coefficient for absorption, \(B_{{12}}^{J}\), and natural lifetime, \({\tau _0}\), of the studied transition Q(42) in A1Π – X1Σ+ (1, 0) of SiO. Careful consideration of these parameters is paramount for accurate absolute mole fraction determination, yet there is uncertainty in these values. Firstly, the Einstein coefficient can be computed in principle for diatomics with a reasonable degree of accuracy, however it is good practice to verify such calculations with experiments. Since SiO is an unstable intermediate, it cannot be easily handled and requires generation in situ. However, such generation requires a priori knowledge of the quantity of SiO produced in order to deduce experimentally determined Einstein coefficients. In this study, we therefore adopt a theoretical value of \(5.313 \times {10^{18}}\) m3/J/s2 derived from parameters given in Refs. [11, 45] as a sufficiently good estimation of \(B_{{12}}^{J}\) for our line Q(42) in view of such experimental limitations. For a diatomic molecule, \(B_{{12}}^{J}\) is derived from the product of three key parameters: the electronic transition moment, the vibrational Frank–Condon factor, and the rotational Hönl–London factor. The largest source of uncertainty in such a calculation is the transition moment being approximately 20%.
Secondly, the natural lifetime, \({\tau _0}\), has also shown to harbor uncertainty, ranging from 9.6 [46] to 49.5 ns [47] in the literature. All the surveyed values, except for one [46], were derived from quenching models [47,48,49,50]. In this work, we adopted the mean average of all natural lifetimes documented in the literature resulting in a value of 24.8 ns. Given the above-mentioned uncertainties in both \(B_{{12}}^{J}\) and \({\tau _0}\), it is clear that the measured SiO mole fraction in this study strongly depends on these two spectroscopic parameters. Nevertheless, with view to more accurate future determinations of these parameters, our results herein can easily be rescaled. Based on our current estimations of \(B_{{12}}^{J}\) and \({\tau _0}\), it is interesting to note that the profile for 2050 ppm HMDSO in Fig. 10b peaks at 146 ppm SiO, which is equivalent to 3.6% of the total silicon loading to the flame in this case. Nearly identical proportions hold true for the other two loadings of 1230 and 530 ppm HMDSO. Correspondingly, uncertainties in the measured absolute SiO mole fraction profiles range 5–20% (1 σ), as depicted for the 1230 ppm HMDSO case in Fig. 10b. These originate chiefly from (1) fluorescence lifetime, (2) SiO signal, and (3) photometric calibration contributions. It should be noted that previous comparisons for various precursors have shown strong variations in the part of the silicon inventory that passes through SiO in the flame. With tetramethylsilane (TMS), for example, up to 8-times higher SiO concentrations per Si inventory were found compared to experiments in similar flames doped with HMDSO indicating that the structural motif of the precursors significantly influences the subsequent chemical pathways [9]. Despite these low concentrations, strong SiO–LIF signals can be observed that enable measurements with good signal-to-noise ratios. This can be attributed to the exceptionally large Einstein coefficients that are, e.g., two orders of magnitude larger compared to NO, and therefore can deliver significant LIF signals.
5 Summary
A strategy for imaging measurements of absolute mole fractions of the intermediate SiO in silica-synthesis flames has been devised using LIF. Absolute measurements are made possible by calibrating the detection system with a reference; here, we compare both Rayleigh and NO LIF for this purpose. It was demonstrated that room-temperature NO LIF was a convenient and suitable pathway for photometric calibration. For all measurements, including that of temperatures and lifetimes, we use a fast-gated ICCD camera to detect two-dimensional maps via light-sheet excitation in a sizeable region of our flame. Using the same setup throughout is convenient, and facilitates data interpretation. Furthermore, native SiO was also used as the thermometric marker to infer the temperature field, along with measurement of its effective lifetime by using the shortest camera gate width of 5 ns. Our measurements also depend on accurate knowledge of both the Einstein absorption coefficient and the natural lifetime of the chosen transition. Future refinements could be envisioned towards more accurate determination of such spectroscopic parameters for greater reliability in the reported mole fraction values.
References
S.E. Pratsinis, Flame aerosol synthesis of ceramic powders. Progr. Energy Combust. Sci. 24(3), 197–219 (1998)
P. Roth, Particle synthesis in flames. Proc. Combust. Inst. 31(2), 1773–1788 (2007)
S. Li et al., Flame aerosol synthesis of nanostructured materials and functional devices: Processing, modeling, and diagnostics. Progr. Energ. Combust. Sci. 55, 1–59 (2016)
C. Schulz et al., Gas-phase synthesis of functional nanomaterials: challenges to kinetics, diagnostics, and process development. Proc. Combust. Inst. 37, 83–108 (2019)
N.G. Glumac, Formation and consumption of SiO in powder synthesis flames. Combust. Flame 124, 702–711 (2001)
H. Janbazi et al., Response surface and group-additivity methodology for estimation of thermodynamic properties of organosilanes. Int. J. Chem. Kin. 50(9), 681–690 (2018)
M.R. Zachariah, D.R.F. Burges, Strategies for laser excited fluorescence spectroscopy. Measurements of gas phase species during particle formation. J. Aerosol Sci. 25(3), 487–497 (1994)
Feroughi, O.M., et al., Experimental and numerical study of a HMDSO-seeded premixed laminar low-pressure flame for SiO2 nanoparticle synthesis. Proc. Combust. Inst. 36, 1045–1053 (2017)
R.S.M. Chrystie et al., Comparative study of flame-based SiO2 nanoparticle synthesis from TMS and HMDSO: SiO–LIF concentration measurement and detailed simulation. Proc. Combust. Inst. 37(1), 1221–1229 (2019)
T. Dreier, C. Schulz, Laser-based diagnostics in the gas-phase synthesis of inorganic nanoparticles. Powder Technol. 287, 226–238 (2016)
P. van de Weijer, B.H. Zwerver, Laser-induced fluorescence of OH and SiO molecules during thermal chemical vapour deposition of SiO2 from silane-oxygen mixtures. Chem. Phys. Lett. 163(1), 48–54 (1989)
A.J. Hynes, Laser-induced fluorescence of silicon monoxide in a glow discharge and an atmospheric pressure flame. Chem. Phys. Lett. 181(2–3), 237–244 (1991)
R. Yamashiro, Y. Matsumoto, K. Honma, Reaction dynamics of Si(PJ3) + O2→ SiO(XΣ + 1) + O studied by a crossed-beam laser-induced fluorescence technique. J. Chem. Phys. 128(8), 084308 (2008)
D. Goodwin, D. Capewell, P. Paul, Planar laser-induced fluorescence diagnostics of pulsed laser ablation of silicon, in MRS Online Proceedings Library Archive (1995), p. 388
R. Walkup, S. Raider, In situ measurements of SiO(g) production during dry oxidation of crystalline silicon. Appl. Phys. Lett. 53(10), 888–890 (1988)
O. Motret, F. Coursimault, J. Pouvesle, Absolute silicon monoxide density measurement by self-absorbed spectroscopy in a non-thermal atmospheric plasma. J. Phys. D Appl. Phys. 37(13), 1750 (2004)
R.S.M. Chrystie et al., SiO multi-line laser-induced fluorescence for quantitative temperature imaging in flame-synthesis of nanoparticles. Appl. Phys. B Lasers Opt. 123(4), 104 (2017)
J.R. Reisel et al., Laser-saturated fluorescence measurements of nitric oxide in laminar, flat, C2H6/O2/N2 flames at atmospheric pressure. Combust. Sci. Technol. 91(4–6), 271–295 (1993)
P. Desgroux, M. Cottereau, Local OH concentration measurement in atmospheric pressure flames by a laser-saturated fluorescence method: two-optical path laser-induced fluorescence. Appl. Opt. 30(1), 90–97 (1991)
A. Koch et al., Planar imaging of a laboratory flame and of internal combustion in an automobile engine using UV Rayleigh and fluorescence light. Appl. Phys. B 56(3), 177–184 (1993)
E. Rothe et al., Effect of laser intensity and of lower-state rotational energy transfer upon temperature measurements made with laser-induced predissociative fluorescence. Appl. Phys. B Lasers Opt. 66(2), 251–258 (1998)
E.W. Rothe et al., Effect of laser intensity and of lower-state rotational energy transfer upon temperature measurements made with laser-induced predissociative fluorescence. Appl. Phys. B 66, 251–258 (1998)
E.W. Rothe, P. Andresen, Application of tunable excimer lasers to combustion diagnostics: a review. Appl. Opt. 36(18), 3971–4033 (1997)
C. Schulz, V. Sick, Tracer-LIF diagnostics: Quantitative measurement of fuel concentration, temperature and air/fuel ratio in practical combustion systems. Prog. Energy Combust. Sci. 31, 75–121 (2005)
W.G. Bessler et al., Quantitative NO–LIF imaging in high-pressure flames. Appl. Phys. B: Lasers Opt. 75(1), 97–102 (2002)
C. Hecht et al., Imaging measurements of atomic iron concentration with laser-induced fluorescence in a nano-particle synthesis flame reactor. Appl. Phys. B 94, 119–125 (2009)
M. Versluis et al., 2-D absolute OH concentration profiles in atmospheric flames using planar LIF in a bi-directional laser beam configuration. Appl. Phys. B Lasers Opt. 65(3), 411–417 (1997)
C. Brackmann et al., Structure of premixed ammonia + air flames at atmospheric pressure: laser diagnostics and kinetic modeling. Combust. Flame 163, 370–381 (2016)
J. Luque et al., Quasi-simultaneous detection of CH2O and CH by cavity ring-down absorption and laser-induced fluorescence in a methane/air low-pressure flame. Appl. Phys. B 73(7), 731–738 (2001)
S.V. Naik, N.M. Laurendeau, Measurements of absolute CH concentrations by cavity ring-down spectroscopy and linear laser-induced fluorescence in laminar, counterflow partially premixed and nonpremixed flames at atmospheric pressure. Appl. Opt. 43(26), 5116–5125 (2004)
J.D. Koch et al., Rayleigh-calibrated fluorescence quantum yield measurements of acetone and 3-pentanone. Appl. Opt. 43(31), 5901–5910 (2004)
C. Kaminski, P. Ewart, Absolute concentration measurements of C2 in a diamond CVD reactor by laser-induced fluorescence. Appl. Phys. B 61(6), 585–592 (1995)
J. Luque, D. Crosley, Absolute CH concentrations in low-pressure flames measured with laser-induced fluorescence. Appl. Phys. B 63(1), 91–98 (1996)
J. Luque et al., Quantitative laser-induced fluorescence of CH in atmospheric pressure flames. Appl. Phys. B 75(6–7), 779–790 (2002)
W. Juchmann et al. Absolute radical concentration measurements and modeling of low-pressure CH4/O2/NO flames, in Symposium (International) on Combustion (Elsevier, 1998)
W.G. Bessler et al., Strategies for laser-induced fluorescence detection of nitric oxide in high-pressure flames: III. Comparison of A−X strategies. Appl. Opt. 42(24), 4922–4936 (2003)
W.G. Bessler et al., Strategies for laser-induced fluorescence detection of nitric oxide in high-pressure flames. I. A−X (0, 0) excitation. Appl. Opt. 41(18), 3547–3557 (2002)
A.C. Eckbreth, Laser Diagnostics for Combustion Temperature and Species, 2 edn. (Gordon and Breach, Amsterdam, 1996)
S.V. Naik, N.M. Laurendeau, Measurements of absolute CH concentrations by cavity ring-down spectroscopy and linear laser-induced fluorescence in laminar, counterflow partially premixed and nonpremixed flames at atmospheric pressure. Appl. Opt. 43, 5116–5125 (2004)
M. Born, E. Wolf, Principles of Optics (Pergamon. New York, 1980) pp. 393–401
I.S. McDermid, J.B. Laudenslager, Radiative lifetimes and electronic quenching rate constants for single-photon-excited rotational levels of NO (A2Σ+, v′ = 0). J. Quant. Spectrosc. Radiat. Transf. 27, 483–492 (1982)
C. Amiot, R. Bacis, G. Guelachvili, Infrared study of the X2Π v = 0, 1, 2 levels of 14N16O. Preliminary results on the v = 0, 1 levels of 14N17O, 14N18O, and 15N16O. Can. J. Phys. 56, 251–265 (1978)
M. Geier, C.B. Dreyer, T.E. Parker, Laser-induced emission spectrum from high-temperature silica-generating flames. J. Quant. Spectr. Radiat. Transf. 109, 822–830 (2008)
P. Andresen et al., Laser-induced fluorescence with tunable excimer lasers as a possible method for instantaneous temperature field measurements at high pressures: checks with an atmospheric flame. Appl. Opt. 27(2), 365–378 (1988)
H.S. Liszt, W.M.H. Smith, RKR Franck–Condon factors for blue and ultraviolet transitions of some molecules of astrophysical interest and some comments on the interstellar abundance of CH, CH+ and SiH+. J. Quant. Spectrosc. Radiat. Trans. 12, 947–958 (1972)
W.H. Smith, H. Liszt, Radiative lifetimes and absolute oscillator strengths for the SiO A1Π-X1Σ + transition. J. Quant. Spectrosc. Radiat. Transf. 12(4), 505–509 (1972)
J. Oddershede, N. Elander, Spectroscopic constants and radiative lifetimes for valence-excited bound states in SiO. J. Chem. Phys. 65(9), 3495–3505 (1976)
S.R. Langhoff, J.O. Arnold, Theoretical study of the X1Σ+, A1Π, C1Σ− and E1Σ + states for the SiO molecule. J. Chem. Phys. 70(2), 852–863 (1979)
S. Chattopadhyaya, A. Chattopadhyay, K.K. Das, Configuration interaction study of the low-lying electronic states of silicon monoxide. J. Phys. Chem. A 107(1), 148–158 (2003)
I. Drira et al., Theoretical study of the A1Π− X1Σ+ and E1Σ+ − X1Σ+ bands of SiO. J. Quant. Spectrosc. Radiat. Transf. 60(1), 1–8 (1998)
Acknowledgements
The financial support of this project by the Deutsche Forschungsgemeinschaft (DFG) within FOR 2284 (contract DR 195/17-2) is gratefully acknowledged. The authors also thank Torsten Endres, Siavash Zabeti and Usama Murtaza for fruitful discussions and supporting experiments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chrystie, R.S.M., Ebertz, F.L., Dreier, T. et al. Absolute SiO concentration imaging in low-pressure nanoparticle-synthesis flames via laser-induced fluorescence. Appl. Phys. B 125, 29 (2019). https://doi.org/10.1007/s00340-019-7137-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-019-7137-8