
Abstract
The methylidyne radical (CH) is a hydrocarbon combustion intermediate of great importance in the prompt formation of NO, a highly regulated pollutant. Recent work in modeling the combustion chemistry of NO has highlighted the need for further study of CH and its associated high-temperature chemistry. This paper details the new development of a fixed-wavelength, cw laser absorption diagnostic for measurements of CH at high temperatures behind reflected shock waves. A detailed spectroscopic model was utilized to predict CH absorption within the A ← X band of CH, and the broadening coefficient of the selected blended transition was measured in a series of fixed-wavelength measurements. A demonstration of the diagnostic was performed in highly Ar-diluted C2H6/O2 and CH4/O2 mixtures between 1750 and 3050 K near 101 kPa. For both mixtures, experimental peak levels of CH were found to be generally overestimated by 40–80% by two chemical kinetics mechanisms. Such discrepancies highlight the need for further refinement of CH chemistry, which could be facilitated through future applications of this diagnostic and through the data provided herein. To the best of the authors’ knowledge, this study provides the first CH profiles measured in C2H6 oxidation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The methylidyne radical (CH) is an intermediate species in hydrocarbon flames [1], where its emission can serve as a marker of heat release. It also participates in the chemical kinetics of reburning [2], an industrial method for reducing emissions of the pollutant NO (and, ultimately, NO2). However, perhaps the most prominent role of CH is that as the key hydrocarbon combustion intermediate in the prompt-NO reaction sequence [3], which rapidly forms NO near the reaction zone of hydrocarbon flames and is an important pathway to NO formation. CH plays a critical role in prompt-NO formation by initiating this sequence via the reaction
where the NCN radical eventually forms NO via reactions with O or OH. The products of R1 were originally postulated to be HCN + N [3], but more recent work has confirmed that the products are NCN + H [4].
The importance of NOx kinetics has instigated numerous measurements of the CH radical, primarily within low-pressure, hydrocarbon flames. Such flame-based measurements of CH have utilized methods including laser-induced fluorescence [5] and cavity ring-down spectroscopy [6]. For a more complete summary of the large number of flame-based CH measurements, which is beyond the scope of interest of this paper, the reader is referred to Appendix B of Glarborg et al. [4]. A handful of CH measurements have also been performed in shock tubes [7,8,9,10,11,12,13,14,15,16]. Shock tubes are ideal devices for producing well-characterized conditions at extreme temperatures (up to ~ 6000 K) and pressures (up to ~ 100 MPa). The flow fields produced behind reflected shock waves are highly homogenous, and thus the chemical kinetics modeling may be decoupled from fluid mechanics effects, making shock tubes invaluable in the study of gas-phase chemical kinetics.
The earliest shock-tube study of CH using laser absorption seems to be the work of Dean and Hanson in 1989 [7]. Dean and Hanson developed a cw, fixed-wavelength laser absorption diagnostic for CH using narrow-linewidth light at 431.1311 nm. Subsequent work by the same group employed this diagnostic behind reflected shock waves to study the CH + N2 reaction [8, 9]; pyrolysis of CH4 and C2H6 [10]; oxidation of CH4 [11]; the CH + O2 reaction [12]; and CH3 decomposition [13]. Concurrent shock-tube work from another group, utilizing the same CH diagnostic method developed by Dean and Hanson, investigated CH3 decomposition [14], CH2 decomposition [15], and the reactions of CH with CO2 and O2 [16]. Note that, to the best of the authors’ knowledge, these studies encompass the whole of the shock-tube literature concerning the measurement of CH radicals.
Despite the significant body of knowledge on prompt-NO chemical kinetics, the 2018 review by Glarborg et al. on nitrogen chemistry in combustion concluded that one of two primary uncertainties in nitrogen chemistry remains the prompt-NO reaction sequence [4]. Given the crucial role of CH in this sequence, there is a need to rectify the small number of CH shock-tube measurements to further improve understanding of CH chemistry. Therefore, the goal of the present study was to detail the development of a CH laser absorption diagnostic for future shock-tube studies and also to provide representative measurements as a demonstration of the diagnostic. This diagnostic is employed herein in the same A2∆(ν = 0) ← X2Π(ν = 0) band of CH utilized previously, albeit at a slightly different wavelength. Presented in this paper are the pertinent theory and a description of the experiment, including a validation of the spectroscopic parameters for the selected CH transition. Sample results from CH measurements in CH4 and C2H6 oxidation are then presented, followed by a brief discussion of the chemical kinetics involved in analyzing such time histories and the opportunities for new insights that they can provide.
2 Experiment
Experiments were performed at the Aerospace Corporation in El Segundo, CA. Given below are descriptions of the shock tube and its associated hardware, as well as the laser diagnostic and its arrangement in conjunction with the shock tube.
2.1 Shock tube
The shock tube used in this study has been discussed in detail previously [17] and is described only briefly herein. The driver section has a length and inner diameter (ID) of 3.5 m and 7.62 cm, respectively, while the driven section has a length and ID of 10.7 m and 16.2 cm; both are composed of stainless steel. Five fast-response, piezoelectric pressure transducers (PCB P113A) were used to monitor the passage of the incident shock wave; the intervals between transducers were timed with four Fluke PM6666/016 timers and used to measure the incident-shock velocity. The incident-shock velocity, including a typical attenuation of 0.5–2.0%/m, was then extrapolated to the endwall and used in conjunction with the 1-D normal shock relations to calculate the temperature (\(T_{5}\)) and pressure (\(P_{5}\)) behind the reflected shock wave, with estimated uncertainties of ± 0.8% and ± 1.0%, respectively, originating from uncertainty in the measured incident-shock velocity [17]. Laser measurements were carried out in a plane located 1.6 cm from the endwall through the use of two CaF2 windows; simultaneous pressure measurements were obtained at this same location using the final PCB transducer. A vacuum system incorporating rotary vane pumps and a turbomolecular pump was used to ensure high-purity conditions inside the shock tube; typical driven-side ultimate pressures were ~ 10−3 Pa, while typical leak rates were 10−2 Pa min−1 or less.
Mixtures were prepared manometrically in a stainless steel mixing tank. Three MKS Baratron capacitance manometers (PDR-C-1B) with ranges of 0–1.3, 0–13, and 0–130 kPa were used to measure the partial pressures of constituents. Due to the high levels of dilution used herein, a double dilution method was utilized. This method involved the creation of a highly concentrated mixture that was allowed to mix for 30 min to ensure homogeneity, followed by further dilution with Ar and another 30 min of mixing prior to experiments. Gases were supplied by the following manufacturers at the following purity levels: C2H6—Matheson, 99.99%; CH4—Matheson, 99.97%; O2—Airgas, 99.6%, extra dry; Ar—Praxair, 99.998%.
2.2 Laser diagnostic
A diode-pumped solid-state laser (Millennia eV 15 from Spectra-Physics) was used to pump the Ti:Sa crystal. The pump laser provided up to 15 W at 532 nm and was sealed, requiring no laser head adjustments in normal operation. While the pump laser did not operate with single-frequency output, the amplitude was stable to within ± 1% and the beam quality was high (M2 < 1.1). The input power level selected for Ti:Sa pumping was 11–12 W. A chiller (RTE-7 from Thermo Neslab) provided temperature-controlled cooling water to the system, which was sensitive to temperature variations.
A cw ring laser in Ti:Sa configuration (899-21 from Coherent) was pumped by the pump laser, generating tunable, single-frequency output that can be locked to a scanning Fabry–Perot reference cavity. A mix of short wave and medium wave optical elements was installed in the ring cavity to enable usable power output over the 780–860 nm wavelength range without changing any optics. Typical power output at 855 nm was 0.8 W. The temperature of the Ti:Sa crystal was controlled by either a dedicated chiller (RTE-7 from Thermo Neslab) or by sharing the unit that cooled the pump laser.
A small portion of the ring laser output was split off to be monitored by a wavemeter (WA-1500 from Burleigh), with an estimated accuracy of ± 0.0047 cm−1 according to the manufacturer specification of ± 0.2 ppm. An optical telescope expanded the beam prior to admission to an external frequency doubler (MBD200 from Coherent). An LBO crystal optimized for 860 nm operation was used as the doubling crystal. Typical output power at the doubled frequency of 23424.924 cm−1 was 13 mW.
While the laser line widths were not measured, the laser system was capable of 1-MHz line widths, and the laser frequency was locked to an external laser cavity with a piezoelectrically controlled cavity mirror to theoretically reduce the line width down to the MHz range. The doubling process increases the line width by less than a factor of 2. The blended spectral feature of CH utilized in this work has a width of about 1.3 × 105 MHz.
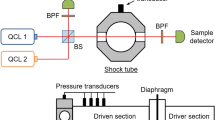
The doubled laser beam was focused into a CeramOptec non-solarizing UV optical fiber with a 200-µm core diameter, for transmission to the shock-tube laboratory about 60 m away. The far end of the optical fiber was mounted on a 4-axis positioning stage, and the emerging light was re-collimated to a beam of about 5-mm diameter. This beam was split into power reference \((I_{0} )\) and signal (\(I_{\text{t}}\)) paths, with the latter passing through CaF2 wedged windows on the shock tube. A differential preamplifier (SR560 from Stanford Research Systems) was used to measure the difference \(I_{0} - I_{\text{t}}\) for improved signal-to-noise ratio, with the \(I_{0}\) beam passing through an adjustable attenuator to permit beam balancing. Both beams were detected by silicon photodetectors (2032 from New Focus) operated with a gain of 105 and a bandwidth of 150 kHz. Apertures were employed to block broadband emission, and both detectors were fitted with optical bandpass filters centered at 430 nm with a full width at half maximum (FWHM) of 10 nm. In accordance with the detector bandwidth, some of the measured time histories were digitally filtered during post-processing using a first-order Butterworth filter with a cutoff of 150 kHz. A schematic of the laser setup is shown in Fig. 1.

Schematic of the laser diagnostic and shock tube. BS signifies a beamsplitter
Figure 2 shows representative laser transmission (\(I_{\text{t}} /I_{0}\)) and sidewall pressure traces. The passage of the incident shock wave raises the pressure (and thus temperature) slightly, and the passage of the reflected shock wave does so to an even greater extent. The pressure then remains nearly constant over the duration of the test time of interest. Both shock waves can also be observed as spikes in the laser transmission trace; these spikes are due to strong density gradients from the shock waves and do not represent actual CH formation. The peak of the spike from the reflected shock wave observed in the laser transmission trace was used to fix time zero for the experiment. The spike has a finite width due to the limited bandwidth of the detectors and is ~ 4 µs (FWHM) in Fig. 2; this introduces a conservative time zero uncertainty of ± 2 µs. For this experiment and all other CH4 and C2H6 oxidation experiments, the laser frequency was fixed at 23424.924 cm−1.

Representative a laser transmission and b sidewall pressure traces. Conditions are 2581 K, 86 kPa in a mixture of 1041 ppm CH4, 1982 ppm O2, balance Ar
The reactive mixtures used herein can cause deviations from the initial \(T_{5}\), as predicted by two chemical kinetic mechanisms. For the case of C2H6, the fuel decomposition causes a predicted drop in temperature of 30–40 K, while the ensuing exothermic combustion causes a rise of very nearly the same amount. For CH4, the predicted drop in temperature due to fuel decomposition is much smaller (~ 5 K), but the temperature rise due to exothermicity is about the same (30–40 K). However, an uncertainty analysis revealed that the CH absorption coefficient is largely insensitive to temperature changes of this magnitude and that the uncertainty is dominated by spectroscopic parameters and baseline uncertainty (see Sect. 4.2).
To check for the possibility of broadband emission entering the detectors and artificially reducing the amount of absorption measured, a series of emission experiments were performed. In these experiments, the laser was blocked from entering the detectors, and a shock experiment was performed at the high-temperature end of the test conditions (emission will be a maximum at higher temperatures). Even for the highest temperatures, the emission was only ~ 7% of the absorption signal, which may cause a slight systemic underestimation of the peak CH level at the highest temperatures herein. The emission drastically decreases with temperature, thus only affecting the higher temperature experiments.
3 Spectroscopic model
The Beer–Lambert law relates the transmission of monochromatic light to the concentration of an absorbing species and can be written as
where \(k_{\text{v}}\) is the absorption coefficient, \(X_{\text{abs}}\) is the mole fraction of the absorbing species, \(P\) is the pressure, and \(L\) is the absorption path length. In the current experiments, \(X_{\text{abs}}\) was the desired outcome of the experiment, while \(I_{\text{t}}\), \(I_{0}\), \(P\), and \(L\) were measured. The absorption coefficient \(k_{v}\), however, required calculation, and this calculation was the goal of the spectroscopic model outlined in this section. Dean and Hanson [7] demonstrated that the A2∆(ν = 0) ← X2Π(ν = 0) band of CH is useful for single-pass (\(L\) ≈ 10 cm) measurements of ~ ppm levels of CH at high temperatures, and thus this band was also selected for use in the present work. The spectroscopic model described herein was based upon the work of Vasudevan et al. [13], who used an updated version of the model proposed by Dean and Hanson.
3.1 Line strengths
The absorption coefficient \(k_{v}\) for an individual transition can be written as the product of the line strength \(S\left( T \right)\) and the line shape \(\phi \left( {\nu - \nu_{0} } \right)\),
where \(T\) is the temperature, \(\nu\) is the frequency, and \(\nu_{0}\) is the frequency of the transition. \(S\left( T \right)\) has units of cm−1 atm−1s−1, while \(\phi \left( {\nu - \nu_{0} } \right)\) has units of s. The line strength \(S\left( T \right)\) can be written as
Here, \(h\) is Planck’s constant, \(c\) is the speed of light, \(f_{B}\) is the Boltzmann fraction of the population in the lower state, \(B_{mn}\) is the Einstein absorption coefficient of the transition, \(N_{\text{A}}\) is Avogadro’s number, and \(R_{u}\) is the universal gas constant. The Boltzmann fraction \(f_{B}\) is given by
In Eq. (4), \(J^{\prime \prime }\) is the rotational quantum number of the lower state, \(F^{\prime \prime }\) is the term value of the lower state, \(k\) is Boltzmann’s constant, \(v^{\prime \prime }\) is the vibrational quantum number of the lower state, \(\omega_{\text{e}}\) is the vibrational frequency, and \(Q\) is the total internal partition function. Note that the second exponential term in the numerator of Eq. (4) does not contribute to \(f_{B}\) in this case since \(v^{\prime \prime } = 0\). The partition function \(Q\) is the product of the rotational, vibrational, and electronic partition functions:
Here, \(B^{\prime \prime }\) is the rotational constant, while \(Q_{el}\) has been assumed to be equal to the ground-state degeneracy of 4, as discussed by Vasudevan et al. [13].
The spectroscopic parameters required to evaluate Eqs. (2–5) were taken from a variety of sources to model a total of 328 lines. Line positions were taken from the LIFBASE database [18] and also from Zachwieja [19] and Brazier and Brown [20]. Einstein absorption coefficients were also taken from LIFBASE. Term values were taken from Zachwieja [19], as were the vibrational frequency and the rotational constant. The symbols, values, and sources for these parameters are summarized in Table 1.
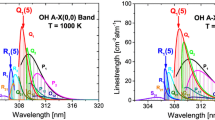
To assess the absorption coefficient across the entire A2∆(ν = 0) ← X2Π(ν = 0) band of CH, one must evaluate Eq. (2) for every single transition and then sum these results over the frequency range of interest to account for line overlap. Performing such a calculation at typical shock-tube conditions for producing measurable amounts of CH yields the spectrum shown in Fig. 3. Three distinct regions corresponding to the R, Q, and P branches can be observed, with occasional peaks occurring due to overlap of two or more lines. Labeled in Fig. 3 are two such peaks. Peak 2 is the line at 431.1311 nm (23194.801 cm−1) corresponding to the overlap of the Q1d(7) and Q2c(7) lines. Dean and Hanson [7] utilized Peak 2, as did all of the subsequent works mentioned previously. Peak 1 occurs at 426.8957 nm (23424.924 cm−1) as a result of the overlap of the R1d(7) and R2d(7) lines. In the present work, the laser configuration used did not allow access to Peak 2 and thus Peak 1 was selected. While Peak 1 has a lower absorption coefficient than Peak 2, it is worth noting that our detection limit (SNR of 3) is estimated to be 0.1 ppm at around 1750 K and 101 kPa, which is slightly lower than the 0.2 ppm value estimated by Dean and Hanson at similar conditions. The increased SNR of the present setup despite the smaller \(k_{v}\) is likely due to more-stable performance of the frequency-doubled Ti:saph setup as compared to the ring-dye configuration used in the earlier studies.

Absorption coefficients of CH in the A2∆(ν = 0) ← X2Π(ν = 0) band. The broadening coefficient for all lines was assumed to be that measured in the present work
3.2 Line shapes
Modeling of the line shape \(\phi \left( {\nu - \nu_{0} } \right)\) was performed using the Voigt profile. The Voigt profile is the convolution of the Gaussian and Lorentzian profiles, which model Doppler and collisional broadening phenomena, respectively. These profiles are respectively described by their FWHM values, \(\Delta v_{\text{D}}\) and \(\Delta v_{\text{c}}\). The Doppler FWHM is given in cm−1 by
where \(M\) is the molecular weight of the absorbing species. The constant coefficient in Eq. (6) assumes that \(\bar\nu_{0}\) and \(T\) have units of cm−1 and K, respectively. The collisional FWHM is given in cm−1 by
where \(\gamma_{i} \left( T \right)\) is the broadening coefficient of the absorbing species by the perturbing species \(i\) and \(X_{i}\) is the mole fraction of the perturbing species \(i\). The summation in Eq. (7) takes places over all \(i\) perturbing species including the absorbing species itself, although self-broadening was neglected in this work due to extremely small CH mole fractions. The temperature dependence of the broadening coefficient is typically given by the power law
where \(\gamma_{i} \left( {T_{0} } \right)\) is the broadening coefficient of the perturbing species \(i\) at reference temperature \(T_{0}\), and \(n_{i}\) is the temperature coefficient of the perturbing species \(i\).
In this work, the broadening coefficient \(\gamma_{\text{Ar}} \left( T \right)\) was assessed by performing a series of fixed-wavelength experiments in a highly dilute C2H6/Ar mixture and varying the laser wavelength incrementally over the selected transitions. Shown in Fig. 4 is a set of four representative absorption time histories from these experiments, obtained at very nearly the same \(T_{5}\) and \(P_{5}\) but with varying laser frequency. Obtaining the peak absorption from each of these time histories and plotting the results versus laser frequency yields Fig. 5. To obtain a line shape independent of temperature and pressure variations, the individual data points in Fig. 5 were corrected for slight deviations from the average temperature and pressure according to the Beer–Lambert law using the spectroscopic model discussed above along with normalized predictions of peak CH levels from the GRI 3.0 mechanism [21], which was used for this purpose since it accurately predicted relative changes in peak CH levels for this mixture. By repeatedly obtaining nearly the same \(T_{5}\) and \(P_{5}\) during the line shape characterization, the line shape displayed in Fig. 5 and the resulting best-fit broadening coefficient are independent of the exact amount of CH present other than the slight uncertainty introduced by the small corrections using the GRI 3.0 mechanism; this uncertainty is small in comparison to that introduced by noise in the laser signal (representative error bars in Fig. 5 are due to laser noise). The line positions were taken from the most recent experimental measurements by Zachwieja [19] and are 23424.7556 cm−1 for the R2d(7) line and 23424.9681 cm−1 for the R1d(7) line.

Representative absorption time histories obtained during C2H6 pyrolysis. All profiles were filtered with a 150-kHz filter. The parameter of interest in these time histories is the peak absorption

Peak absorption measured during pyrolysis of a mixture of 527 ppm C2H6, balance Ar. Voigt line shape modeling was performed using the broadening coefficient of the present study, both with (solid line) and without (dashed line) a line shift of − 0.008 cm−1
The best-fit value for the collisional width \(\Delta v_{c}\) at the conditions of Fig. 5 was found to be 0.020 cm−1 with an estimated uncertainty of 40%. Based on this collisional width and taking Eq. (8) with \(T_{0}\) = 2000 K, the expression for the broadening coefficient \(\gamma_{\text{Ar}} \left( T \right)\), was determined to be
with \(\gamma_{\text{Ar}} \left( T \right)\) in cm−1 atm−1 and \(T\) in K. The coefficient in Eq. (9) is 26% smaller than that measured by Dean and Hanson [7] for the line at 431.1311 nm. The temperature exponent of 0.94 was taken from the work of Dean and Hanson, who based their value on previously measured high-temperature Ar-broadening of the diatomic radicals OH and NH; these values were based on scanned-wavelength measurements that can often provide more accurate determinations of \(n\) than fixed-wavelength diagnostics. A slight line shift of − 0.008 cm−1 was incorporated into the fit shown in Fig. 5 and also into all calculations of \(k_{v}\) herein. This line shift is similar in magnitude and direction to that measured by Vasudevan et al. [13] for the line at 431.1311 nm and might be due to a pressure-induced collisional line shift or to a systematic error in the experimental line positions. It is worth noting that this line shift approaches the ± 0.0047 cm−1 accuracy of the wavemeter. The 40% uncertainty in \(\Delta v_{\text{c}}\), and thus in \(\gamma_{\text{Ar}} \left( T \right)\), stems from the fact that the Voigt profile is dominated by the Doppler FWHM \(\Delta v_{\text{D}}\), which is 10–12 times greater than \(\Delta v_{\text{c}}\) at the conditions of the present study. However, because of the dominance of \(\Delta v_{\text{D}}\) at these conditions, even a 40% uncertainty in \(\gamma_{\text{Ar}} \left( T \right)\) creates only a ~ 2.8% uncertainty in the calculated CH mole fraction (see Sect. 4.2).
4 Sample results
To demonstrate the application of this CH diagnostic to shock-tube chemical kinetics studies, a set of representative tests were performed in oxidative mixtures of CH4 and C2H6. Shown below are sample CH time histories from the C2H6 mixture alongside model predictions from chemical kinetics mechanisms. Peak- and timing-related parameters for both mixtures are also extracted and compared to model predictions, uncertainty levels are estimated, and important reactions are identified via reaction pathway analyses as a demonstration of the capabilities of this diagnostic when paired with such detailed chemical kinetic analyses.
4.1 CH time histories
CH time histories measured during the oxidation of a near-stoichiometric (\(\phi\) = 1.03) C2H6/O2 mixture diluted in ~ 99.5% Ar are shown in Fig. 6 in order of increasing temperature. Following the passage of the reflected shock wave, CH immediately begins to form. At colder temperatures, an interesting two-stage feature manifests itself, where a second regime of CH formation is visible. At higher temperatures, the two stages of CH formation seem to become indistinguishable. Note that, as discussed in Sect. 2.2, the peaks at time zero are artefacts of the reflected shock wave and do not signify any CH formation, although the finite bandwidth of the detectors has caused the actual CH formation to be slightly masked by this phenomenon for the hotter experiments.

Also displayed in Fig. 6 are the predictions of two chemical kinetics mechanisms. These mechanism simulations were performed in the Chemkin Pro software suite using the closed, homogenous batch reactor with the constant-energy, constant-volume assumption (note that for highly dilute mixtures such as this one, assuming constant pressure instead of constant volume would yield identical results). Both mechanisms generally capture the shape of the experimental CH time histories, including the two-stage formation. However, both mechanisms seem to be lacking in their predictions of peak CH levels, particularly at higher temperatures.
The discrepancy between the experimental and predicted peak CH levels is highlighted in Fig. 7a. Both models overestimate the peak CH at higher temperatures by about 40% (Aramco 3.0) and 75% (GRI 3.0). GRI 3.0 also overestimates the peak CH at lower temperatures by about 70%, while Aramco 3.0 actually begins to underestimate the peak CH by about 25%. The interesting behavior between 1800 and 2000 K predicted by both models is due to effects of the two-stage formation of CH, which can be seen in Fig. 6. This two-stage formation can also be observed in the predicted temporal FWHM of the CH profiles (referred to as FWHMCH) shown in Fig. 7b, where both models predict interesting non-monotonic behavior between 1800 and 2000 K. GRI 3.0 seems to correctly predict this behavior, while Aramco 3.0 tends to be slightly too reactive.

Temperature dependence of a peak CH and b FWHMCH in a mixture of 1041 ppm C2H6, 3553 ppm O2, balance Ar. The average pressure is 98 kPa. Respective uncertainty bars of ± 7.5% and ± 0.8% for peak CH (see Sect. 4.2) and temperature are approximately the size of the symbols. Y-axis uncertainty bars at the temperature extrema are shown in b and are experiment-specific. Mechanism predictions were calculated using GRI 3.0 [21] and Aramco 3.0 [22]
Figure 8 shows two CH time histories measured during the oxidation of a near-stoichiometric (\(\phi\) = 0.95) CH4/O2 mixture diluted in ~ 99.7% Ar, along with model predictions from two chemical kinetics mechanisms. These two profiles represent the low- and high-temperature extremes measured in the CH4 mixture. Only two profiles are shown for the CH4 mixture since the CH formation in this mixture displayed only a single stage of CH formation across all temperatures tested, in contrast to the C2H6 mixture. Another difference between the C2H6 and CH4 mixtures is the presence of an induction delay period at colder temperatures in the CH4 mixtures, which can be observed in Fig. 8a.

Figure 9 shows the peak CH and time-to-peak-CH data for the CH4 mixture used herein. The time-to-peak-CH criterion was chosen since the CH time histories from the CH4 mixture showed no signs of two-stage CH formation across the conditions tested. This single-stage behavior can be observed in the monotonic trends predicted by both mechanisms in Fig. 9, as opposed to the piecewise behavior predicted for the C2H6 mixture in Fig. 7. The time to peak is accurately predicted by both mechanisms across all conditions, save a slight deviation by Aramco 3.0 above 2900 K. On the other hand, in a similar manner observed for the C2H6 mixture, peak CH levels are overpredicted by ~ 40% (Aramco 3.0) and ~ 80% (GRI 3.0) across the experimental range of temperatures. All 14 CH time histories recorded in the present study are provided in the Supplementary Material.

Temperature dependence of a peak CH and b time to peak in a mixture of 1041 ppm CH4, 1982 ppm O2, balance Ar. The average pressure is 89 kPa. Respective uncertainty bars of ± 7.5% and ± 0.8% for peak CH (see Sect. 4.2) and temperature are approximately the size of the symbols. Y axis uncertainty in b was estimated at ± 15%. Mechanism predictions were calculated using GRI 3.0 [21] and Aramco 3.0 [22]
4.2 Uncertainty analysis
To quantify the uncertainty in the measured CH mole fraction, detailed uncertainty analyses were performed on two experiments at the high- and low-temperature extremes of this study. A total of 9 independent variables were individually adjusted within their uncertainty limits and the corresponding change in calculated CH mole fraction was recorded; the uncertainty limits for each variable were either retrieved from the appropriate sources or estimated. Finally, the contributions of all uncertainty sources were aggregated using the root-sum-squares method. For the coldest experiment performed in this study (1747 K), the total uncertainty in the CH mole fraction was estimated to be ± 7.46% (Fig. 10). The primary contributors to this uncertainty are the spectroscopic parameters \(Q\), \(\gamma_{\text{Ar}} \left( T \right)\), and \(B_{\text{mn}}\). For the hottest experiment of this study (3048 K), the estimated uncertainty due to the variables considered in Fig. 10 was slightly lower at ± 7.14%. Using the root-sum-squares method to incorporate the additional ~ 7% uncertainty due to the emission present at higher temperatures, the uncertainty at higher temperatures was estimated to be ± 10.0%.

Root-sum-squares uncertainty analysis for an experiment at 1747 K and 107 kPa in a mixture of 1041 ppm C2H6, 3553 ppm O2, balance Ar. The total estimated uncertainty is ± 7.46%. Inset text: assumed uncertainty of each variable, bars: relative contribution to total uncertainty, left column: percent change in \(X_{\text{CH}}\)
4.3 Chemical kinetics analysis
This CH diagnostic permits more than comparisons between experimental data and model predictions, useful though such comparisons are. A detailed chemical analysis of the model predictions can explain qualitative or quantitative differences between models in terms of reaction rates and can also yield valuable chemical kinetic information such as individual reaction rates or branching ratios. Tools that are readily available for such investigations include A-factor sensitivity, rate-of-production, and reaction pathway analyses.
For example, a reaction pathway analysis at the lower temperature conditions of Fig. 6a sheds light on the two-stage formation of CH. The first stage of CH formation takes place due to the rapid formation of CH3 radicals via reaction with a third-body collider M:
These CH3 radicals then go on to form CH through the sequence
The second stage of CH formation visible in Fig. 6a is primarily due to gradual H-abstraction of C2H6 and its products to progressively form C2H5, C2H4, C2H3, and finally C2H2. The C2H2 then forms CH2 through the following sequence:
where the CH2 produced by R7 then forms CH via R4 (CH2* denotes singlet CH2). Another channel for direct CH2 formation from C2H2 is the reaction
but this pathway is relatively minor, consuming C2H2 at ~ 25% of the rate of R5 for this low-temperature case.
At higher temperatures, the CH3 radical consumption from R3 faces competition from an alternate pathway for C2H5 formation,
which accelerates the second-stage formation process, thus blending the two stages together until they coalesce (Fig. 6d). Note that R9 has a significantly larger activation energy than R3, making R9 increasingly more significant than R3 with increasing temperature. As for the differences in predictions of peak CH levels from GRI 3.0 and Aramco 3.0, it was observed that GRI 3.0 does not include the primary pathway for CH consumption,
which is partly the explanation for the higher levels of CH formation predicted by GRI 3.0 (OH* denotes OH that has been excited to the A2Σ+ state).
For the case of the CH4 mixture, CH3 is formed primarily through CH4 decomposition
and subsequently goes on to form CH through R3 and R4. However, the CH3 formation in the CH4 mixture is significantly reduced and delayed as compared to the C2H6 mixture due partly to the fact that R11 produces only one CH3 radical from each CH4 molecule, as opposed to two CH3 radicals from each C2H6 molecule via R2. This delayed CH3 formation is the reason for the induction delay observed in Fig. 8a. As for the single-stage CH formation in the CH4 mixture, while some C2H6 is formed in the CH4 mixture due to the well-known recombination of CH3 radicals, this amount of C2H6 is almost negligible at these lower pressures, and thus the H-abstraction pathway leading to C2H2 (and ultimately CH through R5, R6, and R7) is effectively removed. Therefore, the sequence of R11, R3, and R4 is dominant CH formation pathway in the CH4 mixture at all conditions tested herein, and there is accordingly no two-stage formation of CH in the CH4 mixture.
The preceding analysis is intended neither to make any statements concerning the rates of individual reactions nor to suggest any changes to either mechanism considered; such a goal is beyond the scope of the present paper (although these newly acquired data may aid in such work in the future). Rather, this discussion is meant to briefly demonstrate the utility of this species-specific diagnostic when coupled with detailed chemical kinetics analyses. Such analyses can identify reaction rates that may be responsible for discrepancies between models and data and thus highlight the need for experimental or computational work to further refine these reaction rates. As mentioned above, the prediction of CH concentration is important for the prompt NO formation mechanism in combustion systems. Furthermore, chemical kinetic analyses can also be used to design carefully tailored experiments (i.e., specific mixtures and conditions) using this CH diagnostic that can isolate individual reaction rates or branching ratios.
5 Conclusions
A fixed-wavelength, cw laser diagnostic was developed for the measurement of CH at high temperatures behind reflected shock waves. This diagnostic utilizes frequency-doubled radiation near 427 nm from a diode-pumped Ti:Sa ring laser. The CH absorption coefficient was calculated as a function of temperature and pressure via a detailed spectroscopic model, with an estimated uncertainty of ± 7.5%. The broadening coefficient \(\gamma_{\text{Ar}}\) was measured in a series of separate experiments involving C2H6 pyrolysis. The detection limit of this diagnostic is estimated to be 0.1 ppm at 1750 K and 101 kPa. Representative tests were performed in dilute mixtures of C2H6/O2 and CH4/O2, and the measurements were compared with predictions from chemical kinetics mechanisms. Peak levels of CH were found to be generally overpredicted by the mechanisms, highlighting a need for future work to resolve such discrepancies. This diagnostic can be applied to the measurement of CH time histories in a variety of combustion systems, with specific application to NO chemistry.
References
J. Kojima, Y. Ikeda, T. Nakajima, Proc. Comb. Inst. 28, 1757 (2000)
J.A. Miller, C.T. Bowman, Prog. Ener. Comb. Sci. 15, 287 (1989)
C.P. Fenimore, Symp. (Int.) on Combust. 13, 373 (1971)
P. Glarborg, J.A. Miller, B. Ruscic, S.J. Klippenstein, Prog. Energy Comb. Sci. 67, 31 (2018)
J. Luque, R.J.H. Klein-Douwel, J.B. Jeffries, G.P. Smith, D.R. Crosley, App. Phys. B 75, 779 (2002)
R. Evertsen, R.L. Stolk, J.J. Ter Meulen, Comb. Sci. Tech. 149, 19 (1999)
A.J. Dean, R.K. Hanson, J. Quant. Spec. Rad. Trans. 42, 375 (1989)
A.J. Dean, R.K. Hanson, C.T. Bowman, Symp. (Int.) on Combust. 23, 259 (1991)
V. Vasudevan, R.K. Hanson, C.T. Bowman, D.M. Golden, D.F. Davidson, J. Phys. Chem. A 111, 11818 (2007)
A.J. Dean, R.K. Hanson, Int. J. Chem. Kin. 24, 517 (1992)
D. Woiki, M. Votsmeier, D.F. Davidson, R.K. Hanson, C.T. Bowman, Combust. Flame 113, 624 (1998)
M. Röhrig, E.L. Petersen, D.F. Davidson, R.K. Hanson, C.T. Bowman, Int. J. Chem. Kin. 29, 781 (1997)
V. Vasudevan, R.K. Hanson, D.M. Golden, C.T. Bowman, D.F. Davidson, J. Phys. Chem. A 111, 4062 (2007)
M.W. Markus, D. Woiki, P. Roth, Symp. (Int.) on Combust. 24, 581 (1992)
M.W. Markus, P. Roth, A.M. Tereza, Symp. (Int.) on Combust. 25, 705 (1994)
M.W. Markus, P. Roth, T. Just, Int. J. Chem. Kin. 28, 171 (1996)
E.L. Petersen, M.J. Rickard, M.W. Crofton, E.D. Abbey, M.J. Traum, D.M. Kalitan, Meas. Sci. Tech. 16, 1716 (2005)
J.C. Luque, D.R., (SRI International, Menlo Park, CA, SRI International Report MP 99-0099, 1999)
M. Zachwieja, J. Mol. Spectr. 170, 285 (1995)
C.R. Brazier, J.M. Brown, Canadian. J. Phys. 62, 1563 (1984)
G.P. Smith, D.M. Golden, M. Frenklach, N.W. Moriarty, B. Eiteneer, M. Goldenberg et al., http://www.me.berkeley.edu/gri_mech/(1999)
C.-W. Zhou, Y. Li, U. Burke, C. Banyon, K.P. Somers, S. Ding et al., Combust. Flame 197, 423 (2018)
Acknowledgments
This work was funded in part by the TEES Turbomachinery Laboratory and the Aerospace Corporation’s Internal Research and Development program. A critical portion of the CH diagnostic setup was funded by Siemens Canada, with Dr. Gilles Bourque as project monitor. Additional funding came from the National Science Foundation, Grant Number CBET-1706825. The authors wish to thank J.L. Elms for significant contributions to the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mulvihill, C.R., Crofton, M.W., Arnold, D.G. et al. A laser diagnostic at 427 nm for quantitative measurements of CH in a shock tube. Appl. Phys. B 125, 78 (2019). https://doi.org/10.1007/s00340-019-7188-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-019-7188-x