
Abstract
The temporal variation of chemiluminescence emission from OH∗(A2 Σ +) and CH∗(A2 Δ) in reacting Ar-diluted H2/O2/CH4, C2H2/O2 and C2H2/N2O mixtures was studied in a shock tube for a wide temperature range at atmospheric pressures and various equivalence ratios. Time-resolved emission measurements were used to evaluate the relative importance of different reaction pathways. The main formation channel for OH∗ in hydrocarbon combustion was studied with CH4 as benchmark fuel. Three reaction pathways leading to CH∗ were studied with C2H2 as fuel. Based on well-validated ground-state chemistry models from literature, sub-mechanisms for OH∗ and CH∗ were developed. For the main OH∗-forming reaction CH+O2=OH∗+CO, a rate coefficient of k 2=(8.0±2.6)×1010 cm3 mol−1 s−1 was determined. For CH∗ formation, best agreement was achieved when incorporating reactions C2+OH=CH∗+CO (k 5=2.0×1014 cm3 mol−1 s−1) and C2H+O=CH∗+CO (k 6=3.6×1012exp(−10.9 kJ mol−1/RT) cm3 mol−1 s−1) and neglecting the C2H+O2=CH∗+CO2 reaction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Spontaneous light emission from chemically excited species in combustion processes is frequently used for detecting the location of flame fronts [1], the heat release [2, 3], and the equivalence ratio [4–7]. Blue light emission from the CH (A2 Δ−X2 Π) transition is specific for hydrocarbon combustion while the UV emission from the OH (A2 Σ +−X2 Π) transition appears in hydrocarbon and hydrogen flames. Both, hereafter denoted as CH∗ and OH∗ chemiluminescence (CL), respectively, are frequently used for emission measurements in flame diagnostics. Electronically excited species are more than four orders of magnitude less abundant compared to the corresponding ground-state species. Therefore, they have a negligible influence on the overall reaction progress and are usually not included in conventional combustion kinetics models.
The simplicity of the chemiluminescence signal detection compared to, e.g., laser-based diagnostics makes chemiluminescence emission spectroscopy very attractive for practical applications such as monitoring and controlling of combustion processes. This, however, requires the coupling of chemiluminescence signals with the underlying chemical processes in a quantitative way. However, characterizing the responsible reactions leading to chemiluminescence and determining their associate rate coefficients is challenging.
This study aims at the extension and validation of an existing OH∗ kinetics model [8] in hydrocarbon combustion based on shock-tube experiments with methane-blended hydrogen/oxygen mixtures following the approach of Hall and Petersen [9]. The OH∗ concentration resulting from the hydrocarbon combustion was calibrated with OH∗ signals from the well-known H2/O2 system via the reaction H+O+M=OH∗+M.
Additionally, the formation of CH∗ during acetylene oxidation was investigated by means of time-resolved measurements of CH∗ chemiluminescence in the shock-tube. The experiments were evaluated using a ground-state mechanism upgraded by an additional CH∗ subset. The CH∗ mechanism was optimized with regard to the experimental data and the formation channels with the corresponding rate coefficients leading to CH∗ chemiluminescence were identified.
This paper is organized in four sections. The first section gives a literature review of the kinetics of OH∗ and CH∗. Section 2 describes the shock-tube setup and the experimental conditions. This is followed by the description of the modeling approach, the comparison between experimental and computational results and the conclusions of the OH∗ chemiluminescence in H2/O2/CH4 mixtures in Sect. 3. The study of CH∗ and OH∗ chemiluminescence with the underlying modeling approach, the discussion of experiments and simulations and the conclusions in acetylene oxidation are given in Sect. 4.
2 Kinetics of electronically excited species
2.1 OH∗ kinetics
OH∗ chemiluminescence (CL) has been intensively investigated and the formation pathways leading to OH∗ are fairly well identified. A comprehensive literature review on OH∗ CL in hydrogen oxidation is available in [8]. OH∗ formation was experimentally studied in various H2/O2 systems [9–14]. Theoretical investigations [15–17] of the formation pathways were done by ab-initio analysis by Skrebkov et al. [15, 16] and Smekhov et al. [17]. More recently, Kopp et al. [18] investigated the OH∗ chemiluminescence in various shock-heated H2/O2 mixtures for technical relevant pressures up to 32 bar. They derived a pressure-dependent rate coefficient for reaction (R1) based on a calibration procedure. The rate coefficient was 1-order of magnitude lower than the rate difference in the rate coefficient k 1 from Kathrotia et al. [8]. In a previous study, we investigated the OH∗ production in H2 oxidation by a combined experimental and numerical approach [8]. For calibration of the measured OH∗ signals, high-temperature experiments were used where OH∗ is present in the combustion mixtures at well-known equilibrium concentrations. The signals obtained from subsequent lower-temperature measurements were then converted into absolute OH∗ concentrations with an accuracy of ±20 %. The temperature dependence of OH∗ CL was monitored in shock-tube experiments behind reflected shock waves and was compared with absolute species concentrations computed with a homogeneous reactor model. The mechanism describing the excited-state species was validated against the measured absolute OH∗ concentrations and the ignition delay times. The key reaction of the OH∗ formation for hydrogen oxidation was considered as
with a corresponding rate coefficient k 1=1.5×1013×exp(−25 kJ mol−1/RT) cm6 mol−2 s−1.
While OH∗ CL is weak in H2 oxidation, it is strong in hydrocarbon combustion. OH∗ CL in hydrocarbon combustion has been studied by several groups [11, 12, 19–25]. There is agreement in identifying the key reaction leading to OH∗ emission:
Carl et al. [26] showed that there is a proportionality between OH∗ formation and the product of CH and O2 concentrations by means of molecular-beam-sampling threshold-ionization mass spectrometry (MB-TIMS). Based on this observation, they deduced that reaction (R2) is responsible for the OH∗ emission and recommended a rate coefficient of k 2=4.8×1010 cm3 mol−1 s−1. Smith et al. [27] measured absolute OH∗ and CH∗ concentrations in laminar premixed CH4/air flames with different equivalence ratios. They calibrated their spatially resolved images with respect to computed excited-state species concentrations and determined a rate coefficient k 2=1.8×1011 cm3 mol−1 s−1. Hall and Petersen [9] performed a series of shock-tube experiments with H2/O2 and H2/O2/CH4 mixtures. By fitting modeling results to their experimental observations, they also identified reaction (R2) as main channel with a temperature-dependent value for k 2 of 3.2×1014 T −0.4exp(−17.4 kJ/RT) cm3 mol−1 s−1. Depending on temperature, these kinetics coefficients differ by up to three orders of magnitude. Recently, Kathrotia et al. [28] studied OH∗ chemiluminescence in premixed low-pressure flames for various methane-based mixtures. Based on their flame calculations, they supported reaction (R2) using the rate coefficient from Smith et al. [27].
2.2 CH∗ kinetics
For CH∗ CL there is disagreement in identifying the reaction(s) leading to the formation of CH∗. The dominating pathway(s) and their suggested rate coefficients from literature also vary by several orders of magnitude. Broida et al. [29, 30] identified CH∗ almost half a century ago, but ambiguity about the key reactions for its formation still persists. The reaction producing CH∗ was suggested by Gaydon and Broida [29, 31] as
Porter et al. [20] suggested a rate coefficient of (R5) to be 4.2×1011 cm3 mol−1 s−1. Bleekrode and Nieuwpoort [32] derived from radiative lifetimes of CH a range for k 5 of 6.0×1011 to 6.0×1012 cm3 mol−1 s−1. Bulewicz et al. [33] later supported reaction (R5) and derived a rate coefficient of k 5=4.8×1012 cm3 mol−1 s−1 via absorption and emission experiments for various species (C2, CH, OH) in a low-pressure burner. Based on their premixed flame experiments, Smith et al. [27] recommended the value k 5=1.1×1013 cm3 mol−1 s−1. They pointed out that reaction (R5) plays a minor role for CH∗ formation with a contribution less than 10 %.
Glass et al. [34] rejected the relevance of reaction (R5) as dominating pathway and introduced the reaction of ethynyl (C2H) radicals with atomic oxygen. Also Brennen and Carrington [35] and Grebe and Homann [23] privileged the following reaction as the main channel:
Based on highly resolved spectroscopic investigations of CH∗ CL, Brockhinke et al. [36] also concluded that R5 can only play a minor role in the formation scheme of CH∗. This formation reaction is accompanied by a large excess of free enthalpy, whereas the measured spectra did not show such effects. Therefore, they suggested that CH∗ is formed with little excess energy only (such as from R6). Joklik et al. [37] showed in their low-pressure acetylene flame the dominance of the latter reaction, but suggested a value for k 6=7.0×1012 cm3 mol−1 s−1 which is one order of magnitude higher than the recommended value stated in an earlier work from Grebe and Homann [23]. Reaction (R6) was also supported by Devriendt et al. [38]. They determined the room-temperature rate coefficient by means of a pulsed laser photolysis study of the C2H2/N2O system. The rate coefficient of the reaction (R6) was determined by using the well-quantified chemiluminescence of NO+O (i.e., \(\mathrm{NO}_{2}^{*}\) CL) as reference. The rate coefficient for reaction (R6) was found to be 1.1×1013 cm3 mol−1 s−1. One year later, Devriendt and Peeters [39] presented a study of mixtures containing C2H2/O/H using an isothermal flow reactor coupled with MB-TIMS to quantify C2H and oxygen atoms. Again, CH∗ CL signal intensities were linked to \(\mathrm{NO}_{2}^{*}\) CL as internal standard to derive absolute CH∗ concentrations. They found proportionality between CH∗ emission intensity and [C2H]×[O]. Based on this observation, they estimated a rate coefficient for k 6=1.4×1013exp(−1.9 kJ mol−1/RT) cm3 mol−1 s−1. Based on the calibration procedure of the optical detection system described in the previous section, Smith et al. [27] introduced a rate coefficient k 6=6.2×1012 cm3 mol−1 s−1 which was considered as the main formation channel leading to CH∗ in premixed CH4/air flames. Afterwards, the authors investigated CH∗ formation in CH4/N2O flames in [13] using the same calibration procedure stated in [27]. They found that CH∗ CL cannot solely be described by reaction (R6) with their recommendation of k 6 in [27]. The CH4/N2O flame experiments revealed that a rate coefficient of k 6=2.5×1012 cm3 mol−1 s−1 [13] is required which is slightly lower than their previous recommendation. Based on their shock-tube study, Hall et al. [40] estimated a rate coefficient of k 6=5.2×1011exp(−10.9 kJ mol−1/RT) cm3 mol−1 s−1. The study revealed that the main reaction channel was attributed to the reaction (R5). In the same study, they determined an exceptionally high rate coefficient of k 5=2.0×1014 cm3 mol−1 s−1. Recently, Elsamra et al. [41] deduced from their pulsed laser photolysis study for He-diluted C2H2/O2 mixtures a value of k 6=6.0×1012×exp(−1.9 kJ mol−1/RT) cm3 mol−1 s−1. They considered (R6) as major source of CH∗ production especially for fuel-rich conditions.
A third CH∗ formation pathway was proposed in the early 1960s by Hand and Kistiakowsky [42] which was supported by Gutman and Matsuda [43] and Matsuda et al. [44]. They postulated that the reaction of C2H radicals with O2 strongly contribute to the production of CH∗:
Other studies were devoted to the investigation of reaction (R7). A laser photolysis study by Renlund et al. [45] favored (R7) as important pathway for CH∗ formation. Hwang et al. [46] also supported this recommendation and deduced a rate coefficient (k 7=4.10×1013exp(−4.5 kJ mol−1/RT) cm3 mol−1 s−1) for reaction (R7) based on shock-tube investigations. From time-resolved CH∗ CL after laser photolysis of C2H2/N2O mixtures at room-temperature, Devriendt et al. [38] deduced a rate coefficient k 7=2.2×1010 cm3 mol−1 s−1 which is 500 times lower than their recommendation of k 6. Smith et al. [27] primarily excluded the relevance of (R7) based on their modeling approach for CH4/O2 flames. Later on, however, they found out that the model overpredicts the monitored CH∗ in CH4/N2O flames [13] in cases where only reaction (R6) was considered. Hence, they proposed for k 7=3.2×1011exp(−6.7 kJ mol−1/RT) cm3 mol−1 s−1, which was derived from flame experiments with different [O]/[O2] ratios. Elsamra et al. [41] published a temperature-dependent rate coefficient for (R7) of 6.0×10−4 T 4.4exp(9.6 kJ mol−1/RT) cm3 mol−1 s−1. They denoted the importance of the latter reaction particularly for hot flames under fuel-lean conditions. Recently, Kathrotia et al. [28] investigated CH∗, OH∗ and \(\mathrm{C}_{2}^{*}\) chemiluminescence in various premixed low-pressure flames. Their modeling revealed that the available rate coefficient k 7 from Elsamra et al. [41] strongly overpredicts the CH∗ concentration. Depending on the equivalence ratio they revised the recommended rate coefficients for the three potential reaction pathways (R5)–(R7).
All these studies pointed out the difficulties in undertaking quantitative measurements. There is a large scatter not only in determining the major channel leading to CH∗ CL in various hydrocarbon systems, but also in providing recommendations of their respective rates. Therefore, the aim of our work is to improve the knowledge of the formation pathways leading to CH∗ CL and determining their corresponding kinetics data via linking shock-tube experiments and numerical modeling.
3 Experimental
The experiments were carried out in a stainless-steel shock tube with a constant inner diameter of 79 mm. An aluminum diaphragm (thickness: 50 μm) divides the shock tube into the driver section (length: 3.5 m) and the driven section (length: 5.7 m). Hydrogen was used as driver gas. The driver section was pumped down to 10−2 mbar. Prior to each experiment the driven section was turbo-pumped to final pressures below 3×10−8 mbar.
Gas mixtures were prepared in a stainless-steel cylinder using the partial-pressure method. The cylinder was also evacuated by a separate turbo-molecular pump in between experimental series. The stated purities were Ar≥99.9999 %, N2O≥99.999 %, O2≥99.998 %, CH4≥99.999 % and C2H2≥99.6 %. The shock speed was measured over three intervals using four piezo-electric pressure transducers with a time resolution of the data acquisition of 0.4 μs. The temperature T 5 and pressure p 5 behind the shock wave were computed from measured incident shock-wave velocity and speed attenuation using a one-dimensional shock model (shock tube code of the CHEMKIN Package [47]) with respect to the initial conditions p 1 and T 1. The estimated uncertainty in reflected shock temperature was less than ±15 K in the temperature and time range of our measurements. The pressure p 5 and temperature T 5 values behind the reflected shock wave as well as the mixture compositions for the experiments are given in Table 1 (H2/CH4/O2 experiments) and Table 2 (C2H2/O2/N2O experiments).
Four optical ports were located 40 mm upstream of the end flange and allow the detection of chemiluminescence. Measuring chemiluminescence with high temporal resolution requires limiting the detection to a small volume within the shock tube. Hence, two slits were placed at 15 and 45 mm in front of each detector to limit the detection solid angle. Widths of 0.2 mm and 1 mm were selected to provide an optimal balance between signal strength and time resolution. This setup provided a time resolution of 1 μs as determined from the light collection angle and the passing velocity of the reflected shock wave. Interference filters with center wavelengths of \(\lambda_{\mathrm{OH}^{*}} = 307~\mathrm{nm}\) and \(\lambda_{\mathrm{CH}^{*}} = 430~\mathrm{nm}\) (both 10 nm full width at half maximum) were limiting the spectral detection to OH∗ and CH∗ chemiluminescence from the transitions in the A–X system, respectively. The chemiluminescence radiation was detected by two photomultipliers (OH∗: Hamamatsu 1P28, CH∗: Hamamatsu R955) with constant amplification voltage for all presented measurements. To ensure sufficient time resolution of the signal detection, 10 kΩ and 3.8 kΩ resistors were connected in parallel to the amplifiers for the OH∗ and CH∗ detectors, respectively. The time resolution of each setup was investigated for various resistors by investigating the signal recorded from the input of short square pulses (duration: 1 μs) of an LED. A compromise between time resolution and signal intensity was chosen with selecting a time resolution of 2 μs that matched the time resolution of the optical arrangement. Care was taken not to change the optical configuration during the set of experiments.
Corresponding OH∗ and CH∗ concentrations were simulated using a homogeneous reactor model with regard to the initial mixture composition, temperature and pressure behind the reflected shock wave. Constant volume was assumed and the temperature was calculated for adiabatic conditions using a 0-D model (Aurora in CHEMKIN Package [47]). The underlying reaction mechanisms are described in Sects. 4.1 and 5.1. Forward as well as backward reactions were considered.
4 OH∗ chemiluminescence in CH4/O2 mixtures
4.1 Numerical model
The correct description of the reactions relevant for chemiluminescence crucially depends on the correct description of the underlying ground-state chemistry and the elementary reactions that quench the electronically excited states. In the present work, the GRI3.0 mechanism with 53 species and 325 elementary reactions [48] was used which is extensively validated for methane combustion for a wide range of conditions. A quantitative study of OH∗ requires accurate knowledge of the time-dependent concentration of the corresponding precursor species leading to the formation of OH∗, such as CH. GRI3.0 mechanism precisely predicts absolute CH concentrations, which was previously demonstrated for premixed low-pressure methane/air flames at various equivalence ratios by Berg et al. [49]. Especially for near-stoichiometric conditions, an almost perfect agreement of CH concentration and peak position as function of the height above the burner (HAB) was found. The model for OH∗ CL, consisting of reaction H+O+M=OH∗+M (R1) and the corresponding quenching reactions, was adopted from our previous H2/O2 study [8]. Additionally, the CH+O2=OH∗+CO (R2) reaction was included. The OH∗ formation and consumption reactions and their corresponding rate coefficients are summarized in Table 3. The OH∗ mechanism is validated by comparing the simulations with absolute OH∗ concentration measurements.
4.2 Results and discussion
A calibration factor was determined that connects the measured signal intensity with the corresponding OH∗ concentration. According to the procedure described in [8], high-temperature H2/O2 experiments (T 5>2800 K) were carried out where well-defined OH∗ concentrations can be determined based on an equilibrium resulting from OH+M=OH∗+M. Using this calibration—with an estimated error of ±20 % as stated in [8]—measured OH∗ CL intensities were converted into absolute species concentrations. Figure 1 (grey line) shows a typical temporal variation of the OH∗ concentration for an experiment with a pure H2/O2 mixture. It shows a rapid formation of OH∗, which is exclusively formed by (R1) followed by slow depletion via radiative decay (R3) and collisional quenching (R4). For the CH4-blended H2/O2 mixture (black line), one can see a similar temporal behavior of the OH∗ concentration with a slow depletion phase. But the formation is significantly different compared to the pure H2/O2 case. It shows a characteristic two-stage behavior. In the first stage, the concentration of the CH4-containing mixture shows a first steep increase of OH∗ forming a peak, which afterwards rapidly declines. In the second stage, the OH∗ concentration merges into the slower depletion phase via reaction (R3) and (R4), as also occurs in pure H2/O2 mixtures. This two-phase behavior is attributed to the kinetics of the two reaction channels forming OH∗, (R1) and (R2). The rate-of-production (ROP) analysis (dotted lines shown in Fig. 2) supports the dual-channel behavior. In hydrocarbons, reaction (R2) dominates the OH∗ formation and contributes almost three times more OH∗ than (R1). The consumption of OH∗ in the first phase where reaction CH+O2=OH∗+CO (R2) dominates is mainly attributed to the depletion of the CH concentration. In this phase, the OH∗ profile follows the concentration-time profile of CH. Afterwards, reaction (R1) via H+O+M=OH∗+M arises and contributes to the OH∗-CL emission, which slowly decreases.

Comparison of the temporal variation of the OH∗ concentration from two shock-tube experiments at T 5=1621 K and p 5=1.49 bar with mixture A (grey line) mixture B (black line)

Absolute OH∗ concentration (simulation: solid grey line, experiment: solid black line) with regard to the normalized rate-of-production (ROP) of reaction (R1) H+O+M=OH∗+M (dotted line) and reaction (R2) CH+O2=OH∗+CO (dashed line) forming OH∗. Initial modeling conditions: T 5=1884 and p 5=1.76 bar and mixture B
Based on our shock-tube experiments, the reaction rate coefficient of the OH∗ formation reaction (R2) was optimized with regard to absolute OH∗ concentration, while keeping the other reaction rates constant. An initial value for k 2 is adopted from Smith et al. [27]. This value was varied—for each shock-tube experiment for mixtures B and C—until the experimental and computed concentration profiles matched for the first peak. A rate coefficient equal to 8.0×1010 cm3 mol−1 s−1 was found to give the best-fit to the entire set of experimental data.
Figure 2 shows a comparison of an experimental (solid black line) and simulated (solid grey line) OH∗ concentration profile. The simulation reproduces well the two-stage formation of OH∗, which is primarily formed via CH+O2=OH∗+CO (R2) and then later by H+O+M=OH∗+M (R1). Good agreement was found in terms of profile shapes, absolute species concentration and peak position for a wide range of experimental conditions (for brevity not shown here).
Additionally, rate coefficients of the reaction (R2) taken from literature (Smith et al. [27] and Hall and Petersen [9]) were used to simulate the OH∗ concentration-time behavior based on our experimental data. Figure 3 shows the measured and the simulated OH∗ concentration profiles, based on the conversion of the measured OH∗ intensity into absolute species concentrations and the derived rate coefficients k 1 and k 2 from the present study in comparison to the results of the data of the literature. The peak concentration of OH∗ was notably overpredicted when using the rate coefficients from Hall and Petersen [9]. A difference of measured and simulated concentrations of more than one order of magnitude was obtained for both reaction pathways (R1) and (R2). In cases when simulating the OH∗ concentration with the values from Smith et al. [27], the OH∗ concentration was overpredicted by a factor of two. The first-stage peak concentration via (R2) was about three times higher than the experimental data, whereas the second-stage peak concentration via (R1) was almost two times higher than the experimental data. The best agreement was achieved with the rate coefficients k 1=1.5×1013exp(−25.0 kJ mol−1/RT) cm6 mol−2 s−1 and k 2=8.0×1010 cm3 mol−1 s−1, respectively.

Comparison of the experimental OH∗-concentration profile (symbols) with simulation using rate coefficients for the reactions (R1) and (R2) from the present work (dashed line), from Hall and Petersen [9] (dotted line, data divided by a factor of 10) and Smith et al. [13] (dashed line) for mixture B at T 5=2116 K and p 5=1.22 bar
Rate coefficients for k 2 available in literature are shown in an Arrhenius representation in Fig. 4. Our suggested rate coefficient is slightly higher than the value by Porter et al. [20] and Grebe and Homann [23]. The rate coefficient from Smith et al. [27] is about two times higher than our value. With regard to the underlying experimental and modeling uncertainties, there is an agreement for the range of k 2 around 1011 cm3 mol−1 s−1. The recommendations from Hall and Petersen [9, 50], however, are more than two orders of magnitude higher.

The uncertainty in the rate coefficient k 2 can be divided into systematic and statistical contributions. The determination of the rate coefficient depends on the absolute OH∗ concentration and therefore on the calibration procedure. The error for the calibration of the optical system can be estimated as ±20 % [8]. The computed OH∗ concentration, which was used to fit the profiles with regard to the experimental intensities profiles, strongly depends on the CH concentration. Berg et al. [49] estimated the uncertainty of the computed ground-state CH concentration to be ±15 % for the GRI3.0 mechanism. Additional systematic errors of ±5 % can be connected to the quenching rate coefficients. In the data analysis the modeled OH∗ concentration was fitted with regard to the measured one by varying the rate coefficient k 2. Each individual experiment directly leads to the determination of a k 2 value. A contribution of ±20 % covers this statistical error. The influence of the temperature uncertainty of ±15 K in the determination of the gas temperature was considered and it was found to be negligible compared to the other uncertainties. The errors are combined as square root of sum, including all of the uncertainties in the determination of the k 2 values. Thus, the overall uncertainty in k 2 is ±32 %.
5 OH∗ and CH∗ chemiluminescence in C2H2/O2 mixtures
5.1 Numerical model
The underlying ground-state model from Wang and Laskin [51] used in this work describes the high-temperature oxidation of C2H2 with 75 species and 529 reactions. The mechanism was validated for a wide range of conditions with regard to shock-tube experiments, laminar burning velocity and burner-stabilized flames before. Originally, this mechanism did not contain reactions involving C2 species, which are assumed to be potential precursors for CH∗ formation. Therefore, a C2 sub-mechanism recommended by Williams and Pasternack [52] that consists of 18 reactions was implemented. It contains potential formation and consumption reactions of C2 species. We ensured that these modifications did not affect the base mechanism by comparing the original and the modified mechanisms with regard to the published validation data for the relevant species (OH and CO2) and ignition delay times. No notable differences were observed.
To represent the excited-state chemistry a sub-mechanism was implemented, which incorporates OH∗ and CH∗ formation pathways as well as their corresponding quenching reactions. A detailed description of the OH∗ subset is presented in the previous section and the reaction scheme is given in Table 3. The CH∗ mechanism consists of three formation reactions (R5)–(R7) adopted from [53] and 12 consumption reactions taken from Tamura et al. [54]. As initial values for the simulation, rate coefficients for CH∗ formation reactions were originally taken from Hall et al. [40]. The CH∗ formation and consumption reactions and their corresponding rate coefficients are summarized in Table 4.
The acetylene model was exclusively developed for C2H2 and C2H4 combustion with O2 as primary oxidizer. In the present work, the mechanism of CH∗ CL was primarily validated against the shock-tube experiments with C2H2/O2 mixtures. An optimization is performed by comparing normalized experimental and simulated chemiluminescence intensities and peak times, i.e., the time interval between the arrival of the reflected shock and the maximum of the CH∗-CL emission. Additionally, time-resolved normalized measured emission and simulated concentration traces are compared. Rate coefficients are derived for the CH∗ formation reactions (R5) and (R6).
The CH∗ sub-mechanism derived from the C2H2/O2 experiments was applied to predict the experimental results for C2H2/N2O mixtures. To our knowledge, a validated acetylene ground-state model incorporating O2 oxidation as well as NO x chemistry does not exist so far. Therefore, the acetylene oxidation model described above was extended by an additional subset of NO x chemistry adopted from GRI3.0 [48] in order to reproduce the C2H2/N2O shock-tube experiments. This additional nitrogen-chemistry subset was originally implemented in a CH4-combustion model and therefore contains cross-reactions linking nitrogen with hydrocarbon chemistry. The previously derived CH∗ reaction scheme based on the C2H2/O2 study was kept constant without further changes.
5.2 Results and discussion of C2H2/O2 experiments
Over 80 experiments were carried out under various conditions in a temperature range from 1354 to 2535 K with pressures from 1.1 to 2.0 bar using Ar-diluted C2H2/O2 mixtures. The experimental conditions cover a wide range of temperature, pressure and equivalence ratios from fuel-rich to lean conditions, cf. Table 2.
Figure 5a shows the comparison of typical normalized CH∗ peak intensities from the experiment and simulated CH∗ peak concentrations. The data were normalized with respect to the reference temperature of T 5=1900 K. The corresponding measured and simulated peak times are shown in Fig. 5b. The CH∗ peak intensities increase exponentially with temperature, while the peak times show an opposed effect; namely the induction time decreases for increasing temperatures. This temperature dependence of CH∗ chemiluminescence can be observed for all equivalence ratios (for brevity not shown here).

Measurement (symbols) and simulation (line) of (a) CH∗ chemiluminescence intensities normalized to the corresponding value at T=1900 K and (b) delay times for peak CH∗ for mixtures with 0.10 % C2H2 and 0.25 % O2 in Ar
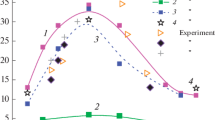
In order to show the importance of the different pathways forming the chemiluminescence intensities (R5)–(R7), the data were evaluated by normalizing the CH∗-CL intensities for lean conditions with respect to the corresponding value for the stoichiometric mixture. These data are plotted for three different temperatures as a function of equivalence ratio. Overall, the normalized intensities decrease with increasing O2 concentration (cf. Fig. 6), which was used to separate the impact of the three formation reactions. This effect was already stated by Hall and Petersen [40] in an earlier CH4/O2 shock-tube study. They observed the decrease of CH∗ for fuel-lean CH4/O2 mixtures and concluded that it was impossible to match the CH∗ reduction with their kinetics model if reaction C2H+O2=CH∗+CO2 (R7) was involved. Based on our modeling, we agree with this conclusion, because our model cannot retrace the decreasing CH∗ CL in the presence of (R7). If the formation reaction of CH∗ via (R7) with a rate coefficient larger than 1.0×1011 cm3 mol−1 s−1 is incorporated, the CH∗ CL linearly increases with increasing O2 concentration (cf. Fig. 6), which is not consistent with our experimental observations. Furthermore, the experimental and modeled CH∗ peak times were slightly underpredicted when taking (R7) into account (for brevity not shown here). Generally, the kinetics model involving reactions (R6) and (R7) produced more CH∗ for higher O2 concentrations. Our model revealed that only reaction C2+OH=CH∗+CO (R5) can account for this reduction behavior.

Experimental (symbols) and simulated (lines) CH∗ chemiluminescence normalized to the corresponding value at ϕ=1.0 for three different temperatures in C2H2, O2, Ar mixtures. The dash-dotted line represents the modeling results when additionally incorporating reaction (R7) with the rate coefficient of 1.0×1011 cm3 mol−1 s−1
Neglecting the formation reaction of CH∗ via C2H+O2=CH∗+CO2 (R7) had not been considered in other recent studies. Therefore, an extensive evaluation of the relevance of reaction (R7) was required. Therefore, additional simulations using the CH∗ sub-mechanism from the present study (cf. Table 4) and additionally incorporating (R7) with recent literature values for k 7 were performed. With the most recent rate coefficient for k 7 from Elsamra et al. [41], the reduction of the CH∗ CL as function of equivalence ratio seen in Fig. 6 cannot be predicted by the model and the main chemiluminescence formation becomes controlled by (R7) which is again in contradiction with the recent study from Kathrotia et al. [28]. They observed an eightfold overprediction of CH∗ CL especially for fuel-rich conditions when considering the rate coefficient from Elsamra et al. [41].
When considering the corresponding rate coefficient k 7 from Devriendt et al. [38], which is about one order of magnitude lower compared to the value from [41], the simulations show a slight reduction of CH∗ CL for excess of O2. However, this signal reduction is not as pronounced as in the experiments. With the values from Devriendt et al. [38], good agreement between the simulated and the measured dependence on the equivalence ratio was achieved when the rate coefficient of the quenching reaction CH∗+O2=CH+O2 (R9,1) is increased by a factor of two which compensates for the slow rate of Devriendt. However, Tamura et al. [54] stated an error limit of ±15 % for the corresponding quenching rate. Therefore, the given rate coefficients k 7 are too high to account for our experimental results. Recently, Kathrotia et al. [28] also pointed out the overprediction of CH∗ CL when considering the available rate coefficients of k 7. Overall, the formation of CH∗ in the present study is mainly dominated by the reactions C2+OH=CH∗+CO (R5) and C2H+O=CH∗+CO (R6).
The equivalence-ratio-dependent reduction in CH∗ was used to identify the main formation reactions (R5) and (R6). The best correlation between measurement and simulation can be obtained with k 5=2.0×1014 cm3 mol−1 s−1 and k 6=3.6×1012exp(−10.9 kJ mol−1/RT) cm3 mol−1 s−1. The starting values for the rate coefficients k 5 and k 6 were originally taken from Hall et al. [40]. However, our modeling could not reproduce the temperature dependence of the normalized CH∗ CL when considering the original values from [40]. Based on a sensitivity analysis the authors showed that the CH∗ formation is highly sensitive to the rate coefficient k 5, whereas k 6 is of minor importance. We kept k 5 constant, while k 6 was increased to fit the experimental data with regard to the recommendation of k 6 from [40]. According to Smith et al. [27] it should be pointed out that any combination of the chosen rate coefficients k 5 and k 6 with the same net rate of production will reproduce the experimental data. To overcome this ambiguity, more direct measurements of absolute CH∗ concentrations are required.
A typical temporal variation of a normalized CH∗-CL signal for C2H2/O2 mixtures is shown in Fig. 7. After an induction time, the CH∗ emission increases, reaches its maximum and afterwards decays. The measured CH∗ concentration is well reproduced by the simulation. Overall, good agreement of the temperature dependence, of the CH∗-CL intensities and peak times (Fig. 5), the CH∗ signal reduction for lean mixtures (Fig. 6), and the temporal variation of the CH∗ emission (Fig. 7) was obtained for a wide range of conditions and for all three investigated equivalence ratios.

Comparison of the temporal variation of the normalized CH∗ chemiluminescence (symbols) and the CH∗ concentration (solid line) simulated with k 5 and k 6 from the present work. The dotted lines represent the modeled CH∗ traces for variations of k 6 by a factor of 2. The mixture composition is 0.10 % C2H2 and 0.33 % O2 diluted in Ar at T 5=2300 K and p 5=1.32 bar
OH∗ CL shows also a temperature dependence of the peak concentrations (Fig. 8a) which is not as pronounced as found for CH∗. The peak times given in Fig. 8b decrease with increasing temperature. Based on our OH∗ sub-mechanism derived in the previous section and tabulated in Table 3, the OH∗ formation in C2H2/O2 system was modeled. Good agreement of the simulated and measured temperature dependence of OH∗-CL intensities and peak times were found (Fig. 8). The simulation accurately predicted the temporal variation of the OH∗ CL profile (Fig. 9). The subset of OH∗ chemistry derived from our H2/CH4/O2 study well predicts the OH∗ profiles. The ROP analysis revealed that only reaction (R2) leads to the OH∗ CL formation, whereas reaction (R1) reverses and consumes OH∗.

Measurement (symbols) and simulation (line) of (a) OH∗ chemiluminescence intensities normalized to the corresponding value at T=1900 K and (b) delay times for the occurrence of the OH∗-CL peak for mixtures with 0.10 % C2H2 and 0.33 % O2 in Ar

Comparison of the temporal variation of the normalized OH∗ chemiluminescence (circles) and the simulated OH∗ concentration (line). The simulation is performed with the rate coefficients k 5 and k 6 suggested in the present work. The mixture composition is 0.10 % C2H2, 0.50 % O2 diluted in Ar. The experimental conditions are T 5=1900 K and p 5=1.32 bar
5.3 Results and discussion: C2H2/N2O experiments
Based on our experimental and modeling study of C2H2/O2 combustion, the CH∗ formation channels via (R5) and (R6) were identified to be responsible for the CH∗ CL in this reaction system. The existing OH∗ kinetics model involving (R1) and (R2) as formation pathways well reproduced the observed OH∗ CL. The relative importance of (R6) vs. (R7) was investigated by increasing the O-atom concentration by adding N2O to the mixture. For this purpose, the acetylene-oxidation and chemiluminescence model was upgraded by an additional subset of NO x chemistry described in Sect. 5.1. It was ensured that the NO x chemistry did not influence the predictions for C2H2- and O2-containing mixtures.
The temporal variation of the CH∗-CL emission is shown in Fig. 10. CH∗ was formed quasi instantaneously behind the reflected shock wave. The fast decomposition of N2O, even for low temperatures, provides a high amount of O atoms which are consumed by subsequent chain-branching reactions forming additional radicals. Therefore, the radical pool further accelerates the CH∗ formation and the experiments show a significantly faster ignition delay and peak time compared to C2H2/O2 system. Figure 11 shows the activation energy E (i.e. activation temperatures multiplied with the gas constant R) of the normalized CH∗ and OH∗ CL intensities—the temperature-dependent intensities and ignition delay times were described empirically by an Arrhenius-like equation (\(\mathrm{CL/CL}_{T=1900~\mathrm{K}} = A \exp(-\mathit{E/RT})\) and \(\tau = A \exp(-\mathit{E/RT})\), respectively). Both species show a strong temperature dependence of the CL intensities. Compared to those experiments with C2H2/O2 (activation energy E=1–2 kJ mol−1) the temperature dependence increased for mixtures with N2O (activation energy E=3 kJ mol−1). The CH∗ and OH∗ kinetics models based on the previous C2H2/O2 study reproduce the normalized CL intensities and peak times for C2H2/N2O without requiring any changes. The kinetics model presented in this study well predicts the T-dependence of the normalized CL intensities (Fig. 11) and the peak times (Fig. 12). There is good agreement in absolute peak times for CH∗ CL (see Fig. 13), while the simulated peak times tend to be too low for OH∗ CL (Fig. 14). This discrepancy can be attributed to the interaction of the acetylene ground-state model and the implemented NO x model, since the NO x mechanism is originally implemented in a CH4 combustion model and it is not validated for acetylene combustion.

Comparison of the temporal variation of the normalized CH∗ chemiluminescence (symbols) and the CH∗ concentration (line) simulated with the rate coefficients k 5 and k 6 from this work. The mixture composition is 0.10 % C2H2, 1.00 % N2O diluted in Ar at T 5=1900 K and p 5=1.32 bar

Measured (symbols) and simulated (lines) activation energies E for the normalized CL intensities of (a) CH∗ and (b) OH∗ using the expression CL/CL T=1900 K=Aexp(−E/RT) for mixtures with C2H2 and O2 (open triangles) and C2H2 and N2O (open circles) as a function of equivalence ratio ϕ

Measured (symbols) and simulated (lines) activation energy E using the expression τ=Aexp(−E/RT) as a function of equivalence ratio ϕ. The open triangles represent mixture compositions with C2H2 and O2. The open circles represent mixtures containing C2H2 and N2O. The plot contains the results for (a) CH∗ and (b) OH∗

Measurement (symbols) and simulation (line) of (a) CH∗-CL intensities as a function of (inverse) temperature normalized to the corresponding value at T=1900 K and (b) delay times for peak CH∗ for mixtures with 0.10 % C2H2 and 0.67 % N2O in Ar

Measurement (symbols) and simulation (line) of (a) OH∗-CL intensities as a function of (inverse) temperature normalized to the corresponding value at T=1900 K and (b) delay times for peak OH∗ for mixtures with 0.10 % C2H2 and 1.00 % N2O in Ar
Recommended rate coefficients for k 5 and k 6 available in the literature are shown in an Arrhenius representation in Fig. 15. The suggested values for k 5 vary by three orders of magnitude, while the range of k 6 spans about two orders of magnitude. k 5 and k 6 were chosen following the procedure discussed previously in order to reproduce the temperature dependence of the normalized CH∗ CL (cf. Fig. 5a) and the dependence on the equivalence ratio (see Fig. 6). Compared to the literature values, the rate coefficient of reaction (R6) is slightly lower than the recommendation of Smith et al. [27].

 Devriendt et al. [
Devriendt et al. [ Bulewicz et al. [
Bulewicz et al. [ present work
present work5.4 Sensitivity consideration of the OH∗ and CH∗ modeling
OH∗ CL was modeled based on the sub-mechanism derived in the present study (cf. Table 3). For pure H2/O2 mixtures (mixture A), OH∗ is exclusively formed via H+O+M=OH∗+M (R1). The OH∗ concentration is strongly affected by variations in the rate coefficient k 1. Varying the corresponding rate coefficient by a factor of 0.5 and 2, respectively, caused a change in the predicted OH∗ concentration of ±200 %. Even small amounts of CH4 with excess H2/O2 (mixture B: 250 ppm CH4 and mixture C: 500 ppm CH4) shifts the chemiluminescence formation channel towards CH+O2=OH∗+CO (R2). Similarly to k 1, variations of the rate coefficient k 2 induce strong deviations of the absolute OH∗ concentration by ±200 %. For mixtures containing acetylene (mixtures D to F), the ROP analysis revealed that OH∗ is exclusively produced via reaction (R2), whereas reaction (R1) reverses and consumes OH∗. The sensitivity of the modeled data with regard to the target rate k 2 was investigated by varying k 2 by a factor 0.5 and 2, respectively. This method caused a strong deviation of the absolute OH∗ concentration of factor ±2. The peak position and profile shape are insensitive to variations of the rate coefficient k 2. These parameters are mainly affected by the underlying ground-state chemistry and the predicted concentrations of the precursor species CH and O2.
For C2H2/O2 mixtures, CH∗ is mainly formed by reaction (R5) (contribution ∼65 %), while (R6) is responsible for the remaining 35 %. The CH∗ intensities was sensitive towards variations in rate coefficients k 5 and k 6. The CH∗ concentration deviates by ±40 % in case of changing k 5 by a factor of 0.5 and 2. Whereas k 6 has an impact of ±20 % for the same variations (dashed lines in Fig. 7). For mixtures containing N2O as oxidizer, reactions (R5) and (R6) both contribute about 50 % to the formation of the CH∗ CL. The intensities showed a sensitivity with regard to the CH∗ concentration when varying k 5 and k 6. These variations influenced the simulated CH∗ concentration by ±30 % for both reaction channels.
The sensitivity of k 5 and k 6 with regard to the equivalence-ratio-dependent decrease of the CH∗ CL was checked by varying the rate coefficients by a factor of 0.5 and 2, respectively. Experimental and simulated data were evaluated by normalizing the intensity and concentration with regard to the corresponding value for the stoichiometric condition as shown in Fig. 6. It was found that variations of k 5 and k 6 have significant impact (deviation about ±20 %) on the equivalence-ratio-dependent CH∗ reduction shown in Fig. 6. The modeling results cannot reproduce the experimental data when varying the rate coefficients.
6 Conclusions
The kinetics of OH∗ formation was studied by a combined modeling and shock-tube approach. Shock-tube measurements were performed at temperatures between 1300–3200 K at around atmospheric pressures in H2/O2/CH4 systems. The existing kinetics model describing the OH∗ formation in hydrogen oxidation [8] was upgraded by taking the hydrocarbon formation reaction into account. The comparison of measured emission signals and computed OH∗ concentrations reveals that the reaction CH+O2=OH∗+CO (R2) dominates OH∗ chemiluminescence (CL) in hydrocarbon combustion. Good agreement between the model and experiment was found with a rate coefficient of (8.0±2.6)×1010 cm3 mol−1 s−1 for reaction (R2).
Shock-tube experiments with C2H2/O2 and C2H2/N2O mixtures were performed to study OH∗ chemiluminescence. The OH∗ mechanism was applied to predict OH∗ formation in various acetylene mixtures from fuel-rich to lean mixture compositions. The comparison of measured and computed data showed good agreement in terms of T-dependence of chemiluminescence for both oxidizers. The computed peak times of OH∗ for N2O-containing mixtures are underpredicted due to uncertainties in the interaction of C2H2 and NO x chemistry, whereas they match the experimental data for C2H2/O2 mixtures.
CH∗ CL was monitored in reacting C2H2/O2 mixtures under various conditions. Based on the experiments using the C2H2/O2 systems, the reactions C2+OH=CH∗+CO (R5) and C2H+O=CH∗+CO (R6) are identified as main channels leading to CH∗. A kinetics model was developed and optimized to reproduce the experimental data and corresponding rate coefficients of k 5=2.0×1014 cm3 mol−1 s−1 and k 6=3.6×1012exp(−10.9 kJ mol−1/RT) cm3 mol−1 s−1 are determined. The CH∗ sub-mechanism is applied to describe the CH∗ chemiluminescence in C2H2/N2O mixtures.
References
J. Kojima, Y. Ikeda, T. Nakajima, Combust. Flame 140, 34 (2005)
H.N. Najm, P.H. Paul, C.J. Mueller, P.S. Wyckoff, Combust. Flame 113, 312 (1998)
J.G. Lee, D.A. Santavicca, J. Propuls. Power 19, 735 (2003)
N. Docquier, S. Candel, Prog. Energy Combust. Sci. 28, 107 (2002)
Y. Hardalupas, M. Orain, Combust. Flame 139, 188 (2004)
P.G. Aleiferis, Y. Hardalupas, A.M.K.P. Taylor, K. Ishii, Y. Urata, Exp. Fluids 39, 789 (2005)
T.M. Muruganandam, B.H. Kim, M.R. Morrell, V. Nori, M. Patel, B.W. Romig, J.M. Seitzman, Proc. Combust. Inst. 30, 1601 (2005)
T. Kathrotia, M. Fikri, M. Bozkurt, M. Hartmann, U. Riedel, C. Schulz, Combust. Flame 157, 1261 (2010)
J.M. Hall, E.L. Petersen, Int. J. Chem. Kinet. 38, 714 (2006)
W.E. Kaskan, J. Chem. Phys. 31, 944 (1959)
T. Koike, K. Morinaga, Bull. Chem. Soc. Jpn. 55, 52 (1982)
Y. Hidaka, S. Takahashi, H. Kawano, M. Suga, W.C. Gardiner Jr., J. Phys. Chem. 86, 1429 (1982)
G.P. Smith, C. Park, J. Luque, Combust. Flame 140, 385 (2005)
E.L. Petersen, D.M. Kalitan, M.J.A. Rickard, in 39th AIAA Joint Propulsion Conference and Exhibit, AIAA 2003-4493, Huntsville, AL, 2003
O.V. Skrebkov, S.P. Karkach, V.M. Vasil’ev, A.L. Smirnov, Chem. Phys. Lett. 375, 413 (2003)
O.V. Skrebkov, Y.P. Myagkov, S.P. Karkach, V.M. Vasil’ev, A.L. Smirnov, Dokl., Phys. Chem. 383, 93 (2002)
G.D. Smekhov, L.B. Ibraguimova, S.P. Karkach, O.V. Skrebkov, O.P. Shatalov, High Temp. 45, 395 (2007)
M. Kopp, N. Donato, M. Brower, E.L. Petersen, Appl. Phys. B (2012, in press)
F.E. Belles, M.R. Lauver, J. Chem. Phys. 40, 415 (1964)
R.P. Porter, A.H. Clark, W.E. Kaskan, W.E. Browne, Proc. Combust. Inst. 11, 907 (1967)
D. Gutman, R.W. Lutz, N.F. Jacobs, E.A. Hardwidge, G.L. Schott, J. Chem. Phys. 48, 5689 (1968)
W.C. Gardiner Jr., K. Morinaga, D.L. Ripley, T. Takeyama, Phys. Fluids 12, 120 (1969)
J. Grebe, K.H. Homann, Ber. Bunsenges. Phys. Chem. 86, 587 (1982)
D.A. Lichtin, M.R. Berman, M.C. Lin, Chem. Phys. Lett. 108, 18 (1984)
P.H. Paul, J.L. Durant Jr., J.A. Gray, M.R. Furlanetto, J. Chem. Phys. 102, 8378 (1995)
S.A. Carl, M. Van Poppel, J. Peeters, J. Phys. Chem. A 107, 11001 (2003)
G.P. Smith, J. Luque, C. Park, J.B. Jeffries, D.R. Crosley, Combust. Flame 131, 59 (2002)
T. Kathrotia, U. Riedel, A. Seipel, K. Moshammer, A. Brockhinke, Appl. Phys. B (2012, in press). doi:10.1007/s00340-012-5002-0
H.P. Broida, A.G. Gaydon, Proc. R. Soc. Lond. Ser. A, Math. Phys. Sci. 218, 60 (1953)
A.M. Bass, H.P. Broida, Natl. Bur. Stand. Monogr. 24, 20 (1961)
A.G. Gaydon, The Spectroscopy of Flames (Wiley, New York, 1957)
R. Bleekrode, W.C. Nieuwpoort, J. Chem. Phys. 43, 3680 (1965)
E.M. Bulewicz, P.J. Padley, R.E. Smith, Proc. R. Soc. Lond. Ser. A, Math. Phys. Sci. 315, 129 (1970)
G.P. Glass, G.B. Kistiakowsky, J.V. Michael, H. Niki, J. Chem. Phys. 42, 608 (1965)
W. Brennen, T. Carrington, J. Chem. Phys. 46, 7 (1967)
A. Brockhinke, M. Heusing, J. Krüger, M. Letzgus, Appl. Phys. B (2012, in press)
R.G. Joklik, J.W. Daily, W.J. Pitz, Proc. Combust. Inst. 21, 895 (1988)
K. Devriendt, H. Van Look, B. Ceursters, J. Peeters, Chem. Phys. Lett. 261, 450 (1996)
K. Devriendt, J. Peeters, J. Phys. Chem. 101, 2546 (1997)
J.M. Hall, J. de Vries, A.R. Amadio, E.L. Petersen, in 43th AIAA Aerospace Sciences Meeting and Exhibit, AIAA 2005-1318, Reno, NV, 2005
R.M.I. Elsamra, S. Vranckx, S.A. Carl, J. Phys. Chem. A 109, 10287 (2005)
C.W. Hand, G.B. Kistiakowsky, J. Chem. Phys. 37, 1239 (1962)
D. Gutman, S. Matsuda, J. Chem. Phys. 52, 4122 (1970)
S. Matsuda, I.R. Slagle, D.J. Fife, J.R. Marquart, D. Gutman, J. Chem. Phys. 57, 5277 (1972)
A.M. Renlund, F. Shokoohi, H. Reisler, C. Wittig, Chem. Phys. Lett. 84, 293 (1981)
S.M. Hwang, W.C. Gardiner Jr., M. Frenklach, Y. Hidaka, Combust. Flame 67, 65 (1987)
R.J. Kee, F.M. Rupley, J.A. Miller, M.E. Coltrin, J.F. Grcar, E. Meeks, H.K. Moffat, A.E. Lutz, G. Dixon-Lewis, M.D. Smooke, J. Warnatz, G.H. Evans, R.S. Larson, R.E. Mitchell, L.R. Petzold, W.C. Reynolds, M. Caracotsios, W.E. Stewart, P. Glarborg, C. Wang, O. Adigun, in CHEMKIN Collection, San Diego, CA, 2000; vol. Release 3.6
M. Frenklach, M. Goldenberg, N. Moriarty, C.T. Bowman, R.K. Hanson, D.F. Davidson, W.C. Gardiner Jr., V. Lissianski, G.P. Smith, D.M. Golden, R.V. Serauskas, in Proc. Int. Gas Res. Conf. (1998), pp. 329–336
P.A. Berg, D.A. Hill, A.R. Noble, G.P. Smith, J.B. Jeffries, D.R. Crosley, Combust. Flame 121, 223 (2000)
J.M. Hall, E.L. Petersen, in 40th AIAA Joint Propulsion and Exhibit, AIAA 2005-4164, Fort Lauderdale, 2004
H. Wang, A. Laskin, in A Comprehensive Kinetic Model of Ethylene and Acetylene Oxidation at High Temperatures (Department of Mechanical Engineering, University of Delaware, 1998)
B.A. Williams, L. Pasternack, Combust. Flame 111, 87 (1997)
T. Kathrotia, Reaction kinetics modeling of OH∗, CH∗, and \(\mathrm{C}_{2}^{*}\) chemiluminescence, PhD Thesis, Heidelberg, 2011
M. Tamura, P.A. Berg, J.E. Harrington, J. Luque, J.B. Jeffries, G.P. Smith, D.R. Crosley, Combust. Flame 114, 502 (1998)
N.L. Garland, D.R. Crosley, Chem. Phys. Lett. 134, 189 (1987)
Acknowledgements
The authors gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG) for financial support within the cooperative project “Chemiluminescence and heat release”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bozkurt, M., Fikri, M. & Schulz, C. Investigation of the kinetics of OH∗ and CH∗ chemiluminescence in hydrocarbon oxidation behind reflected shock waves. Appl. Phys. B 107, 515–527 (2012). https://doi.org/10.1007/s00340-012-5012-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-012-5012-y