
Abstract
Time-resolved fluorescence spectra of gas-phase toluene and naphthalene were investigated upon picosecond laser excitation at 266 nm as a function of temperature (toluene 296–1,025 K, naphthalene 374–1,123 K), pressure (1–10 bar), and bath gas composition (varying concentrations of N2, O2, and CO2) with a temporal resolution of 50 ps. In the investigated temperature range, the fluorescence spectra of both toluene and naphthalene show a significant red-shift, whereas the fluorescence lifetime decreases with increasing temperature, more pronounced for toluene than for naphthalene. Increasing the total pressure of either N2 or CO2 from atmospheric to 10 bar leads to an increase by about 20 % (naphthalene at 373 K) and a decrease by 60 % (toluene at 575 K) in fluorescence lifetimes, respectively. As expected, at atmospheric pressure collisions with O2 shorten the fluorescence lifetime of both toluene and naphthalene significantly, e.g., by a factor of 30 and 90 when changing O2 partial pressure at 373 K from 0 to 0.21 bar, respectively. The fluorescence model of Koban et al. (Appl Phys B 80: 777, 2005) for the dependence of the toluene quantum yield on temperature and O2 partial pressure at atmospheric pressure describes toluene fluorescence lifetimes well within its range of validity. The model is modified to satisfactorily predict effective toluene fluorescence lifetimes in N2 at pressures up to 10 bar. However, it still fails to predict the dependence at simultaneously elevated temperatures and pressures in air as bath gas. Similarly, an empirical model is presented for predicting (relative) fluorescence quantum yields and lifetimes of naphthalene. Although the fitting models have their shortcomings this publication presents a data set of great importance for practical LIF applications, e.g., in-cylinder mixture formation diagnostics in internal combustion engines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Organic molecules, such as ketones as well as mono-aromatics (e.g., toluene) and di-aromatics (e.g., naphthalene), are frequently used as fluorescent tracers for visualizing gas-phase mixing processes and for gas-phase thermometry using laser-induced fluorescence (LIF). The tracers are added at low concentration (1–10 %) to a non-fluorescing surrogate fuel [1]. Due to their matching evaporation characteristics, toluene and naphthalene are often used as representative for gasoline [2] and Diesel or kerosene, respectively [3, 4]. For quantitative interpretation of signal intensities in practical applications their dependence on environmental conditions, i.e., temperature, pressure, and bath gas composition must be understood. Variations in the absolute fluorescence intensity of these tracer species are determined by the respective absorption cross-section [5] and the fluorescence quantum yield (FQY) [3, 6, 7]. With increasing temperature and O2 concentration a red-shift in the absorption and emission spectra are reported [3, 5, 6]. In addition, the fluorescence quantum yield varies with increasing temperature and O2 content and is mirrored by respective changes of the effective fluorescence lifetime. For practical applications of fluorescence tracers it is desirable to describe these effects with models that cover the relevant range of temperature, pressure, and bath gas composition [6, 8–10].
The photophysical properties of the S0 → S1 (π, π*) transition of gas-phase toluene and naphthalene have been investigated for decades [11–14]. The broad and almost structureless absorption spectra, located between 230 and 270 nm (toluene) and between 240 and 290 nm (naphthalene) at room temperature, are easily accessible with typical high-power UV lasers (e.g., of KrF* excimer and quadrupled Nd:YAG lasers at 248 and 266 nm, respectively). Their red-shifted fluorescence spectra cover the range 265–350 nm (toluene) and 305–400 nm (naphthalene). The high fluorescence quantum yield [5, 15, 16] makes toluene and naphthalene attractive as fluorescent tracers for LIF imaging diagnostics of gaseous mixing processes [17] and temperature in, e.g., in internal combustion (IC) engines [18, 19], standard burners [20, 21], and shock tubes [22]. In all these applications, the variation of the time-integrated LIF-signal intensity (or its relative variation in selected wavelength sections of the emission spectrum) with total pressure, O2 partial pressure, and temperature is exploited to measure the respective quantity. Therefore, for extracting quantitative results from such measurements the dependence of the LIF signal on these parameters has to be explored under well-controlled environmental conditions.
Temperature-dependent fluorescence quantum yields have been determined by measuring absolute signal intensities by calibration of the optical setup, e.g., with Rayleigh scattering [3, 5]. Based on such data, for the case of toluene excited at 248 or 266 nm, the fluorescence quantum yield was described as a function of temperature and O2 partial pressure by a phenomenological model introduced by Koban et al. [6]. This approach uses analytical functions fitted to temporally and spectrally integrated fluorescence intensities. This model was able to reproduce measured LIF intensities with 10 % accuracy in the temperature range of 300–700 K at 1 bar [2, 6], in the absence of O2 even up to 950 K. The model was also applied with limited success to predict signal intensities in internal combustion engine applications where O2 partial pressures and temperature vary during the compression stroke [23, 24]. For naphthalene, however, such a phenomenological model has not yet been published.
An alternative, less common but more direct approach for the determination of the (relative) fluorescence quantum yield is the measurement of effective fluorescence lifetimes [21, 25, 26]. For toluene and naphthalene, published data are limited mostly to atmospheric pressure ([25–29] and references therein), few collision partners [30–32] and for a restricted temperature range [33]. Measurements of toluene and naphthalene fluorescence lifetimes at simultaneously elevated temperatures and pressures have never been published before. For air as bath gas even no quantum yield data are available. The present work aims at extending the available data base into regions of higher pressures and temperatures which are of great interest for quantitative engine, shock-tube, or gas-turbine diagnostics. The new effective fluorescence lifetime information will also be of importance when extending existing and developing new photo-physical models for the prediction of fluorescence quantum yields of these species. In the present work, we analyze the dependence of toluene and naphthalene fluorescence lifetimes in the 296–1,200 K temperature range with excitation at 266 nm. Measurements are carried out in pure N2, CO2, and in air at total pressures between 1 and 10 bar as well as at 1 bar in N2/O2 mixtures with varying O2 partial pressures. For toluene, the fluorescence quantum yields are compared to predictions of the Koban model [6].
2 Theoretical background and model description
In aromatic molecules several excited singlet states exist. In naphthalene upon 266 nm excitation, besides populating S1 the second state S2 can be populated at 373 K (at higher temperatures even the S3 is reached), followed by very fast non-radiative vibrational relaxation to S1. In toluene, most excitation takes place into S1, only at higher temperatures (>600 K) the S2 state can be populated. For simplification we assume the presence of only one excited singlet state S1 and ignore radiative processes originating from higher singlet states and from triplet states. Although this seems to be a strong limitation, our results show that within our experimental uncertainty this assumption is justified. Neglecting energy transfer during photoabsorption, the intra-molecular deactivation of the excited molecules [M*] can be described by the following rate equation [1, 34].
where k fl, k ISC, k IC are the rates (all in s−1) of fluorescence emission, intersystem crossing, and internal conversion, respectively. The total depopulation rate, k tot, is then connected to the experimentally accessible effective fluorescence lifetime, τ eff, via the following relation:
where k nr is the non-radiative decay rate (s−1) as the sum of all decay rates except k fl. k q,j is the respective quenching rate (in cm3/s) with n q,j (in m−3) as the concentration of the respective quencher species Q j . The relation between τ eff and the fluorescence quantum yield, ϕ fl, is given by
with τ rad, the radiative lifetime [34]. Thus, the fluorescence quantum yield is proportional to the effective fluorescence lifetime.
The deactivation processes can be separated in intra-molecular (fluorescence, internal conversion, intersystem crossing) and inter-molecular processes (collisional quenching). In the latter case, energy transfer from the excited molecule to the collider species occurs. The quenching efficiency depends on the collision frequency and the quenching cross-section which depends on the species itself. O2 is known as a highly efficient fluorescence quencher for aromatic molecules due to its triplet structure in the ground state [1]. For the investigated species toluene and naphthalene a Stern–Volmer relation
can be used [3, 6, 20], i.e., a linear dependence of the ratio of signal intensity S fl (and thus τ eff) without (index 0) and with presence of the quencher with number density n q. The respective Stern–Volmer factor, k SV, can be determined experimentally by measuring signal intensities (or lifetimes) in environments with varying quencher concentration.
Koban et al. [6] built a phenomenological model for predicting toluene-LIF signal intensities, i.e., fluorescence quantum yields, as a function of excitation wavelength, λ exc, temperature, T, and O2 number density, \( n_{{{\text{O}}_{ 2} }} \) relative to a calibration performed at known conditions based on the simplifications mentioned above. The model was validated against experimental data obtained in a cell at 1 bar total pressure for temperatures between 300 and 950 K in pure N2, and between 300 and 700 K in the presence of 0–300 mbar O2. According to Eq. (3), the model should be applicable to the present lifetime measurements, if the latter are normalized to the fluorescence quantum yield at calibration conditions. As outlined by Koban et al. [2, 6], the model describes the time-integrated LIF-signal intensity (cf. Eq. (5)) as being proportional to two factors:
The first part considers the number density, n tol, of toluene and the temperature dependence of the absorption cross-section, σ abs, which is necessary to predict signal intensities as a function of temperature. However, since the variations of the absorption cross-section are not part of the calculation of the fluorescence quantum yield, it needs not be taken into account for comparison to the present lifetime measurements. The second factor describes the relative quantum yield as a function of temperature and O2 partial pressure. It considers two separate de-excitation pathways for molecules excited to the S1 state represented by two separate Stern–Volmer factors [6], which, however, is just an empirical finding without a clear photophysical background. A shortcoming of the model is that it does not consider effects of total pressure in, e.g., N2 bath.
3 Experiment
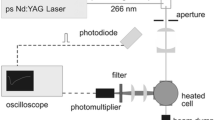
The experiment is described in detail in [25], therefore, only a brief overview is given here (cf. Fig. 1). Fluorescence measurements were carried out in a mixture of the tracer and the respective bath gas flowing continuously through a cylindrical ceramic flow chamber (outer diameter 90 mm, length 250 mm) with four holes on its circumference for optical access, and placed inside a 400-mm-diameter stainless steel pressure vessel. Heating elements placed around the flow chamber and thermally insulated against the outer vessel walls by ceramic foam elements allow heating of the gas flow up to 1,400 K at a maximum gas pressure of 10 bar while keeping the pressure vessel and its window flanges at below 370 K. Gas temperature and pressure in the flow chamber were measured with an R-type thermocouple located close to the measurement volume and a pressure transducer, respectively. Constant pressure was achieved by a back-pressure regulator in the exhaust line of the flow system. The stainless steel vessel has four optical ports sealed with quartz windows of 30 mm clear aperture in line with the openings of the ceramic cell.

Experimental arrangement for spectrally and time-resolved fluorescence measurements, CCD CCD camera, DG pulse delay generator
Toluene (for analysis grade, Merck Chemicals) was used as a pure substance without further purification, while naphthalene (for analysis grade, Merck Chemicals) was dissolved in dodecane (>99 %, purity, Sigma-Aldrich; 5 % naphthalene by weight). Pure dodecane was tested to show no fluorescence upon 266 nm excitation. N2 (purity >5.0) and air were provided from vaporized liquid N2 and compressed air for pressures up to 6 bar, respectively. N2 and air for higher pressures (purity 5.0) and CO2 for all pressures (purity 4.6) were provided from gas cylinders (Air Liquide). For providing gas mixtures with well-controlled composition the liquids were metered by a high-pressure pump (HPLC Compact Pump 2250, Bischoff-Chrom) and injected into a heated chamber (CEM, Bronkhorst) where they were mixed with the carrier gas. Different bath gas compositions were provided via mass flow controllers (Bronkhorst) and a small gas mixing chamber. To avoid condensation all tubes were resistively heated up to 470 K. The O2 partial pressure in the chamber was varied by diluting a flow of air with varying amounts of N2.
The optical setup is shown in Fig. 1. A flashlamp-pumped, frequency-quadrupled Nd:YAG laser (PL2143B, Ekspla) delivered a pulse energy of maximum 7 mJ at 266 nm with a pulse width of 26 ps at a repetition rate of 10 Hz. The diameter of the circular laser beam was reduced by a Galilei telescope to 3 mm before entering the cell. The signal was collected perpendicular to the laser beam by a quartz lens and then focused with a second quartz lens via an UV-enhanced aluminum mirror on the entrance slit of a spectrograph (f = 300 mm, 150 grooves/mm grating, Acton SP2300, Princeton Instruments). The fluorescence spectrum was then imaged on the entrance slit of a streak camera (C5680-24C, Hamamatsu Photonics) with a streak module (M5677-01) and a CCD camera (Orca R2), which provides a maximum temporal resolution of 20 ps. The system thus delivers two-dimensional images with a wavelength and a time axis, respectively. Laser and camera were synchronized with trigger pulses from the laser via a pulse delay generator (DG645, Stanford Research Systems).
The fluorescence intensity was investigated as a function of laser fluence to determine the limits of the linear regime (not shown here) which was found to be 35 mJ/cm2 at 296 K for toluene and 374 K for naphthalene, respectively. This laser fluence was used for temperatures up to 900 K. Because the saturation limit is known to shift to higher fluences at elevated temperatures [23], for reasons of gaining a better signal-to-noise ratio at higher temperatures the fluence level was increased stepwise to 70 mJ/cm2 at the highest temperature investigated which was checked to be in the linear regime. The partial pressures of toluene and naphthalene were 4 and 0.4 mbar, respectively, at the lowest used temperature (296 K for toluene, 374 K for naphthalene, respectively). At higher temperatures the partial pressure was increased to maintain a nearly constant number density of tracer molecules in the probe volume. In additional measurements it was tested that at this number density self-quenching can be neglected.
4 Data evaluation
Depending on the signal-to-noise (S/N) ratio, for each condition 300–1,000 individual single-shot measurements were acquired and stored. The respective images were then averaged after adjusting the time axis with a jitter-correction routine provided by the streak camera software to minimize smearing of the temporal profiles. A background image was subtracted which was obtained while the laser was firing with pure N2 flowing through the cell. Resulting sample images for both tracers are depicted in Fig. 2 with wavelength along the horizontal and time along the vertical axis. These images deliver spectrally selected temporal profiles (horizontal binning of columns) and fluorescence spectra for specific time intervals (vertical binning of rows) after laser excitation. It was found that within the error limits the number of binned columns did not influence the evaluated effective fluorescence lifetimes, i.e., there was no significant dependence of the lifetime on emission wavelength. Thus, for S/N enhancement the signal within columns covering the spectral range between 270 and 320 nm (toluene) and 305 and 375 nm (naphthalene) was integrated. Spectrally resolved fluorescence was determined by binning a number of rows in the range of significant pixel counts (red rectangles in Fig. 2). The resulting spectral profiles were corrected for the instrument spectral response by recording the emission of a deuterium lamp with known spectral radiance using the same optical setup. The recorded spectra also proved useful to detect the onset of tracer degradation at high sample temperatures that is clearly visible from a significant change in shape of the fluorescence spectra.

Sample images (averaged over 300 single-shot images) of time- and spectrally resolved toluene (left) and naphthalene (right) LIF after excitation at 266 nm. Conditions: 373 K, 2 bar total pressure of CO2, toluene partial pressure 4 mbar, naphthalene partial pressure 0.4 mbar. The signal integration ranges along the time axis for the evaluated spectral profiles are marked as a red, and along the spectral axis for the decay profiles as green rectangles, respectively
Figure 3 presents three examples for LIF excitation/decay profiles for different gas temperatures together with the instrument response function recorded in the spectral region of the laser stray light at 266 nm in the absence of tracers. Because decay times strongly depend on the environmental conditions, the temporal resolution of the streak tube was adjusted for each condition to optimize the time resolution to take advantage of the full detector size. For each condition the data trace covers a different time span. Thus, separate instrument functions were recorded.

Time-resolved LIF intensity for toluene (left) and naphthalene (right) in 1 bar N2 with excitation at 266 nm at various temperatures. For 296 K (toluene) and 374 K (naphthalene), the fitted function and the residuals are shown as blue and green curve, respectively. The depicted instrument response curves (red dashed lines) were recorded without tracer for the streak speeds appropriate for the respective 296 K and 374 K measurements
Signal decay times, τ, were determined using the convolve-and-compare method described by Settersten et al. [35, 36]. A convolution of a single- or double-exponential decay with the measured instrument response function was fitted to the experimental excitation/decay profiles by minimizing the sum of squared residuals between both curves. Figure 3 shows an example of such a convoluted signal profile for each substance (blue lines in Fig. 3) fitted to the 296 K LIF trace of toluene and the 374 K trace of naphthalene, respectively (black dots), with the residuals shown as green curves and the respective instrument function as red curve. The motivation for the respective fitting functions will be discussed in Sects. 5 and 6. In the fits the variable parameters were the decay time and a shift on the time axis to optimize the instance where experimental and simulated signal intensities reach their respective peak values. For the fastest decays the minimum temporal resolution of the detection system was 50 ps.
5 Results
Measurements for toluene are presented for temperatures from 296 to 1,026 K in N2 and CO2 bath gas. Due to the low signal intensity and tracer oxidation, the upper temperature for measurements in air was limited to about 800 K. Naphthalene was investigated from 373 to 1,137 K in both N2 and CO2. At lower temperatures, the naphthalene vapor pressure is too low to obtain useful data with the present setup, while at higher temperatures significant naphthalene pyrolysis occurs. As with toluene, oxidation of naphthalene in air limited the upper temperature to around 900 K.
5.1 Fluorescence spectra
Figure 4 depicts the temperature dependence of toluene fluorescence spectra in 1 bar N2 normalized to their respective peak intensities. Spectra recorded in CO2 (not shown) exhibit similar shapes and positions. Spectra were smoothed by an adjacent-average filter with a window size of 3 nm. At room temperature emission extends from roughly 265 to 340 nm with a maximum at around 280 nm. With increasing temperature, a slight red-shift of this maximum of about 2 nm per 100 K can be observed, and the tail on the long wavelength side of the spectra becomes stronger relative to the peak. Due to the limited spectral resolution of the present experiment of about 3 nm (indicated by the arrows in Fig. 4), a possible “fine structure” is not observed in the spectra at lower temperatures. The spectral shape does not change with increasing pressure, even when air is used as bath gas. This finding is in accord with Koban et al. [6] and Rossow [8] who found that upon 266 nm excitation no spectral shift could be observed with either increasing O2 partial pressure or total pressure.

Peak-normalized toluene fluorescence spectra at various temperatures in 1 bar N2 bath at a toluene number density of ~2 × 1023 m−3. Spectra in CO2 (not shown) are similar. The arrows indicate the spectral resolution of the detection system by the FWHM of a recorded mercury line at 254 nm from a low-pressure discharge lamp
Figure 5 shows the temperature dependence of naphthalene fluorescence spectra diluted in N2 bath gas at a total pressure of 1 bar and normalized to their respective peak intensities. As for toluene, within the measurement accuracy spectra in CO2 (not shown) exhibit indistinguishable shapes and positions. At 374 K, emission extends from roughly 305 to 400 nm with a maximum at around 335 nm. For naphthalene, the red-shift of the emission spectrum with increasing temperature (5 nm per 100 K) is more pronounced than for toluene. Again, the tail on the long wavelength side of the spectrum becomes stronger relative to the peak emission. These results are consistent with work by other groups [3, 20, 26], regarding the general position and the FWHM. Orain et al. [3], however, report a different fine structure of the emission peak, where the first vibrational peak is larger than the second.

Peak-normalized fluorescence spectra of naphthalene upon 266 nm excitation at various temperatures in 1 bar N2 at a naphthalene number density of ~2 × 1022 m−3. The spectra in CO2 are similar. The arrows indicate the spectral resolution of the detection system
5.2 Effective fluorescence lifetimes
5.2.1 Toluene
The present work enlarges the range of data from our previous work [25] on the temperature and bath gas dependence of effective fluorescence lifetimes of toluene to pressures higher than atmospheric. Evaluated fluorescence lifetimes of toluene in N2 as bath gas are shown in Fig. 6 recorded for various temperatures versus total pressure between 1 and 10 bar. Generally the lifetimes strongly decrease with temperature, slightly more at higher pressures. At fixed temperatures larger than 296 K the lifetimes slightly decrease with increasing pressure. A similar quenching effect with pressure in N2 was also observed by Rossow [8] for time-integrated signal intensities determined at 450 and 700 K. When normalizing his data with the respective lifetimes at 1 bar at the nearest temperatures from our measurements they show good agreement (cf. Fig. 6). In CO2 (not shown), the quenching effect is slightly more pronounced (by about 5 %) compared to N2. A comparison with the model of Koban et al. is not possible because it does not include the effect of total pressure. In Sect. 6, we will expand Koban’s model to higher pressures in N2. The results of this modified model are included in Fig. 6.

Effective fluorescence lifetimes of toluene in N2 (symbols) as a function of total pressure at various temperatures. For comparison, normalized signal intensity values from Rossow [8] are included (cross symbols). Lines are from our modified fluorescence model. See text for details
Figure 7 shows effective fluorescence lifetimes of toluene in air (symbols) as a function of temperature for pressures from 1 to 10 bar. Due to the high O2 partial pressures, fluorescence lifetimes are much shorter than in N2 (cf. Fig. 6). For constant temperature the fluorescence lifetime decreases with increasing air pressure, more significantly at lower temperatures. The effect of O2, however, saturates at an air pressure of around 10 bar (i.e., a O2 partial pressure of around 2 bar). The temperature drop-off is shifted to higher temperatures with increasing pressure due to increased quenching by the higher O2 concentrations. For comparison, predictions from the Koban model [6] are included in Fig. 7 (lines) by normalizing each pressure to the respective measured lifetimes at room temperature. The model shows large deviations at total pressures higher than 1 bar, especially at higher temperatures, which is due to the fact that the model was not developed for this pressure regime.

Effective fluorescence lifetimes of toluene as a function of temperature at various air pressures. Solid line relative fluorescence quantum yield calculated from the Koban model [6] normalized to the respective values at room temperature for each pressure
5.2.2 Naphthalene
Previous work by Ossler et al. [26] has shown that the decay of naphthalene fluorescence can be described by two lifetime components, representing two different deactivation pathways of the excited molecule. In contrast to toluene, the 266 nm excitation in naphthalene already provides enough excess energy at low temperatures (~6,000 cm−1 at 400 K) to activate an additional deactivation pathway [31, 37]. Therefore, double-exponential functions were applied when fitting all our naphthalene fluorescence decay profiles. Section 7 will discuss this topic in more detail.
In Fig. 8, the evolution of both components of naphthalene fluorescence lifetime with temperature is shown for pressures of N2 between 1 and 10 bar. At temperatures higher than 1,000 K the limited signal-to-noise ratio of the fluorescence decay profiles prevented double-exponential fits from extracting the short lifetime component. The fluorescence lifetime decreases by a factor of 370 when temperature increases from 373 to 1,123 K, i.e., from 104 ± 6 ns down to 0.28 ± 0.05 ns. Noteworthy in Fig. 8 is the marked increase of the negative slope of the data points above about 800 K. Compared with results from toluene (cf. Fig. 6) for naphthalene the effect of pressure is not only much reduced, but it increases instead of decreases with increasing pressure (cf. inset in upper panel of Fig. 8), although this trend levels out within the error limits at temperatures higher than 800 K. This difference in behavior will be discussed in Sect. 7. At temperatures below 800 K our data at 1 bar agree well with results from Ossler et al. [26]. However, at higher temperatures our evaluated lifetimes are significantly shorter than those reported by Ossler et al. which might be attributed to their limited temporal resolution of 2.5 ns. In CO2 lifetimes at 1 bar (not shown here) are the same as those in N2 within experimental error. At pressures above atmospheric lifetimes in CO2 are marginally longer (by about 5 %) than in N2. The results of a phenomenological model for naphthalene introduced in Sect. 6 are included in Fig. 8 for a total pressure of 1 bar.

Effective fluorescence lifetimes of naphthalene as a function of temperature for several total pressures in N2. Upper part long lifetime component, lower part short lifetime component. Comparison to literature data from Ossler et al. [26]. Inset in the upper part long lifetime component versus total pressure of N2 for two temperatures. The long lifetime component is compared to calculated values (for 1 bar N2) from our model presented in Sect. 6
As shown in the lower panel in Fig. 8, the short lifetime component shows a similar behavior with temperature, although with much larger experimental scatter. At temperatures up to 600 K the lifetime nearly stays constant at around 7 ns, while at higher temperatures it decreases to 0.6 ns at 923 K. A pressure effect could not be observed within the error limits. Within the measurement uncertainty, our values at 1 bar total pressure are consistent with those of Ossler et al. [26], while at temperatures beyond 800 K our lifetimes are smaller by about a factor of 2.
In air the effect of temperature on the fluorescence lifetime is quite different from the behavior in N2 (cf. Fig. 9). For constant temperature, increased air pressure causes a strong decrease of fluorescence lifetime due to the increased O2 number density. The quenching effect for naphthalene is stronger than for toluene (cf. Fig. 7). For naphthalene the on average lifetime decrease ranges from about 1.1 ns at 1 bar to about 0.05 ns at 10 bar, while for toluene at 373 K these values change from 0.7 ns to 0.09 ns in the same pressure interval. For each air pressure the fluorescence lifetime only slightly changes with temperature in the range from 373 to 700 K, and then drops off slightly, before at the highest temperatures a strong decrease is observed. This behavior is similar to that reported by Orain et al. [3], who also found a plateau region for time-integrated spectral intensities at 1 bar total pressure. For air pressures above 2 bar the range of constant lifetimes (within experimental error) is extended to even higher temperatures. In our range of conditions the amplitude of the short lifetime component is too short to be determined with sufficient accuracy. Our measurements show that for naphthalene quenching by O2 almost dominates over the temperature effect—in contrast to toluene, where a pronounced temperature effect is still present in air up to a pressure of about 8 bar (cf. Fig. 7). Therefore, our results support the conclusion of Kaiser and Long [20] that naphthalene should be a suitable tracer for fuel/air-ratio measurements. Figure 9 also displays calculated lifetime values for various total pressures normalized to the respective value at 373 K for all air pressures above atmospheric using a phenomenological fitting function described below. As these functions are derived from data at 1 bar total pressure, differences at elevated pressures might occur due to effects not covered by this functional dependence, e.g., non-linear Stern–Volmer behavior at higher total pressures. More realistic models based on the photo-physical background of these aromatic species, e.g., considering the detailed energy transfer and relaxation processes in the S1 state, will be necessary to improve the consistency with experimental data.

Effective fluorescence lifetimes of naphthalene as a function of temperature for several air pressures. Lines are lifetimes calculated from a fitting function introduced in Sect. 6 normalized to the respective value at 373 K
The lifetimes at 875 K as the highest temperature are significantly smaller than the modeled ones. This discrepancy is attributed to the onset of pyrolysis and oxidation of naphthalene in the gas flow which makes it uncertain as to whether signal originates from naphthalene or contributions from pyrolysis and oxidation products.
5.3 Stern–Volmer coefficients
As temporally integrated fluorescence intensities of toluene show Stern–Volmer behavior [6, 8], according to Eq. (4), this should also be applicable to our measured effective fluorescence lifetimes. To validate this, Stern–Volmer coefficients for toluene were determined from the slopes of previous lifetime data [25] plotted for constant atmospheric pressure (with N2) versus O2 partial pressures and several temperatures (cf. Eq. 4). The resulting temperature-dependent Stern–Volmer coefficients for quenching of toluene by O2 are depicted in Fig. 10 (filled squares) together with values from Koban et al. [6] and Rossow [8]. There is overall agreement, especially with the values of Koban et al., while up to 550 K the Stern–Volmer coefficients of Rossow are systematically higher by about 30 %. Shifting his data by about 25 K towards lower temperatures brings the values in agreement with our data. Koban's and our coefficients follow the same single-exponential trend line (solid line in Fig. 10) and are close to each other within their combined measurement uncertainty. Thus, it is more likely that the values of Rossow are too high than that both our and Koban's values are too low.

Stern–Volmer coefficients for toluene and naphthalene at various temperatures for 1 bar total pressure (with N2). Values of this work (filled and open squares) compared to values for toluene by Koban et al. (filled stars) [6] and Rossow (plus signs) [8], and for naphthalene by Orain et al. (crosses) [3] and Kaiser and Long (open rhomb) [20]. Lines are mono-exponential fits to our data
We also investigated collisional quenching of naphthalene by O2 at various temperatures by varying the O2 partial pressure (between 0 and 210 mbar) at a total cell pressure of 1 bar (held with N2). From previous work it is known that collisional quenching of naphthalene by O2 follows Stern–Volmer behavior [3, 20], cf. Eq. (4). As shown in Fig. 10, within the overlapping temperature range our evaluated Stern–Volmer coefficients (filled squares and fitted exponential) are systematically lower and with a somewhat larger slope than the results from Orain et al. [3]. The result from Kaiser and Long [20] at 500 K (open diamond) is lower by 40 % relative to the function fitted to our data. The Stern–Volmer coefficients for naphthalene are larger than those of toluene, indicating higher fluorescence quenching efficiency in collisions with O2. This is in accord with above that in air the naphthalene fluorescence lifetime is nearly unaffected by temperature.
6 Model functions
One simplification made in Koban’s fluorescence model for toluene [6] (cf. Eq. 5) is the neglect of the effect of total pressure on the relative fluorescence quantum yield. To adapt the model to our measurements at higher pressures in N2, an additional multiplicative pressure-dependent factor was introduced, as shown in Eq. (6)
where p tot is the total pressure in bar. To obtain the new fitting coefficients B 1, B 2, and B 3, the pressure-dependent lifetime data in Fig. 6 were first fitted with exponential functions for each temperature, normalized to their value at 1 bar for each temperature. Then the coefficients of the exponential fits were plotted against temperature and fitted with polynomials to yield the expressions in Table 1. All other parameters in Eq. 6 can be found in [6].
A comparison of calculated fluorescence lifetimes to the experimental data is presented in Fig. 6 showing overall good agreement when reproducing the dependence on total pressure with N2 as buffer gas. At atmospheric pressure the model is reduced to Koban’s expression. It should be noted that these modifications are only valid for the conditions of the present experiments, i.e., 266 nm excitation, temperatures between room temperature and 950 K, and for total pressures of N2 ranging from 1 to 10 bar. Additional quenching effects, that occur in the presence of O2 (cf. Fig. 7) at pressures above atmospheric, are not covered by Eq. (6).
The model for naphthalene is a parameterized functional fit to the experimental lifetime data. As naphthalene fluorescence decay curves contain two lifetime components it is more complex to create a model to predict relative quantum yields. The short lifetime component, however, is low in amplitude and not measureable in air. Therefore, for simplification, we neglect this component and focus on the long lifetime component.
The fitting procedure was such that, initially the temperature dependence in pure N2 at 1 bar was fitted, then the influence of total pressure in a similar way as for toluene, and then the sensitivity to O2 quenching was taken into account by considering a temperature-dependent Stern–Volmer expression as a multiplicative factor. Thus, the model (Eq. 7) consists of three factors (distinguished by square brackets): The first one describes the temperature dependence of the relative naphthalene quantum yield (ϕ rel) at 1 bar in N2. The second factor represents the pressure dependence in N2 bath in the absence of O2; it equals unity at 1 bar total pressure. The third term considers O2 quenching with a Stern–Volmer relationship, with the temperature dependence of the Stern–Volmer coefficient is derived from the exponential fit in Fig. 10.
Table 2 lists the respective fit-parameters of the model. Fluorescence lifetimes can be calculated by multiplying ϕ rel with a constant factor of 104 ns as shown in the last row of Table 2.
The derived model is valid from 373 to 1,100 K without O2, and up to 900 K in the presence of O2. The results of the model are compared with the temperature dependence of the data in N2 at 1 bar in Fig. 8, and with the temperature dependence in air at various total pressures in Fig. 9, respectively. At air pressures above 1 bar the agreement between experimental data and model predictions is reduced, especially since the modeled lifetimes evolve through a shallow maximum with increasing temperature, which is not reflected in the data.
7 Discussion
The decrease in fluorescence lifetime of toluene (cf. Fig. 6) with increasing pressure is in contrast to pressure effects known from, e.g., ketones (e.g., [38, 39]) and naphthalene (e.g., [3] and this work). This phenomenon is thought to arise from the fact that upon 266 nm excitation toluene is excited near the 0–0 band (~580 cm−1 at 300 K) of the S1 origin, where the stabilization effect of collisions with the bath gas is inverted into a destabilization effect. This was interpreted as a so-called photo-induced cooling effect of the excited-state vibrational distribution, subsequently followed by collisional heating by the bath gas towards thermal equilibrium [40, 41]. This pressure-induced destabilization effect is also found for other aromatic species when excited near the vibrationless S1–S0 transition by photon absorption, e.g., anthracene excited at 365 nm [42]. In contrast to toluene, excitation of naphthalene at 266 nm deposits energy far above (~6,000 cm−1 at 400 K) its S1 0–0 band origin. Therefore, collisions cause stabilization of population in S1 due to vibrational relaxation, and thus an increase of the fluorescence lifetime with increasing pressure. This stabilization effect was first described for naphthalene by Beddard et al. [43] theoretically and proven experimentally.
With respect to excited-state energy transfer and relaxation in previous literature for toluene [31], a so-called “third-decay channel” is discussed if a certain amount (~2,150 cm−1) of excess energy in the S1 state is reached. For benzene this third-decay channel opens up because of enhanced internal conversion due to nonadiabatic transitions via conical intersections [44, 45]. It is postulated that for toluene similar processes occur [8, 31]. This additional loss channel also causes the appearance of a second lifetime component in the fluorescence decay curves, e.g., when at room temperature excitation is accomplished with wavelengths shorter than 250 nm. In addition to the typical fluorescence lifetimes measured in the present work, Zimmermann et al. [29] found a short lifetime component when excitation of toluene is performed at 248 nm at room temperature. Alternatively, for 266 nm excitation this deactivation channel is opened for temperatures higher than ~480 K. At temperatures above ~500 K our toluene decay curves can be fitted equally well using double-exponential decays. However, the fast decay component in toluene fluorescence has a low amplitude making meaningful data evaluation difficult.
For naphthalene, a similar “third-decay channel” is reported at a threshold of 10,000 cm−1 [46]. When exciting beyond this threshold the non-radiative decay rate increases rapidly. This energy corresponds to a temperature of about 750 K. Thus, we can explain our change in logarithmic slope in Fig. 8 for the long lifetime component and the beginning decrease in the short lifetime component at around 800 K. Above this limit the non-radiative processes are enhanced and thus, the lifetime is shortened “faster” than below this level. The short lifetime component in naphthalene, however, cannot be explained by this third-decay channel, as it was above in the case of toluene because a short lifetime component can already be observed at excess energies in S1 below this threshold. One possible alternative found in literature is fluorescence from higher excited states like S2 [26] competing with vibrational relaxation to S1, but satisfactory explanations have not been published yet.
The presented effective fluorescence lifetimes of toluene were compared to the existing fluorescence yield model of Koban et al. [6]. In the regime of conditions where this model was validated (i.e., 300–700 K, 1 bar, p O2 between 0 and 300 mbar) we found good agreement between the calculated (from the quantum yield) and the measured fluorescence lifetimes. However, when extrapolating calculated values of the original Koban model to higher pressures of air (cf. Fig. 7), significant discrepancies occur at simultaneously elevated temperatures and pressures, even when normalizing the model function to the respective room temperature value which even gets worse when the modified model is applied. As it was shown previously, the Stern–Volmer assumption for O2 quenching is no longer valid for high temperatures (>600 K) and O2 partial pressures beyond 300 mbar at 1 bar total pressure [6]. Therefore, it seems not unlikely that this non-Stern–Volmer behavior is responsible for the discrepancies observed between the modeled and measured values at simultaneously higher air pressures and temperatures.
For naphthalene the applicability of the model is satisfactory in N2 at various total pressures and in air at 1 bar. At higher pressures of air discrepancies occur. In the literature a non-Stern–Volmer behavior of O2 quenching is reported [26] at simultaneously elevated temperatures and pressures, similar as found for toluene, which might also be an explanation for the model shortcomings for naphthalene found in this work.
8 Conclusions
Effective fluorescence lifetimes of gas-phase toluene and naphthalene were determined upon picosecond laser excitation at 266 nm in a heated ceramic flow cell as a function of temperature (toluene: 296–1,026 K, naphthalene: 374–1,137 K), pressure (1–10 bar), and bath gas composition (N2, N2/O2, air, and CO2). Wavelength- and time-resolved fluorescence detection was accomplished with a streak camera coupled to a spectrometer. Normalized fluorescence spectra are presented for both aromatics as a function of temperature. At 1 bar total pressure the decay time of the spectrally integrated fluorescence was identical in N2 and CO2 within experimental uncertainty. The fluorescence lifetime decreases significantly with increasing temperature, for toluene more pronounced than for naphthalene. At room temperature the presence of O2 strongly reduces the fluorescence lifetime, and the derived Stern–Volmer factors for naphthalene are larger than those of toluene. However, for both aromatics the susceptibility to O2 quenching is reduced at higher temperatures.
For tracer LIF applications in engine studies our finding that both N2 and CO2 almost equally affect the fluorescence lifetime of toluene and naphthalene may render these tracers suitable alternatives to, e.g., 3-pentanone [47]. This would simplify the interpretation of time-integrated LIF-signal-intensity measurements in mixing studies and thermometry during the compression stroke when significant amounts of exhaust gas residuals are present under conditions of, e.g., high exhaust gas recirculation.
For toluene the measurements at 1 bar total pressure of N2 confirmed that the fluorescence lifetime is proportional to the fluorescence quantum yield predicted by the phenomenological model of Koban et al. [6]. The model was modified by an additional multiplicative exponential factor to take into account the pressure dependence of the fluorescence quantum yield in the absence of O2. In total, for 266 nm excitation, and within the validated range of temperature, N2 pressure, and O2 partial pressure (at atmospheric pressure), effective fluorescence lifetimes of toluene can be predicted with an overall uncertainty of about 10 %. For naphthalene a similar phenomenological model was set up for predicting fluorescence lifetimes as a function of temperature, total pressure, and O2 partial pressure. However, as for toluene the model fails in correctly predicting fluorescence lifetimes (i.e., quantum yields) in air at simultaneously elevated temperatures and pressures. Our results show that more sophisticated phenomenological models such as the “step-ladder” model introduced by Thurber et al. [9] for acetone, and recently applied to toluene and naphthalene by Rossow [8] will be more appropriate for modeling effective fluorescence lifetimes of these two aromatic species, thereby also providing a deeper understanding of their photophysics for varying environmental conditions.
Although the modeling can be further optimized beyond the current fitting strategies by using approaches such as [8, 9, 38] the data and its fitting functions are highly relevant for quantitative LIF diagnostics. They close an important gap for conditions with simultaneously elevated temperatures and pressures, and hence support quantitative LIF diagnostics in, e.g., IC engines which otherwise would have to be covered by uncertain extrapolations from low-pressure, low-temperature data. As examples, for cases of constant oxygen concentration and independent on local tracer concentration novel measurement strategies would enable “on-the-fly” temperature determinations from effective fluorescence lifetimes during engine compression strokes [48]. Also, at constant temperature oxygen concentrations can be determined [49], even in two-dimensional approaches [49, 50].
References
C. Schulz, V. Sick, Prog. Energy Combust. Sci. 31, 75 (2005)
W. Koban, C. Schulz, SAE technical paper series 2005-01-2091 (2005)
M. Orain, P. Baranger, B. Rossow, F. Grisch, Appl. Phys. B 102, 163 (2011)
P. Baranger, M. Orain, F. Grisch, AIAA paper 2005–828 (2005)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Phys. Chem. Chem. Phys. 6, 2940 (2004)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 777 (2005)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Appl. Phys. B 80, 147 (2005)
B. Rossow, Photophysical processes of organic fluorescent molecules and kerosene—application to combustion engines, in dissertation thesis. Institut des Sciences Moléculaires d’Orsay, Université Paris-Sud 11, Paris, France (2011)
M.C. Thurber, F. Grisch, B.J. Kirby, M. Votsmeier, R.K. Hanson, Appl. Opt. 37, 4963 (1998)
B.H. Cheung, R.K. Hanson, Appl. Phys. B 106, 755 (2012)
A.M. Bass, J. Chem. Phys. 18, 1403 (1950)
J. Savard, Ann. Chim. 10, 287 (1929)
E. Wiedemann, G.C. Schmidt, Ann. Phys. 56, 18 (1895)
A. Reimann, Ann. Phys. 80, 43 (1926)
C.S. Burton, W.A. Noyes, J. Chem. Phys. 49, 1705 (1968)
N. Nijegorodov, V. Vasilenko, P. Monowe, M. Masale, Spectrochim. Acta A 74, 188 (2009)
K. Mohri, M. Luong, G. Vanhove, T. Dreier, C. Schulz, Appl. Phys. B 103, 707 (2011)
J. E. Dec, W. Hwang, SAE technical paper series 2009-01-0650 (2009)
M. Luong, R. Zhang, C. Schulz, V. Sick, Appl. Phys. B 91, 669 (2008)
S.A. Kaiser, M.B. Long, Proc. Combust. Inst. 30, 1555 (2005)
F. Ossler, T. Metz, M. Aldén, Appl. Phys. B 72, 479 (2001)
J. Yoo, D. Mitchell, D.F. Davidson, R.K. Hanson, Exp. Fluids 49, 751 (2010)
R. Devillers, G. Bruneaux, C. Schulz, Appl. Phys. B 96, 735 (2009)
W. Koban, J.D. Koch, V. Sick, N. Wermuth, R.K. Hanson, C. Schulz, Proc. Combust. Inst. 30, 1545 (2005)
S. Faust, T. Dreier, C. Schulz, Chem. Phys. 383, 6 (2011)
F. Ossler, T. Metz, M. Aldén, Appl. Phys. B 72, 465 (2001)
P. Avouris, W.M. Gelbart, M.A. El-Sayed, Chem. Rev. 77, 793 (1977)
A. Ehn, B. Kaldvee, J. Bood, M. Aldén, Appl. Opt. 48, 2373 (2009)
F.P. Zimmermann, W. Koban, C.M. Roth, D.-P. Herten, C. Schulz, Chem. Phys. Lett. 426, 248 (2006)
G.M. Breuer, E.K.C. Lee, Chem. Phys. Lett. 14, 404 (1972)
M. Jacon, C. Lardeux, R. Lopez-Delgado, A. Tramer, Chem. Phys. 24, 145 (1977)
E.H. Kincaid, V. Worah, M.D. Schuh, J. Chem. Phys. 94, 4842 (1991)
C.G. Hickman, J.R. Gascooke, W.D. Lawrance, J. Chem. Phys. 104, 4887 (1996)
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn. (Springer Science + Business Media, LLC, New York, 2006)
T.B. Settersten, A. Dreizler, R.L. Farrow, J. Chem. Phys. 117, 3173 (2002)
T.B. Settersten, B.D. Patterson, J.A. Gray, J. Chem. Phys. 124, 234308 (2006)
J.C. Hsieh, C.-S. Huang, E.C. Lim, J. Chem. Phys. 60, 4345 (1974)
J. D. Koch, Fuel Tracer Photophysics for Quantitative Planar Laser-Induced Fluorescence, in dissertation thesis. Stanford University, Stanford, California, USA, 2005
V. Modica, C. Morin, P. Guibert, Appl. Phys. B 87, 193 (2007)
Y. He, E. Pollak, J. Chem. Phys. 116, 6088 (2002)
H. Wadi, E. Pollak, J. Chem. Phys. 110, 11890 (1999)
L.A. Barkova, V.V. Gruzinskii, M.N. Kaputerko, J. Appl. Spectr. 47, 186 (1987)
G.S. Beddard, G.R. Fleming, O.L.J. Gijzeman, G. Porter, Proc. R. Soc. Lond. A 340, 519 (1974)
R.E. Smalley, Annu. Rev. Phys. Chem. 34, 129 (1983)
C.E. Otis, J.L. Knee, P.M. Johnson, J. Chem. Phys. 78, 2091 (1983)
S.F. Fischer, A.L. Stanford, E.C. Lim, J. Chem. Phys. 61, 582 (1974)
D. A. Rothamer, Developement and Application of Infrared and Tracer-Based Planar Laser-Induced Fluorescence Imaging Diagnostics, in dissertation thesis. Stanford University, Department of Mechanical Engineering, Stanford, California, USA, 2007
E. Friesen, C. Gessenhardt, S. Kaiser, T. Dreier, C. Schulz, in LACSEA2012. San Diego, CA, USA, 2012
A. Ehn, O. Johansson, A. Arvidsson, M. Aldén, J. Bood, Opt. Expr. 20, 3043 (2012)
T. Ni, L.A. Melton, Appl. Spectr. 50, 1112 (1996)
Acknowledgments
The authors acknowledge funding of this work by the Deutsche Forschungsgemeinschaft. The authors thank Quyen Dinh (University of Pennsylvania, USA) for experimental assistance during a summer internship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Faust, S., Tea, G., Dreier, T. et al. Temperature, pressure, and bath gas composition dependence of fluorescence spectra and fluorescence lifetimes of toluene and naphthalene. Appl. Phys. B 110, 81–93 (2013). https://doi.org/10.1007/s00340-012-5254-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-012-5254-8