
Abstract
Anisole is a promising candidate for use as fluorescent tracer for gas-phase imaging diagnostics. Its high-fluorescence quantum yield (FQY) and its large Stokes shift lead to improved signal intensity (up to 100 times stronger) compared with the often used toluene. Fluorescence spectra and effective fluorescence lifetimes of gaseous anisole were investigated after picosecond laser excitation at 266 nm as a function of temperature (296–977 K) and bath gas composition (varying amounts of N2 and O2) at total pressures in the range of 1–10 bar to provide spectroscopic data and FQY for applications, e.g., in in-cylinder measurements in internal combustion engines. Fluorescence spectra of anisole extend from roughly 270–360 nm with a peak close to 290 nm at 296 K. The spectra show a red-shift with increasing temperature (0.03 nm/K) and O2 partial pressure (5 nm from N2 to air). In the investigated temperature range and in pure N2 at 1 bar total pressure the effective fluorescence lifetime drops with increasing temperature from 13.3 ± 0.5 to 0.05 ± 0.01 ns. Increasing the total pressure of N2 leads to a small decrease of the lifetime at temperatures above 400 K (e.g., at 525 K from 4.2 ± 0.2 ns at 1 bar to 2.7 ± 0.2 ns at 10 bar). At constant temperature and in the presence of O2 the lifetimes decrease significantly (e.g., at 296 K from 13.3 ± 0.5 ns in N2 to 0.40 ± 0.02 ns in air), with this trend diminishing with increasing temperature (e.g., at 675 K from 1.02 ± 0.08 ns in N2 to 0.25 ± 0.05 ns in air). A phenomenological model that predicts fluorescence lifetimes, i.e., relative quantum yields as a function of temperature, pressure, and O2 concentration is presented. The photophysics of anisole is discussed in comparison with other aromatics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Laser-induced fluorescence (LIF) is a powerful and often used technique for non-intrusive visualization of, e.g., mixing gaseous [1–4] and liquid [5] flows and gas temperature [6–9], with extensions to diagnostics in internal combustion engines [10–12]. In many cases a so-called fluorescence tracer is added to a non-fluorescing fluid to visualize the desired property, e.g., fuel distribution or temperature, from a species with known fluorescence properties. Typical organic tracers for gas-phase diagnostics include aromatics, ketones, and less frequently, amines and ethers.
Small aromatic molecules are preferred tracer species because of their high-fluorescence quantum yield (FQY), with toluene and naphthalene being used in the majority of cases [1, 4, 9, 12–18] because of the availability of previous experience and photophysical data. These conventional candidates, however, have not been chosen based on an exhaustive screening, and depending on the measurement situation it makes sense to select other candidates with improved properties. Anisole (methoxybenzene, C6H5OCH3) is a single-ring aromatic hydrocarbon [19] that has the potential to provide up to 100 times stronger signal under otherwise identical conditions in typical measurement situations compared with toluene because of a larger FQY, a larger absorption cross-section, and a larger Stokes shift. Table 1 summarizes the main thermodynamic and photo-physical properties of anisole at room temperature in comparison with those of toluene and naphthalene.
Practical tracer-LIF-imaging applications often use excitation at 266 nm from quadrupled Nd:YAG lasers. The fluorescence is discriminated against elastically scattered laser light with long-pass filters. In this case, toluene has the disadvantage of a small Stokes shift that causes the excitation wavelength to overlap with the fluorescence spectrum. Stray light suppression, therefore, also rejects about 30 % of the fluorescence light. In contrast, anisole has a larger Stokes shift with the peak of the fluorescence spectrum of anisole being 10–15 nm red-shifted compared with toluene (cf., Fig. 6). The same cut-off filter now rejects less than 5 % of the fluorescence signal. Together with the 63 % larger FQY and the about 50 times larger absorption cross section, a significantly improved signal strength can be expected under otherwise identical conditions which makes anisole an attractive alternative for typical LIF imaging applications.
The Stokes shift of naphthalene upon UV excitation is even larger compared with that of anisole, and the dependence of its FQY on temperature, pressure and O2 concentration has been investigated in some detail (e.g., [1, 16, 24]). Its use as an alternative to toluene, however, is hampered by three factors: First, the vapor pressure of naphthalene is very low (0.024 mbar at 293 K), leading to limited concentrations in the gas phase and thus low LIF signal intensities at low temperatures. Second, naphthalene is solid at room temperature and can, therefore, be handled as solution only. Third, the carcinogenic properties of naphthalene make it less attractive for applications in flow and combustion studies open to the atmosphere.
Anisole is a cheap, non-toxic, non-carcinogenic liquid with a vapor pressure of 3.6 mbar at room temperature. Its fluorescence was first investigated in 1924 [25] and the application as a tracer has been discussed earlier [19]. However, there are no systematic investigations of fluorescence spectra or fluorescence lifetimes (i.e., relative FQY) available within a wider range of conditions like temperature, pressure, or bath gas composition, which are a prerequisite for quantitative interpretations of anisole LIF in diagnostic applications in, e.g., gas flow mixing or internal combustion engines.
For fundamental studies of energy-transfer processes in the S1 state it is also of interest that the 0–0 energy gap between S0 and S1 in anisole (36,394 cm−1 [26]) is in between those of toluene (37,476 cm−1 [27]) and naphthalene (32,000 cm−1 [28]). The radiative and non-radiative processes following laser excitation depend severely on the initially populated vibronic energy level in S1 relative to the vibrationless S1 origin [29]. Especially toluene and naphthalene differ significantly in their temperature and pressure dependence of the FQY [24, 29]. With fixed excitation wavelength as in our experiment it, therefore, is of great interest to study a molecule with electronic structure between those of toluene and naphthalene to further understand the interplay between energy-dependent vibrational relaxation towards thermal equilibrium, non-radiative transitions, and fluorescence emission of aromatic species.
In this work we present a systematic investigation of the fluorescence spectra and lifetimes (and thus, relative FQY [30]) of anisole as a function of temperature, pressure, and O2 partial pressure. This supplements the sparse experimental database on the photo-physics of this species for a wide range of typical conditions encountered in tracer-LIF-based diagnostics applications. In a second step, we will discuss the photo-physical properties in the context of anisole having an energetic structure in between those of toluene and naphthalene with respect of the size of its π-electron system.
2 Theoretical background
To simplify the description of the photo-physical processes in aromatic molecules one assumes infinitely fast excitation into just one singlet state S1 and ignores radiative processes originating from triplet states [30]. The intra-molecular deactivation processes of the excited molecules [M*] can then be described by the kinetic equation
[18, 30] where k fl, k ISC, and k IC are the rates (all in s−1) of fluorescence emission (radiative decay), intersystem crossing, and internal conversion, respectively (cf., Fig. 1).

Schematic overview of photophysical processes in organic molecules (Jablonski diagram, adapted from [31]): fluorescence as radiative process is marked by wavy arrows and non-radiative processes (ISC inter system crossing, IC internal conversion, quenching processes) are marked by straight arrows. The horizontal arrow length indicates the relative respective rate constants. ΔE thermal is the thermalization level in each state
The total depopulation rate, k tot, is then connected to the experimentally accessible effective fluorescence lifetime, τ eff, via
where k nr is the non-radiative decay rate (s−1) as the sum of all decay rates except k fl. k q is the quenching rate (in cm3/s) with [Q] as the concentration of the quencher species. The relation between τ eff and the FQY, ϕ fl, is given by [30] with τ rad, the radiative lifetime:
Thus, the FQY is proportional to the effective fluorescence lifetime. On the other hand, if the quantum yield is known, it is possible to calculate the non-radiative decay rate, k nr. Using Eqs. (2) and (3) followed by rearrangement yields the relation between k nr, ϕ fl and τ eff
The various deactivation processes can be distinguished in intra-molecular (fluorescence, internal conversion, intersystem crossing) and inter-molecular processes (collisional quenching, cf., Fig. 1). For the second case energy transfer from the excited molecule to the collision partner occurs. The quenching rate depends on the collider concentration, the total collision rate, and the quenching cross-section—a property of the intermolecular interaction potential. Molecular oxygen is known as a highly efficient fluorescence quencher for aromatic molecules due to its triplet ground-state structure [14]. For most aromatic species a Stern–Volmer relation
exists, i.e., a linear relationship between the ratio of signal intensities S fl (and thus τ eff) without (index 0) and with presence of the quencher with number density n q. The usually temperature-dependent Stern–Volmer factor, k SV, can be determined experimentally by measuring signal intensities (or lifetimes) as a function of quencher number density [18].
3 Experiment
Measurements were performed with the experiment described in detail in [32]; therefore, only a brief overview is given here. Fluorescence measurements were carried out in a gaseous mixture of anisole and the respective bath gas continuously flowing through a cylindrical ceramic flow chamber (cf., Fig. 2) (outer diameter: 90 mm, length: 250 mm) with four viewing holes on its circumference that is placed inside a 400-mm-diameter stainless-steel pressure vessel. The fresh gas mixture enters the steel vessel through its bottom flange via a concentric tube-in-tube feed-through. This is connected with the outer larger diameter ceramic chamber. At its upper end it reaches an upper chamber where the flow direction is reversed and from where the heated gases flow downwards through the inner (measurement) chamber. The gas flow then exits through the inner tube through the feed-through in the bottom plate of the vessel. Ceramic heating elements, placed around the flow chamber, and thermally insulated against the outer vessel walls, heat the gas flow to up to 1,400 K at a maximum gas pressure of 10 bar. Gas temperature and pressure in the flow chamber were measured with an R-type thermocouple and a pressure transducer, respectively. A constant cell pressure is achieved by a back-pressure regulator in the exhaust line of the flow chamber. The stainless steel vessel has four optical ports sealed with quartz windows of 30 mm clear aperture in line with openings of the insulating material and the ceramic flow cell.

Schematic cut through the center plane of the high-temperature flow cell mounted in the center of the pressure vessel. Arrows indicate the gas flow. See text for more details
Anisole (>99 % purity, Sigma-Aldrich) was used as pure substance without further purification. Nitrogen and air were provided from vaporized liquid nitrogen and compressed air for pressures up to 6 bar, respectively. For higher pressures nitrogen was provided from a gas cylinder (Air Liquide). Liquid anisole was metered by a high-pressure pump (HPLC Compact Pump 2250, Bischoff-Chrom) before it was evaporated through a capillary and mixed with part of the main carrier gas in a heated chamber (CEM, Bronkhorst). Mixtures of various bath gases were delivered via mass flow controllers (Bronkhorst) and a small gas mixing chamber. To avoid condensation all tubes after the CEM were resistively heated to 470 K (max.). The O2 partial pressure in the chamber was varied by diluting a flow of air with variable amounts of N2.
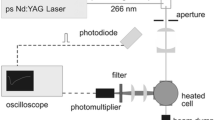
The optical setup is shown in Fig. 3. A flashlamp-pumped quadrupled Nd:YAG laser (PL2143B, Ekspla) delivered a maximum pulse energy of 7 mJ at 266 nm with a pulse width of 26 ps (FWHM of a temporal Gaussian beam profile) at a repetition rate of 10 Hz. The beam diameter was reduced by a Galilei telescope to 3 mm before entering the cell. The signal was collected perpendicular to the laser beam by a quartz lens and then focused with a second quartz lens and an UV-enhanced aluminum mirror on the entrance slit of a spectrograph (Acton SP2300, Princeton Instruments, f = 300 mm, 150 l/mm grating). The spectrally resolved signal was then imaged on the entrance slit of a streak camera (C5680-24C, Hamamatsu Photonics) with a streak module (M5677-01) and a CCD camera (Orca R2), which provides a maximum time resolution of 20 ps. Laser and camera were synchronized with trigger pulses from the laser via a pulse delay generator (DG645, Stanford Research Systems).

Experimental arrangement for spectrally and time-resolved fluorescence measurements of tracer species, CCD CCD camera, DG pulse delay generator
In test measurements the regime of linear fluorescence response as a function of laser fluence was investigated. In the present experiments a laser fluence of 33 mJ/cm2 was found to be below the saturation limit at room temperature (1 bar of N2 as bath gas). Above the limits of the linear regime, the fluorescence lifetime decreased with increasing excitation pulse energy. Thus, for gaining reliable photophysical data, excitation was kept in the linear regime. Because for aromatic species the saturation limit is known to shift to higher energies at elevated temperatures [13], for maintaining acceptable signal-to-noise ratios, with increasing temperature the fluence level was raised stepwise to 50 mJ/cm2. The anisole partial pressure was 2 mbar at 296 K and thus below its vapor pressure (cf., Table 1). At higher temperatures the partial pressure was increased to maintain a nearly constant anisole number density of 5 × 1022 m−3 in the measurement volume. Test measurements revealed that at this number density self-quenching of anisole fluorescence can be neglected.
4 Data evaluation
Depending on the signal-to-noise ratio, for each condition 500 to 1,000 individual measurements were carried out. This set of single-shot images was treated with a jitter-correction routine provided by the streak camera software to minimize smearing of the temporal profiles. Finally, an average background image was taken with the laser firing and pure N2 flowing through the cell. A resulting averaged image after background subtraction is depicted in Fig. 4, where wavelength is shown along the horizontal axis and time along the vertical axis. This image delivers either spectrally selected temporal profiles (horizontal binning of columns) or fluorescence spectra for specific time intervals after laser excitation (vertical binning of rows). Fluorescence spectra were extracted by binning a number of rows in the time range with significant pixel counts (red rectangle in Fig. 4). The resulting spectral profiles were then corrected for the instrument spectral response by recording the emission of a deuterium lamp with known spectral radiance using the same optical setup. At higher temperatures the fluorescence spectra also indicated whether tracer degradation occurred.

Sample image (average over 500 individual laser shots) of time-resolved anisole fluorescence spectra (296 K, 1 bar, N2 bath); the binning ranges for obtaining spectral and temporal profiles are marked by horizontal red and vertical green lines, respectively
Following the abovementioned procedure, Fig. 5 presents sample LIF excitation/decay profiles for three gas temperatures, together with the instrument response function, determined by recording the temporal profile of the laser stray light at 266 nm in the absence of anisole. Within the error limits the number of binned columns did not influence the evaluated effective fluorescence lifetimes; thus for S/N enhancement columns were binned covering the spectral range between 275 and 330 nm. Because decay times vary substantially with, e.g., temperature, for optimized temporal resolution the sweep speed was adjusted to use the full detector format. Therefore, for each temperature the recorded data trace may cover a different time span, and separate instrument functions need to be recorded.

Time-resolved LIF intensity for anisole in 1 bar N2 with excitation at 266 nm at various temperatures. The first 15 ns for each curve are shown. The signal recorded without tracer (stray light, red curve) is shown for the streak unit setting when recording the signal trace at 296 K (black symbols and blue line)
Signal decay times, τ, were determined using the convolve-and-compare technique [33, 34] by forming a convolution of a single-exponential decay with the measured instrument response function and least-squares fitting this to the experimental excitation/decay profiles. Figure 5 shows an example of a model signal profile (blue line) convoluted with the corresponding instrument response profile (red curve in Fig. 5) and fitted to the 296-K experimental fluorescence trace (black squares), with the residuals shown in the bottom green curve. Variable fitting parameters were the decay time(s) and a shift on the time axis to optimize the instance where experimental and simulated signal intensities reach their peak values. The maximum temporal resolution of the detection system was 50 ps.
5 Results
In the following the dependence of fluorescence spectra and effective fluorescence lifetimes of anisole are presented in the range 296 up to 977 K in N2, and at total pressures between 1 and 10 bar. At temperatures beyond 1,000 K significant pyrolysis takes place during the residence time of the gas mixture in the heated zone. The measurements are completed by studying the effect of O2 on the fluorescence emission in N2 with varying amounts of O2 at 1 bar total pressure. As the mixture ignites in air at high temperature, the maximum temperature was limited to 775 K.
5.1 Fluorescence spectra
Figure 6 shows the change with temperature of anisole fluorescence spectra in 1 bar N2 normalized to their respective peak intensities. The spectra at 875 and 977 K were smoothed with an adjacent-averaging filter with a window size of 3 nm. At room temperature emission extends from roughly 270–360 nm with a maximum close to 290 nm. Within the spectral resolution of about 3 nm no fine structure is observed, even at low temperatures, and the spectral shape does not change with increasing pressure. The peak maximum shifts by about 3 nm to the red per 100 K temperature increase and the signal in the long-wavelength tail becomes stronger resulting in an increased FWHM (cf., Fig. 7). The spectral shift was defined as the shift of the 50 % (relative to the peak) intensity point in the long-wavelength tail of the spectrum because the shift of the peak position cannot be accurately determined due to variations in the relative peak maxima with temperature. The FWHM was determined in the conventional way by forming the wavelength difference between the 50 % points of the short- and the long wavelength tail of the spectrum. The spectrum at room temperature is in good agreement with the result of Hirasawa et al. [19] for anisole vapor at room temperature upon 266 nm excitation when comparing the peak position (292 nm) and the FWHM (32 nm) with our values of 292 nm and 30 nm, respectively.

Peak-normalized fluorescence spectra of anisole at various temperatures in 1 bar N2 with 5 × 1022 m−3 anisole. For comparison a toluene LIF spectrum (in 1 bar N2 at 296 K) is also shown (thin solid line; from [24])

Spectral shift and width (FWHM) of the anisole fluorescence spectrum versus temperature in 1 bar N2. The spectral position is defined as the wavelength of the 50 % intensity point in the red tail of the spectrum
At temperatures above 850 K the spectra significantly change their shape (cf. Fig. 6) going along with increased noise due to low signal intensities. Thus, visible “fine structures” at these temperatures (especially at 977 K) are more due to noise than reality. We attribute this change in spectral shape to ongoing pyrolysis processes during the gas flow through the heated section of the cell, such that anisole and its pyrolysis products are excited by the laser radiation, and their respective fluorescence spectra overlap in the imaged wavelength region. We assume that the spectrum at 977 K is dominated by fragments after partial or complete decomposition of anisole, resulting in a significant increase in spectral red-shift and FWHM (cf., Fig. 7). For detailed discussion about the thermal decomposition of anisole and its initial pyrolysis product, the phenoxy radical, see [35] and references therein.
In contrast to toluene, where the O2 partial pressure does not influence the spectral shape after 266 nm excitation [14, 24], Fig. 8 shows a red shift of the anisole LIF spectra with increasing O2 partial pressure. The FWHM is unaffected by O2. The red shift is large at low O2 partial pressures and the effect saturates at around 100 mbar of O2 (cf. Fig. 9). The overall red-shift is about 5 nm at 296 K when changing from pure N2 to air as bath gas. The spectral shift as defined in this work indicates no temperature dependence of the O2-dependent spectral shift within experimental error up to temperatures of 600 K. At higher temperatures the red-shift is less pronounced.

Peak-normalized fluorescence spectra of anisole diluted in N2/O2 mixtures for various O2 partial pressures at 1 bar total pressure and 296 K with an anisole partial pressure of 2 mbar

Spectral shift of anisole fluorescence spectrum versus O2 partial pressure for several temperatures at 1 bar total pressure. As in Fig. 7, the spectral position is defined as the wavelength of the 50 % intensity drop in the long-wavelength tail of the fluorescence spectrum
5.2 Effective fluorescence lifetimes
For measuring fluorescence lifetimes with best temporal resolution, the streak speed was adjusted for the various experimental conditions to the expected fluorescence lifetimes (cf., Fig. 5). Figure 10 shows evaluated effective fluorescence lifetimes of anisole as a function of temperature for several pressures in N2. In the investigated temperature range the lifetime decreases from 13.3 ± 0.5 ns at 296 K to 0.05 ± 0.01 ns at 875 K, with an increasing slope at higher temperatures. In N2, the influence of the total pressure is small at low temperature, but it increase with temperature up to around 600 K. In total, the pressure influence is lower than observed for toluene [24].

Effective fluorescence lifetimes of anisole in N2 as a function of temperature for various pressures
At room temperature, the effective fluorescence lifetime of vapor-phase anisole of 13.3 ns is close to that for anisole dissolved in liquid cyclohexane of 11.1 ns [23]. For anisole a bi-exponential fluorescence decay was observed already at room temperature while for toluene a second lifetime component is observed only at temperatures above 500 K for the same excitation wavelength [36]. The fast decay component—being around 1 ns—however, has a small amplitude preventing a meaningful evaluation.
Molecular oxygen is a strong fluorescence quencher of the S1 state in aromatic molecules, such as toluene and naphthalene (e.g., [14, 24, 29, 32]). Figure 11 shows how O2 affects the effective fluorescence lifetime of anisole. At room temperature the fluorescence lifetime in air is reduced to 0.40 ± 0.02 ns (compared to 13.3 ± 0.5 ns in N2). The effect of O2 quenching decreases with increasing temperature, which is in line with observations for toluene and naphthalene [14, 24, 29, 32]. In the presence of O2, with increasing temperature the anisole fluorescence lifetime shows a weak increase at low temperatures due to the decreasing O2 collision rates after which the lifetime-reducing effect of the temperature dominates (inset in Fig. 11). At 775 K, the course of the lifetime with O2 partial pressure differs from that at lower temperatures which is attributed to ignition of the mixture (the auto ignition temperature of anisole is about 750 K).

Effective fluorescence lifetimes of anisole diluted in N2/O2 mixtures as a function of O2 partial pressure at 1 bar total pressure and for various temperatures. Measurements at 325 and 424 K are not shown to improve the clarity of the figure because they nearly match with the data at 196 and 373 K, respectively. Inset fluorescence lifetimes of anisole as a function of temperature in air at 1 bar total pressure
From the dependence of effective lifetimes on O2 concentration, temperature-dependent Stern–Volmer coefficients (cf., Sect. 2) were derived. For this purpose, the fluorescence lifetime in the absence of O2 divided by the lifetime in the presence of a certain O2 partial pressure is subtracted by 1 and plotted against the O2 partial pressure. Linear regressions are calculated and the slope is the Stern–Volmer coefficient. Figure 12 displays this for selected temperatures.

Stern–Volmer plots of anisole fluorescence quenched by O2 for selected temperatures at 1 bar total pressure
Results are presented in Fig. 13, together with those of toluene and naphthalene from our previous work [24], from Koban et al. [14] and Rossow [29] for toluene, and from Orain et al. [16] and Kaiser and Long [1] for naphthalene. The comparison shows that the O2 quenching is less effective by about one order of magnitude for anisole compared with naphthalene, but of the same order as that of toluene over the entire temperature range. On the other hand, the temperature sensitivity of the Stern–Volmer coefficient is almost the same as that of naphthalene but somewhat smaller than that of toluene. This leads to the conclusion that for 300–450 K toluene is more efficiently quenched by O2 than anisole, whereas above 500 K this order is slightly reversed.

Stern–Volmer coefficients for anisole, toluene, and naphthalene at various temperatures for 1 bar total pressure. Values for anisole of this work (solid squares) are compared with previous results from toluene by Faust et al. (filled circles [24]), Koban et al. (open stars [14]), and Rossow (crosses [29]), and for naphthalene by Faust et al. (filled triangles [24]), Orain et al. (crosses [16]), and Kaiser and Long (open diamond [1])
6 Phenomenological model functions
For practical applications of tracer LIF diagnostics under variable environmental conditions of pressure temperature and O2 partial pressure it is desirable to evaluate effective FQY from a physical model or analytical functions. Here, for convenience we fitted an empirical mathematical model to the obtained experimental fluorescence lifetime data. The model equation (Eq. 6) consists of three terms and calculates a relative FQY, ϕ rel, which equals unity at 1 bar in N2 at 296 K. The first term describes the temperature dependence of the relative anisole FQY at 1 bar in N2. The second term represents the pressure dependence in N2 in the absence of O2, and the third term describes the effect of O2 on the quantum yield with a Stern–Volmer relationship.
The respective parameters in the model function are listed in Table 2. Fluorescence lifetimes can be calculated by multiplying ϕ rel with a constant factor of 13.3 ns, as shown in the last row of Table 2.
The model is validated for environments with O2 partial pressures up to 210 mbar from room temperature to 750 K. For N2 atmospheres at 1 bar the model is valid up to 850 K. For higher pressures of N2 the range of validity is limited to 750 K because at higher temperatures calculated values differ by more than twice the error limits from the measured data.
Figure 14 shows the comparison between the fitting functions (lines) and experimental data (symbols) as a function of temperature for three pressures in N2. Within the scatter of the data the overall agreement is very good up to about 750 K for all pressures, and for 1 bar even up to 850 K, while at higher temperature deviations occur.

Measured effective fluorescence lifetimes (symbols) of anisole and predictions for relative FQY and fluorescence lifetimes by Eq. (5) (lines) as a function of temperature for three pressures in N2
Figure 15 depicts a similar comparison between experimental fluorescence lifetimes (symbols) and the model output (lines) for several temperatures as a function of O2 partial pressure and for air as a function of temperature (inset in Fig. 15). The agreement is satisfactory. At temperatures beyond 700 K, the Stern–Volmer relation with one single Stern–Volmer coefficient is not fully sufficient to describe the dependence of anisole lifetime on O2 partial pressure, a similar observation as known from toluene at high temperatures [14]. It will be part of future work to set up a model describing all phenomena in a photophysically correct way.

Measured effective fluorescence lifetimes (symbols) of anisole and predictions for relative FQY and fluorescence lifetimes by Eq. (5) (lines) as a function of O2 partial pressure at various temperatures at 1 bar total pressure
7 Discussion
The present work has shown that, with 266 nm excitation the fluorescence spectrum of anisole at room temperature is red-shifted by 10–15 nm with respect to that of toluene. This is not unexpected because the position of the emission spectrum depends on the S0–S1 energy gap which is defined by the size and the electron density of the aromatic π system [18, 23, 37].
The decrease in fluorescence lifetime of anisole (and more pronounced with toluene) with increasing pressure is in contrast to the pressure effects known from, e.g., ketones [31, 38] and naphthalene [16, 24]. This phenomenon can be explained by the fact that with 266 nm photons anisole is excited close to the 0–0 band origin [29] (~1,600 cm−1 at 300 K), which means that vibronic levels populated in the S1 state after photon absorption are located below the thermalization level. In this case, bath-gas collisions lead to a “destabilization” in the sense that population is transferred into higher lying vibrational levels of S1, where fast IC and ISC processes reduce the probability for fluorescence emission, thus reducing the FQY. In the literature this is interpreted as “photo-induced cooling” of the excited state vibrational distribution, followed by collisional heating via bath-gas collisions [39, 40]. This pressure destabilization effect is also found for other aromatic species when excited near the vibrationless S1–S0 transition, e.g., anthracene excited at 365 nm [41]. In contrast to toluene the destabilization effect in anisole is less pronounced, leading to reduced pressure sensitivity when determining fluorescence intensities or lifetimes.
The measured fluorescence spectra in Fig. 6 suggest that anisole is a suitable tracer for two-color temperature imaging with constant O2 content in typical LIF applications as demonstrated previously for toluene (e.g., [10]). Additionally, in the presence of O2, anisole shows a red-shift of the fluorescence spectrum with increasing O2 partial pressure, similar to toluene excited at 248 nm [14]. Therefore, for constant temperature the sensitivity of anisole towards O2 could be used for O2-concentration measurements via two-color 2D-LIF imaging, as was shown previously for toluene upon 248 nm excitation [15]. The sensitivity, however, would be slightly reduced compared with toluene due to the reduced red-shift, but the applicability of 266 nm excitation can be advantageous. 2D imaging of O2 concentrations via fluorescence lifetime has been demonstrated for toluene [42]. Hence it is easy to imagine that this procedure can be adapted for anisole to determine either O2 concentrations or temperatures via the (spatially resolved) measurement of fluorescence lifetimes.
It must be emphasized that the fitting model allows the prediction of fluorescence lifetimes and relative FQY within a limited range of conditions relevant for typical tracer LIF applications. It will be subject of forthcoming work to set up a photo-physical model for anisole similar to the step-ladder model first introduced by Thurber et al. [43] for acetone. This would then take into account the vibrational level-dependent internal energy flow and relaxation processes after photo-excitation and would enable a better prediction of fluorescence intensities and lifetimes for temperature and pressure ranges as well as bath gas compositions of interest in future anisole LIF applications.
8 Conclusions
Fluorescence spectra and effective fluorescence lifetimes of gas-phase anisole were determined after picosecond laser excitation at 266 nm in a heated ceramic flow cell as a function of temperature, bath gas composition, and total pressure. Wavelength- and time-resolved fluorescence detection was accomplished with a streak camera coupled to a spectrometer. For all conditions the decay time of the spectrally integrated fluorescence decreases with increasing temperature, e.g., from 13.3 ± 0.5 ns at 296 K to 0.05 ± 0.01 ns at 925 K in pure N2. At room temperature the presence of O2 strongly reduces the fluorescence lifetime from the value in N2 to 0.40 ± 0.02 ns in air, with this tendency diminishing at higher temperatures. Normalized fluorescence spectra are presented as a function of temperature, and as a function of O2 partial pressure for several temperatures. In both cases, a red-shift as well as a broadening of the spectrum is observed. This behavior opens possibilities for quantitative imaging diagnostics of temperature and oxygen concentration [9, 12] using this novel tracer species.
Analytical functions were fitted to the present fluorescence lifetime data for a convenient representation of the temperature, pressure, and O2 concentration dependence of anisole. The existing data base can further be used for developing more sophisticated photo-physical models to predict FQY of anisole. It is also suggested that, depending on environmental conditions, time-resolved LIF of anisole is a promising method to perform, e.g., LIF thermometry in the absence of O2, independent of local tracer concentration.
References
S.A. Kaiser, M.B. Long, Quantitative planar laser-induced fluorescence of naphthalenes as fuel tracers. Proc. Combust. Inst. 30, 1555 (2005)
W. Koban, J. Schorr, C. Schulz, Oxygen-distribution imaging with a novel two-tracer laser-induced fluorescence technique. Appl. Phys. B 74, 111 (2002)
M. Orain, F. Grisch, E. Joudanneau, B. Rossow, C. Guin, B. Trétout, Simultaneous measurements of equivalence ratio and flame structure in multipoint injectors using PLIF. C. R. Mecanique 337, 373 (2009)
B.H. Cheung, R.K. Hanson, CW laser-induced fluorescence of toluene for time-resolved imaging of gaseous flows. Appl. Phys. B 98, 581 (2010)
T. Hagemeier, M. Hartmann, M. Kühle, D. Thévenin, K. Zähringer, Experimental characterization of thin films, droplets and rivulets using LED fluorescence. Exp. Fluids 52, 361 (2012)
M. Orain, P. Baranger, B. Rossow, F. Grisch, Fluorescence spectroscopy of 1,2,4-trimethylbenzene at high temperatures and pressures: application to temperature measurements. Appl. Phys. B 100, 945 (2010)
M. Luong, W. Koban, C. Schulz, Novel strategies for imaging temperature distribution using Toluene LIF. J. Phys.: Conf. Ser. 45, 133 (2006)
J. Yoo, D. Mitchell, D.F. Davidson, R.K. Hanson, Planar laser-induced fluorescence imaging in shock tube flows. Exp. Fluids 49, 751 (2010)
M. Cundy, P. Trunk, A. Dreizler, V. Sick, Gas-phase toluene LIF temperature imaging near surfaces at 10 kHz. Exp. Fluids 51, 1169 (2011)
M. Luong, R. Zhang, C. Schulz, V. Sick, Toluene laser-induced fluorescence for in-cylinder temperature imaging in internal combustion engines. Appl. Phys. B 91, 669 (2008)
W. Koban, J.D. Koch, V. Sick, N. Wermuth, R.K. Hanson, C. Schulz, Predicting LIF signal strength for toluene and 3-pentanone under engine-related temperature and pressure conditions. Proc. Combust. Inst. 30, 1545 (2005)
J.E. Dec, W. Hwang, Characterizing the development of thermal stratification in an HCCI engine using planar-imaging thermometry. SAE Technical Paper Series (2009), p. 2009-01-0650
R. Devillers, G. Bruneaux, C. Schulz, Investigation of toluene LIF at high pressure and high temperature in an optical engine. Appl. Phys. B 96, 735 (2009)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Oxygen quenching of toluene fluorescence at elevated temperatures. Appl. Phys. B 80, 777 (2005)
K. Mohri, M. Luong, G. Vanhove, T. Dreier, C. Schulz, Imaging of the oxygen distribution in an isothermal turbulent free jet using two-color toluene LIF imaging. Appl. Phys. B 103, 707 (2011)
M. Orain, P. Baranger, B. Rossow, F. Grisch, Fluorescence spectroscopy of naphthalene at high temperatures and pressures: implications for fuel-concentration measurements. Appl. Phys. B 102, 163 (2011)
C. Strozzi, J. Sotton, A. Mura, M. Bellenue, Characterization of a two-dimensional temperature field within a rapid compression machine using a toluene planar laser-induced fluorescence imaging technique. Meas. Sci. Technol. 20, 125403 (2009)
C. Schulz, V. Sick, Tracer-LIF diagnostics: quantitative measurement of fuel concentration, temperature and fuel/air ratio in practical combustion systems. Prog. Energy Combust. Sci. 31, 75 (2005)
T. Hirasawa, T. Kaneba, Y. Kamata, K. Muraoka, Y. Nakamura, Temperature dependence of intensities of laser-induced fluorescences of ethylbenzene and naphthalene seeded in gas flow at atmospheric pressure: implications for quantitative visualization of gas temperature. J. Vis. 10(2), 197 (2007)
T. Etzkorn, B. Klotz, S. Sörensen, I.V. Patroescu, I. Barnes, K.H. Becker, U. Platt, Gas-phase absorption cross sections of 24 monocyclic aromatic hydrocarbons in UV and IR spectral range. Atmos. Environ. 33, 525 (1999)
W. Koban, J.D. Koch, R.K. Hanson, C. Schulz, Absorption and fluorescence of toluene vapor at elevated temperatures. Phys. Chem. Chem. Phys. 6, 2940 (2004)
M. Suto, X. Wang, J. Shan, L.C. Lee, Quantitative photoabsorption and fluorescence spectroscopy of benzene, naphthalene, and some derivatives at 106–295 nm. J. Quant. Spectrosc. Radiat. Transf. 48(1), 79 (1992)
N. Nijegorodov, R. Mabbs, D.P. Winkoun, Influence of weak and strong donor groups on the fluorescence parameters and the intersystem crossing rate constant. Spectrochim. Acta A 59, 595 (2003)
S. Faust, G. Tea, T. Dreier, C. Schulz, Temperature, pressure, and bath gas composition dependence of fluorescence spectra and lifetimes of toluene and naphthalene. Appl. Phys. B 110, 81–93 (2013)
J.K. Marsh, Studies in fluorescence spectra. Part II. Phenol and phenolic ether vapours. J. Chem. Soc. Trans. 125, 418 (1924)
L.J.H. Hoffmann, S. Marquardt, A.S. Gemechu, H. Baumgärtel, The absorption spectra of anisole-h8, anisole-d3 and anisole-d8. The assignment of fundamental vibrations in the S0 and the S1 states. Phys. Chem. Chem. Phys. 8, 2360 (2006)
A. Bolovinos, J. Philis, E. Pantos, P. Tsekeris, G. Andritsopoulos, The methylbenzenes vis-à-vis benzenes: comparison of their spectra in the valence-shell transitions region. J. Mol. Spec. 94, 55 (1982)
M. Stockburger, H. Gattermann, W. Klusmann, Spectroscopic studies on naphthalene in the vapor phase. II. Fluorescence quantum yields from single vibronic levels in the first excited singlet state—behavior of higher excited singlet states. J. Chem. Phys. 63, 4529 (1975)
B. Rossow, Photophysical processes of organic fluorescent molecules and kerosene—application to combustion engines, Dissertation Thesis. Institut des Sciences Moléculaires d’Orsay, Université Paris-Sud 11, Paris, 2011
J.R. Lakowicz, Principles of Fluorescence Spectroscopy, 3rd edn. (Springer, New York, 2006)
J.D. Koch, Fuel tracer photophysics for quantitative planar laser-induced fluorescence, Dissertation Thesis. Stanford University, California, 2005
S. Faust, T. Dreier, C. Schulz, Temperature and bath gas composition dependence of effective fluorescence lifetimes of toluene excited at 266 nm. Chem. Phys. 383, 6 (2011)
T.B. Settersten, A. Dreizler, R.L. Farrow, Temperature- and species-dependent quenching of CO B probed by twophoton laser-induced fluorescence using a picosecond laser. J. Chem. Phys. 117(7), 3173 (2002)
T.B. Settersten, B.D. Patterson, J.A. Gray, Temperature- and species-dependent quenching of NO A2 Σ+ (v′ = 0) probed by two-photon laser-induced fluorescence using a picosecond laser. J. Chem. Phys. 124, 234308 (2006)
A.M. Scheer, C. Mukarakate, D.J. Robichaud, G.B. Ellison, M.R. Nimlos, Radical chemistry in the thermal decomposition of anisole and deuterated anisoles: an investigation of aromatic growth. J. Phys. Chem. A 114(34), 9043 (2010)
M. Jacon, C. Lardeux, R. Lopez-Delgado, A. Tramer, On the “third decay channel” and vibrational redistribution problems in benzene derivatives. Chem. Phys. 24, 145 (1977)
M. Hesse, H. Meier, B. Zeeh, Spektroskopische Methoden in der organsischen Chemie, 7th edn. (Thieme, Stuttgart, 2005)
V. Modica, C. Morin, P. Guibert, 3-Pentanone LIF at elevated temperatures and pressures: measurement and modeling. Appl. Phys. B 87, 193 (2007)
Y. He, E. Pollak, Theory of fluorescence decay of naphthalene: was photoinduced cooling observed experimentally? J. Chem. Phys. 116, 6088 (2002)
H. Wadi, E. Pollak, Theory of laser cooling of polyatomic molecules in an electronically excited state. J. Chem. Phys. 110, 11890 (1999)
L.A. Barkova, V.V. Gruzinskii, M.N. Kaputerko, Quenching and stabilization of the fluorescence of anthracene vapor by xenon. J. Appl. Spectrosc. 47, 186 (1987)
A. Ehn, O. Johansson, A. Arvidsson, M. Aldén, J. Bood, Single-laser shot fluorescence lifetime imaging on the nanosecond timescale using a dual image and modeling algorithm. Opt. Exp. 20(3), 3043 (2012)
M.C. Thurber, F. Grisch, B.J. Kirby, M. Votsmeier, R.K. Hanson, Measurements and modeling of acetone laser-induced fluorescence with implications for temperature-imaging diagnostics. Appl. Opt. 37(21), 4963 (1998)
Acknowledgments
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Faust, S., Dreier, T. & Schulz, C. Photo-physical properties of anisole: temperature, pressure, and bath gas composition dependence of fluorescence spectra and lifetimes. Appl. Phys. B 112, 203–213 (2013). https://doi.org/10.1007/s00340-013-5420-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-013-5420-7