
Abstract
1,4-Difluorobenzene (p-DFB) is a promising aromatic tracer for determining concentration, temperature, and O2 partial pressure in mixing gas flows based on laser-induced fluorescence (LIF). Signal quantification requires the knowledge of absorption and fluorescence properties as a function of environmental conditions. We report absorption and fluorescence spectra as well as fluorescence lifetimes of p-DFB in the temperature, pressure, and oxygen partial pressure range that is relevant for many applications including internal combustion engines. The UV absorption cross section, investigated between 296 and 675 K, has a peak value close to 266 nm and decreases with temperature, while still exceeding other single-ring aromatics. Time-resolved fluorescence spectra were recorded after picosecond laser excitation at 266 nm as a function of temperature (296–1180 K), pressure (1–10 bar), and O2 partial pressure (0–210 mbar) using a streak camera (temporal resolution 50 ps) coupled to a spectrometer. The fluorescence spectra red-shift (~2 nm/100 K) and broaden (increase in full width at half maximum by 58% in the investigated temperature range) with temperature. In N2 as bath gas (1 bar), the fluorescence lifetime τ eff decreases with temperature by a factor of about 20 (from 7 ns at 298 K down to 0.32 ns at 1180 K), while at 8 bar the shortest lifetime at 975 K is 0.4 ns. A noticeable pressure dependence (i.e., reduced τ eff) is only visible at 675 K and above. Quenching of p-DFB LIF by O2 (for partial pressures up to 210 mbar) shortens the fluorescence lifetime significantly at room temperature (by a factor of 8), but much less at higher temperatures (by a factor of 1.8 at 970 K). For fixed O2 partial pressures (52 mbar and above), τ eff shows a plateau region with temperature which shifts toward higher temperatures at the higher O2 partial pressures. O2 quenching is less prominent for p-DFB compared to other aromatic compounds investigated so far. The temperature dependence of O2 quenching can be approximately expressed by an exponential function. The influence of temperature, total pressure, and O2 partial pressure on absorption cross sections and fluorescence quantum yields are given as empirical functions that allow for interpolation. For typical applications, p-DFB LIF provides up to three orders of magnitude stronger signal compared to toluene LIF.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Single-ring aromatics are frequently employed as tracers in laser-induced fluorescence (LIF) imaging studies of gaseous systems for visualizing concentration, temperature, oxygen concentration, or fuel/air equivalence ratio [1]. The tracers are added to the respective UV-transparent base fuel (or bath gas) in concentrations low enough to provide sufficient LIF signal and still represent the physical behavior of the base gas composition. The fluorescence signal intensity depends on the tracer concentration, the absorption cross section, and the fluorescence quantum yield ϕ fl. Because the latter two strongly depend on temperature, pressure, and bath-gas composition, quantitative interpretation of signal intensities requires the knowledge of these dependences.
Toluene is one of the best studied fluorescence tracers and widely used for applications [2–6]. Its fluorescence strongly decreases with increasing temperature and O2 partial pressure which leads to a challenge for detecting signal under the respective conditions with high enough signal-to-noise ratio, especially in cases where methods are used that determine the desired quantity (such as temperature [7, 8] or O2 partial pressure [9, 10]) from the ratio of two measured intensities. This study will show that para-difluorobenzene (p-DFB) can provide significantly stronger signal already at room temperature and shows a significantly reduced response to temperature and oxygen quenching leading to a gain in signal intensity compared to toluene by two to three orders of magnitude. Due to its thermophysical properties (boiling point: 88 °C, viscosity: 0.6390 Ns/m2 at 298 K, vapor pressure 72 mbar at room temperature), p-DFB is ideally suited as tracer for gasoline [11] or as representative for the light fraction of multi-component fuels [12].
Investigations of the photophysical properties of the S0 → S1 (π, π*) transition of gas-phase p-DFB are scarce. Since different absorption bands are located between 246 and 273 nm (with a peak at 266 and 271 nm) [13], p-DFB can be excited conveniently with 266 nm radiation from a frequency-quadrupled Nd:YAG laser. Fluorescence spectra cover the 260–350 nm range. The high fluorescence quantum yield of 0.43 (for 266 nm excitation in N2 at room temperature) as well as the large absorption cross section in the range of 1.2 × 10−17–0.4×10−17 cm2 makes p-DFB attractive as fluorescence tracer for LIF imaging diagnostics.
Because of its promising properties, there is a strong need for further characterization of the photophysical behavior of p-DFB. Such data can also contribute to a better understanding of energy redistribution and relaxation processes in p-DFB.
Therefore, we have investigated absorption cross sections and fluorescence spectra as a function of temperature, and effective fluorescence lifetimes τ eff of p-DFB are evaluated at various temperatures in N2 and air as bath gas at various total pressures. Relative fluorescence quantum yields, i.e., normalized to a reference condition, can be quantified through τ eff measured after short-pulse excitation to its first excited electronic state [14]. In the present work, we analyze the dependence of p-DFB absorption cross sections, fluorescence spectra, fluorescence lifetimes, and quantum yields in the 298–1180 K temperature range with excitation at 266 nm. Measurements are carried out in pure N2 and in air at total pressures between 1 and 10 bar. In addition, we present functional relationships for calculating the signal intensity per molecule in dependence of temperature, total pressure, and O2 partial pressure for 266 nm excitation.
For application of fluorescence tracers at high temperature, it is also of interest to study the thermal stability limits. This can be of specific interest for fluorinated tracers because the decomposition can lead to the formation of strongly corrosive hydrofluoric acid.
2 Theoretical background
At room temperature, 266 nm excitation of the single-ring aromatic p-DFB species can populate the S1 state, which generally is followed by very fast non-radiative vibrational relaxation (VR), internal conversion (IC) to S0 and intersystem crossing (ISC) to a triplet state, 7800 cm−1 lower in energy [15] than the S 1 zero-point energy. The simplified rate equation [1, 14]
describes the intramolecular deactivation of the excited molecules M*. Here k fl, k ISC, k IC are the rates (all in s−1) of fluorescence emission, intersystem crossing, and internal conversion, respectively.
Since O2 is known as a highly efficient fluorescence quencher for aromatic molecules due to its triplet structure in the ground state [1], a Stern–Volmer relation
can be used [9, 16, 17], i.e., a linear dependence of the ratio of signal intensity S fl (and thus fluorescence lifetime τ eff) without (index 0) and with the presence of the quencher with number density n q.
Generally, the relation between τ eff and the fluorescence quantum yield ϕ fl is given by
with τ rad, the radiative lifetime [14]. Thus, the fluorescence quantum yield is proportional to τ eff. Measuring the fluorescence decay after picosecond laser excitation therefore provides a more direct access for measuring (relative) ϕ fl [8, 18] and to the energy conversion processes taking place in the electronically excited state than do intensity-based measurements because it is independent on absolute intensity calibration, which needs careful control of instrumental parameters (e.g., laser intensity and tracer concentration).
3 Experiment
1,4-Difluorobenzene (Merck Chemicals, for analysis grade) was used as a pure substance without further purification and stored in a glass container pressurized with N2. N2 (purity >5.0) and air were provided from vaporized liquid N2 and compressed air for pressures up to 6 bar. N2 and air for higher pressures were provided from gas cylinders (Air Liquide, purity 5.0). For gas mixtures with well-controlled composition, p-DFB was metered by a high-pressure pump (Bischoff-Chrom, HPLC Compact Pump 2250) and injected into a heated chamber (Bronkhorst, CEM) where it was evaporated and mixed with a part of the carrier gas. Variable O2 partial pressures were provided via mass flow controllers (Bronkhorst) metering the flows of N2 and air and a small gas mixing chamber. p-DFB partial pressures p DFB were 3 to 12 mbar, well below the room-temperature vapor pressure of 72 mbar. To prevent condensation, all tubes were resistively heated to 470 K.
3.1 Absorption experiment
A stainless steel high-temperature absorption flow cell (flow rate 2 slm) with an absorption length of 110 mm [19] was placed in an oven and used at atmospheric pressure. The temperature was monitored by a thermocouple placed close to the measurement volume.
Absorption spectra were measured against a broadband UV light source (LOT Oriel deuterium lamp) collimated with a quartz lens. A beam splitter was placed in front of the cell entrance window. This reference signal as well as the transmitted light was coupled into a y-fiber connected to a spectrometer (Horiba, iHR20, f = 320 mm, 1200 groves/mm grating) and detected simultaneously by a CCD camera (Horiba, Syncerity CCD, 256 × 1024 pixels). An exposure time of 1 ms and an entrance slit width of 200 µm were used to adjust to the 16-bit dynamic range of the camera with a spectral resolution of 0.47 nm FWHM. Spectra were then generated by binning over 50 pixels for both the transmitted light and the reference. The p-DFB partial pressure was varied (p DFB = 0.1–1 mbar) for each temperature to ensure that the measurements took place in the linear absorption regime and to average over various experimental conditions.
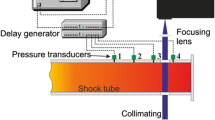
The absorption spectra are structured at low and intermediate temperatures. To ensure that the absorption cross sections relevant for laser experiments with excitation at 266 nm are not affected by under-resolved spectral detection, additional absorption measurements were carried out with light from a Nd:YAG-laser system (BM industries, Series 5000, 5-ns pulse duration). Reference and transmission signals were detected by photo diodes (Thorlabs, DET10A) and read out by an oscilloscope (Teledyne LeCroy, Wavesurfer 3034). Both arrangements are shown in Fig. 1.

Experimental arrangement for absorption cross-sectional measurements with a a deuterium lamp and b a Nd:YAG laser
3.2 Fluorescence experiment
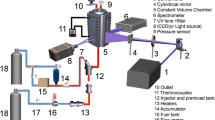
Spectrally and temporally resolved fluorescence measurements were carried out with an arrangement described in Ref. [8]; therefore, only a brief overview is given here. A mixture of evaporated p-DFB and the respective bath gas was flowing continuously from top to bottom through a cylindrical ceramic flow chamber (inner diameter 45 mm, length 250 mm) with four holes on its circumference for optical access. This assembly is contained in a 400-mm-diameter stainless-steel pressure vessel where the incoming and outgoing gas flows through two concentric tubes connected with the flow chamber from the bottom of the vessel. Heating elements placed around the flow chamber and thermally insulated against the outer vessel walls by ceramic foam elements allow heating of the gas flow up to 1400 K at a maximum gas pressure of 10 bar, while keeping the pressure vessel and its window flanges below 370 K. Gas temperature and pressure in the flow chamber were measured with a R-type thermocouple located close to the measurement volume and a pressure transducer, respectively. Constant pressure was achieved by a back-pressure regulator in the exhaust line of the flow system. The residence time of the gases in the heated zone of the cell is ~2 s.
The quadrupled output of a ps-Nd:YAG laser (Ekspla, PL2143B, 266 nm, pulse duration: 26 ps) was collimated to a 3-mm-diameter beam and directed horizontally through the center of the cell. The fluorescence was collected perpendicular and focused on the 150-µm entrance slit of a spectrometer (Acton SP2300, f = 300 mm, 150 grooves/mm grating, spectral resolution: 1.92 nm). The spectrally dispersed light was imaged on the entrance slit of a streak camera (Hamamatsu, C5680-24C, temporal resolution: 50 ps). The system thus simultaneously delivers fluorescence spectra (binning of rows in the time domain) and fluorescence decay profiles (binning of columns in the spectral domain) (Fig. 2).
The fluorescence intensity was investigated as a function of laser fluence to determine the limits of the linear regime (not shown here) which was approached at around 15 mJ/cm2 at 296 K. This value was utilized for temperatures up to 900 K. Because the saturation limit is known to shift to higher fluences at elevated temperatures [20], the laser fluence was increased to 25 mJ/cm2 at >900 K to maintain an acceptable signal-to-noise ratio. p DFB was 3 mbar at the lowest temperature (296 K) and was correspondingly increased at higher temperatures to maintain a nearly constant tracer number densities in the probe volume. In additional measurements, it was tested that at this number density self-quenching [21] can be neglected.
4 Data evaluation
4.1 Absorption experiments
For each measurement of absorption spectra, 200 images were averaged to minimize noise. Measurements were taken with and without p-DFB in the cell for otherwise identical conditions, and the absorption was determined from the difference between both. The result was corrected for potential variations in intensity and lamp spectra using the reference measurement from the beam splitter. The absorption cross section was then calculated using Beer–Lambert’s law
with I 0 and I as intensity without and with p-DFB, respectively, d as the distance between the quartz windows (110 mm), and n as the tracer number density.
For the laser absorption measurements, 1000 pulses were averaged. Afterward, the same calculation with Lambert–Beer’s law has been done for the laser with peak-to-peak measured signals to subtract the background.

Experimental arrangement for spectrally and temporally resolved LIF measurements, CCD CCD camera, DG pulse delay generator
4.2 Fluorescence experiments
For each condition, 1000 individual single-shot measurements were acquired and stored. The respective images were then averaged after adjusting the time axis with a jitter-correction routine provided by the streak camera software to minimize smearing of the temporal profiles. A background image was subtracted that was obtained while the laser was firing with N2 flowing through the cell only. Figure 3 shows a sample image with wavelength along the horizontal and time along the vertical axis. In the CCD image, columns or rows can be binned to generate temporal or spectral profiles of the recorded fluorescence signals, respectively. It was found that within the error limits the number of binned columns did not influence the evaluated effective fluorescence lifetimes, i.e., there was no significant dependence of the lifetime with emission wavelength. Thus for SNR enhancement, the signal between 265 and 350 nm was integrated when determining fluorescence decay curves.

Sample image (averaged over 1000 single-shot images) of time- and spectrally resolved p-DFB LIF after 266 nm excitation. Conditions: 325 K, 1 bar total pressure of N2, p DFB = 3 mbar
Fluorescence spectra were generated by binning all rows with significant pixel counts. The resulting spectra were corrected for the instrument spectral response by recording the spectrum from a deuterium lamp with known spectral irradiance using the same optical setup. Figure 4 presents typical LIF excitation/decay profiles for 298, 475, and 675 K together with the instrument response function determined from stray light in the empty cell. Here the streak camera was set to 20-ns sweep duration. Because the fluorescence lifetimes vary depending on the environmental conditions, the temporal resolution (i.e., the sweep duration) was adjusted, requiring separate instrument functions to be recorded.

Time-resolved p-DFB-LIF intensity in 1 bar N2 with excitation at 266 nm at various temperatures. For 298 K, the fitted function (with τ = 6.6 ns) is shown as dotted curve. The instrument response curve (magenta line) was recorded based on 266-nm stray light. Sweep duration 20 ns
Signal decay times τ were determined using the convolve-and-compare method described by Settersten et al. [22, 23]. A convolution of a single- or double-exponential decay with the measured instrument response function was fitted to the experimental excitation/decay profiles by minimizing the sum of squared residuals between both curves. Figure 4 shows an example of such a convoluted signal profile (black dotted line in Fig. 4) fitted to the 298 K LIF signal trace (red) and the respective instrument function (black). In the fits, the variable parameters were the decay time τ and a shift of the time axis to optimize the instance where experimental and simulated signal intensities reach their respective peak values. For the fastest measured decays, the minimum temporal resolution of the detection system was 50 ps.
5 Results
Absorption measurements were performed for temperatures between 296 and 675 K in 1 bar N2 as bath gas. Fluorescence measurements were done at 296–1175 K and at 1–10 bar in N2. At higher temperatures, no fluorescence was detectable apart from a significant increase in laser stray light, most probably due to pyrolysis during the ~2-s residence time in the heated cell. When O2 was present, possible tracer degradation/oxidation limited the upper temperature to 975 K. At higher temperatures, again, strong stray light was detected, together with a small increase in gas temperature. Compared to other tracers investigated by us so far (e.g., toluene, naphthalene, anisole), p-DFB showed the strongest resistance to oxidation (i.e., the highest limiting temperature within the residence time of ~2 s in our cell).
5.1 Absorption cross sections
Figure 5 (left panel) shows the absorption spectra of p-DFB for various temperatures. The spectra show a characteristic band structure that is most prominent at the lower temperatures but persists up to the highest temperatures investigated here. The center of gravity of the spectra red-shifts with increasing temperature. The peak absorption was close to 266 nm, and the individual peaks show a minor red-shift only. The right panel in Fig. 5 shows the data for 266 nm from the lamp measurements compared to laser-based absorption measurements. The absorption cross sections decrease with increasing temperature as the band structure vanishes. Although the spectral resolution of the lamp measurement is quite good (0.47 nm FWHM, cf. arrows in Fig. 5 as determined by the FWHM of a recorded mercury line at 254 nm from a low-pressure discharge lamp), the spectra are still under-resolved for low temperatures, leading to an increased deviation between the lamp and laser measurements toward lower temperatures in the right panel in Fig. 5. The room-temperature values of the laser measurement are reassured by results from Volk et al. [24]. At <450 K, the fine structure is almost lost and the lamp and laser measurements approach each other.

Absorption spectra of p-DFB at various temperatures in 1 bar N2 (left). The arrows indicate the spectral resolution. Right absorption cross sections at 266 nm determined from the spectra (black squares) and the laser measurements (red dots) with fit. The fit is based on the function given in Table 1. Room-temperature literature value from Volk et al. [24]
5.2 Fluorescence spectra
The left panel in Fig. 6 shows the temperature dependence of p-DFB fluorescence spectra in 1 bar N2 normalized to their respective peak intensities. The spectra were smoothed by an adjacent-average filter with a window size of 1.5 nm. At room temperature, the emission extends from roughly 265 to 340 nm with a maximum at around 280 nm. With increasing temperature, a slight red-shift of this maximum (~2 nm/100 K) can be observed, and the tail on the long-wavelength side of the spectra becomes stronger relative to the peak. Due to the limited spectral resolution of the present experiment of about 1.9 nm (FWHM, indicated by the arrows in Fig. 6, determined from the FWHM of a recorded mercury line at 254 nm from a low-pressure discharge lamp), the band structure at low temperatures may be under-resolved. The observed spectral shape does not change with increasing total pressure. When adding increasing amounts of O2, a weak red-shift as well as a relative intensity change of the different peaks can be observed (Fig. 6, right).

Left peak-normalized p-DFB fluorescence spectra at various temperatures in 1 bar N2 bath. Right spectra at different O2 partial pressures at 298 K and 1 bar total pressure. p DFB = 3 mbar. The arrows indicate the spectral resolution of the detection system (1.92 nm FWHM)
5.3 Effective fluorescence lifetimes
5.3.1 Nitrogen atmosphere
The present work complements data from our previous works on the temperature, O2, and bath-gas pressure dependences on effective fluorescence lifetimes of toluene and naphthalene [10] and anisole [25]. Evaluated fluorescence lifetimes for p-DFB in N2 are shown in Fig. 7 as a function of temperature for a total pressure of 1 and 8 bar. For both pressures and lower temperatures, the lifetimes are almost equal within the experimental uncertainty and linearly decrease with temperature on a logarithmic scale up to a certain temperature before they continue to decrease more strongly with a different slope. This change in slope takes place at ~700 K at 1 bar and at ~580 K at 8 bar. The stronger slope at 8 bar (38% reduction in τ per 100 K) is about 26% larger than the slope at 1 bar (31% reduction in τ eff per 100 K), and both are larger than the common slope (17% reduction in τ per 100 K) in the lower temperature range (298–580 K). Exponential fits are shown as solid lines in Fig. 7 for both pressures.

Effective fluorescence lifetimes of p-DFB in N2 as a function of temperature for 1 and 8 bar together with the temperature fit for the subsequent pressures found in Table 1
Figure 8 shows effective fluorescence lifetimes of p-DFB in N2 as a function of total pressure and for 298–875 K. As already noticed in Fig. 7, for the two lower temperatures of 298 and 475 K, the fluorescence lifetimes are almost independent of the gas pressure within the experimental uncertainty while for the higher temperatures a decrease with pressure in the lower-pressure region is noticeable, which flattens out into a plateau region up to the highest pressures investigated. As temperature rises, the onset of the plateau region seems to start at increasingly higher pressures.

Effective fluorescence lifetimes of p-DFB as a function of N2 pressure at various temperatures together with the pressure fit for the subsequent temperatures found in Table 1
5.3.2 Oxygen quenching
It is known that O2 with its triplet ground state is an efficient fluorescence quencher in aromatic species due to very fast collision-induced singlet–triplet energy transfer. Therefore, it is expected that (1) the p-DFB fluorescence lifetime strongly depends on \(p_{{{\text{O}}_{2} }}\), and (2) the presence of O2 affects the temperature and pressure dependence of ϕ fl relative to measurements in a N2 atmosphere. For constant temperature, increasing \(p_{{{\text{O}}_{2} }}\) causes a decrease in τ eff. However, as can be seen in Fig. 9, the reduction is most prominent at room temperature (factor of 8.3 between 0 and 210 mbar \(p_{{{\text{O}}_{2} }}\)) and diminishes at higher temperatures. Depending on \(p_{{{\text{O}}_{2} }}\), τ eff exhibit a shallow maximum with increasing temperature, which shifts toward higher temperatures at higher \(p_{{{\text{O}}_{2} }}\), as also seen with other aromatic tracers [10, 25].

Effective fluorescence lifetimes of p-DFB as a function of temperature for several O2 partial pressures
The fluorescence lifetimes at >800 K might be less trustworthy since oxidation might already take place on the timescale of our experiment. However, p-DFB showed no noticeable degradation due to pyrolysis or oxidation within the residence times (~2 s) in the heated zone of the flow system and at the highest temperatures investigated here, as can be deduced from the appearance of the fluorescence spectra presented earlier. In N2, p-DFB is thermally as stable as naphthalene, and in air, it exceeds the stability of naphthalene that has been investigated before in the same flow cell [25].
5.3.3 Stern–Volmer coefficients
The quenching behavior of the fluorescence of aromatic species by O2 can often be expressed by the Stern–Volmer coefficient (Eq. 2). If temporally and spectrally integrated fluorescence intensities show Stern–Volmer behavior, this should apply as well to measured fluorescence lifetimes. To validate this, Stern–Volmer coefficients for p-DFB were determined from the slopes of fluorescence lifetime data plotted for a given total pressure (with N2) and temperature versus \(p_{{{\text{O}}_{2} }}\) (Fig. 10, left). The resulting Stern–Volmer coefficients are plotted as a function of temperature in the right panel of Fig. 10. In comparison with anisole [25] and toluene [10], O2 quenching of p-DFB is comparably weak.

5.4 LIF intensity model
In the previous sections, the dependences of the absorption cross section and the fluorescence quantum yield of p-DFB on the environmental conditions have been described. To enable the application of these data to other experiments, the dependences need to be combined, and then, the results can be compared to measurements in more applied cases, like, e.g., in Faust et al. [25]. With exponential fits to the temperature-dependent absorption cross section (Fig. 5, right panel) and the temperature (Fig. 7 [13])-, pressure (Fig. 8)-, and O2 partial pressure (in the form of the Stern–Volmer coefficients Fig. 10, right)-dependent fluorescence lifetime, it is possible to predict fluorescence intensities per unit number density or volume in a given situation. The respective fit functions are given in Table 1.
The signal intensity per molecule can then be calculated by:
This formula is the result of a simplification of a rate-constant approach to determining the fluorescence quantum yield, ϕ fl (similar to the strategy used by Koban et al. for toluene in Ref. [9]) multiplied by the absorption cross section. Multiplying the result with the number density and the total pressure yields the estimated signal per volume.
A comparison for expected p-DFB and toluene signal are shown in the left panel in Fig. 11 per molecule (i.e., unit number density) and in the right panel per volume. The toluene data are from the extended Koban model together with measured absorption cross sections [7, 10, 11] (note the error in the model description in Ref. pressure [10] on page 90 where the T 4 term in the description of B 3 has the wrong sign). This comparison illustrates the signal improvement by two to three orders of magnitude gained by p-DFB in comparison with toluene depending on the environmental conditions.

6 Discussion
The data presented in Sect. 5 indicate high signal intensities for p-DFB for a wide range of environmental conditions. The structure of the absorption spectra matches the one reported by Volk et al. [24] for room temperature. However, the gas pressure in their experiment was close to vacuum (0.066 mbar air) which explains the broader band structure that we showed in Fig. 5 as well as the difference in lamp and laser measurements. The value at 266 nm from the laser measurements of 1.26 × 10−17 cm2 at room temperature also matches the one determined by Volk of 1.28×10−17 cm2.
The decrease in fluorescence lifetime of p-DFB (Fig. 8) with increasing pressure fits into the explanation we gave for the pressure dependence based on low-pressure experiments for various tracer species [26]. We showed there that the pressure dependence slowly changes from increasing the fluorescence lifetime to decreasing it with increasing temperature. This is due to the change in the Boltzmann distribution in the S 0 and S 1 state and whether the first populated state after excitation is above or below the thermal energy level in the S 1 state. This is further emphasized by the fact that in p-DFB the 0–0 band gap is ~760 cm−1 smaller than 266 nm excitation [13]. This is very close to the difference in the population-weighted thermal energy of the Boltzmann distribution in the S 0 and S 1 vibrational manifolds for this temperature in similar molecules; therefore, no vibrational relaxation and therefore no pressure dependence of the fluorescence quantum yield are to be expected for low temperatures, which is confirmed by the data in Fig. 8. At higher temperatures, the difference in thermal energy increases and the effect of photo-induced cooling takes place, which induces collisional heating by the bath gas toward thermal equilibrium [27, 28]. The presented effective fluorescence lifetimes and spectra of p-DFB were compared to existing data obtained for similar experimental conditions and showed only minor differences [13, 29]. The strong increase in laser stray light for temperatures above 1175 K in N2 and 975 K in air indicates pyrolysis processes to take place; however, it is uncertain if it is due to soot particles or eroded cell material due to the possible formation of reactive fluorine-containing species.
The measured Stern–Volmer coefficients are the lowest that that have been reported for aromatic fluorescence tracers so far. This means that quenching of this fluorinated single-ring aromatic by O2 is comparatively weak, resulting in higher fluorescence intensities for otherwise equal conditions when the tracer is applied in air. The functional forms for calculating signal intensities of p-DFB and toluene from the Koban model suggest that due to the low overall temperature dependence of p-DFB the signal per molecule of p-DFB is more sensitive on quenching by O2 than toluene for temperatures above 600 K.
7 Conclusions
Temperature-dependent absorption spectra in the first electronic singlet absorption band of p-DFB were measured, and absorption cross sections were derived at moderately high spectral resolution. For low and intermediate temperatures, the absorption cross sections derived from laser measurements at 266 nm differ from the ones determined from the recorded spectra. Despite the comparatively high spectral resolution of our setup (0.47 nm FWHM), the measurements are partly under-resolved leading to underprediction of the absorption cross sections by up to 30% for room temperature.
Fluorescence spectra and effective fluorescence lifetimes after picosecond laser excitation of gas-phase p-DFB were determined for a large range of gas-phase conditions commonly faced in LIF applications. Effective fluorescence lifetimes are used to determine relative fluorescence quantum yields. Vaporized p-DFB mixed with appropriate buffer gas was continuously flown through a heated ceramic cell inside a high-pressure vessel at temperatures in the range 296–1200 K, while the pressure could be varied between 1 and 10 bar. Normalized fluorescence spectra are presented as a function of temperature and O2 partial pressure. On a logarithmic scale, the evaluated fluorescence lifetimes decrease with a smaller and larger slope in a lower and upper temperature regime, respectively, whose relative magnitudes also depend on the total pressure. For lower temperatures (<475 K for the data presented here), the fluorescence lifetime τ is independent of the total pressure, while at higher temperatures τ first decreases at lower pressures before becoming pressure independent.
The effect of O2 on the fluorescence lifetime of p-DFB was also investigated, and the fluorescence quantum yields are reduced less drastically than for other aromatic tracers (e.g., toluene, anisole, naphthalene) which makes p-DFB an interesting candidate tracer for measurements in air. Functional forms for predicting the relative signal intensity behavior with environmental conditions were derived from the available experimental data and are compared to similar data for toluene derived from the Koban model, which showed that the expected DFB signal per molecule is about two to three orders of magnitude stronger than that for toluene in many conditions typical for combustion experiments.
The large absorption cross sections, high fluorescence yield, weak quenching behavior, together with its comparatively low trend toward thermal degradation or oxidative reactivity make p-DFB a valuable candidate as a gas-phase fluorescence tracer in LIF diagnostics for the study of mixing processes and for imaging of, e.g., temperature and mass fraction, which may be extended to temperature regions not accessible with other commonly employed aromatic fluorescence tracers. However, if the test conditions exceed 980 K in air, 1150 K in N2 or include combustion, further kinetics studies involving this species are needed to provide data on the thermal decomposition, oxidation or formation of hydrofluoric acid and further explore its limits of applicability in high-temperature reactive systems.
References
C. Schulz, V. Sick, Prog. Energy Combust. Sci. 31, 75 (2005)
C.S. Burton, W.A. Noyes, J. Chem. Phys. 49, 1705 (1968)
C.G. Hickman, J.R. Gascooke, W.D. Lawrance, J. Chem. Phys. 104, 4887 (1996)
F.P. Zimmermann, W. Koban, C.M. Roth, D.-P. Herten, C. Schulz, Chem. Phys. Lett. 426, 248 (2006)
E. Friesen, C. Gessenhardt, S. Kaiser, T. Dreier, C. Schulz, In-cylinder temperature measurements via fiber-based toluene-LIF time-correlated single-photon counting (LACSEA2012, San Diego, CA, USA, 2012)
M. Luong, R. Zhang, C. Schulz, V. Sick, Appl. Phys. B. 91, 669 (2008)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Phys. Chem. Chem. Phys. 6, 2940 (2004)
S. Faust, T. Dreier, C. Schulz, Chem. Phys. 383, 6 (2011)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 777 (2005)
S. Faust, G. Tea, T. Dreier, C. Schulz, Appl. Phys. B 110, 81 (2013)
W. Koban, C. Schulz: SAE technical paper series 2005-01-2091, 2005 (2005)
L.M. Itani, G. Bruneaux, A. Di Lella, C. Schulz, Proc. Combust. Inst. 35, 2915 (2015)
J.M. Hollas, T. Cvitas, Mol. Phys. 18, 793 (1970)
J.R. Lakowicz, Principles of Fluorescence Spectroscopy (Springer, New York, 2006)
M.G. Rockley, J. Metcalfe, D. Phillips, J. Chem. Soc. Faraday Trans. 2(70), 1660 (1974)
S.A. Kaiser, M.B. Long, Proc. Combust. Inst. 30, 1555 (2005)
M. Orain, P. Baranger, B. Rossow, F. Grisch, Appl. Phys. B 102, 163 (2011)
F. Ossler, T. Metz, M. Aldén, Appl. Phys. B 72, 465 (2001)
W. Koban, J. Schorr, C. Schulz, Appl. Phys. B 74, 111 (2002)
R. Devillers, G. Bruneaux, C. Schulz, Appl. Phys. B 96, 735 (2009)
D. Fuhrmann, T. Benzler, S. Fernando, T. Endres, T. Dreier, S. A. Kaiser, C. Schulz: Proc. Combust. Inst. 36 (2016). doi:10.1016/j.proci.2016.06.045
T.B. Settersten, A. Dreizler, R.L. Farrow, J. Chem. Phys. 117, 3173 (2002)
T.B. Settersten, B.D. Patterson, J.A. Gray, J. Chem. Phys. 124, 234308 (2006)
E.K.C. Lee, L.J. Volk, J. Chem. Phys. 67, 236 (1977)
S. Faust, T. Dreier, C. Schulz, Appl. Phys. B 112, 203 (2013)
T. Benzler, S. Faust, T. Dreier, C. Schulz, Appl. Phys. B 121, 549 (2015)
Y. He, E. Pollak, J. Chem. Phys. 116, 6088 (2002)
H. Wadi, E. Pollak, J. Chem. Phys. 110, 11890 (1999)
R.A. Coveleskie, D.A. Dolson, C.S. Parmenter, J. Phys. Chem. 89, 645 (1985)
Acknowledgements
The authors acknowledge funding of this work by the Deutsche Forschungsgemeinschaft (DFG) within SCHU 1369/28. The authors also acknowledge valuable discussions with Björn Rossow (Université de Rouen) on DFB absorption cross sections and fluorescence spectra.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Benzler, T., Dreier, T. & Schulz, C. UV absorption and fluorescence properties of gas-phase p-difluorobenzene. Appl. Phys. B 123, 39 (2017). https://doi.org/10.1007/s00340-016-6612-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-016-6612-8