
Abstract
Key message
CRISPR/Cas9-based multiplexed editing of SlHyPRP1 resulted in precise deletions of its functional motif(s), thereby resulting in salt stress-tolerant events in cultivated tomato.
Abstract
Crop genetic improvement to address environmental stresses for sustainable food production has been in high demand, especially given the current situation of global climate changes and reduction of the global food production rate/population rate. Recently, the emerging clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas)-based targeted mutagenesis has provided a revolutionary approach to crop improvement. The major application of CRISPR/Cas in plant genome editing has been the generation of indel mutations via error-prone nonhomologous end joining (NHEJ) repair of DNA DSBs. In this study, we examined the power of the CRISPR/Cas9-based novel approach in the precise manipulation of protein domains of tomato hybrid proline-rich protein 1 (HyPRP1), which is a negative regulator of salt stress responses. We revealed that the precise elimination of SlHyPRP1 negative-response domain(s) led to high salinity tolerance at the germination and vegetative stages in our experimental conditions. CRISPR/Cas9-based domain editing may be an efficient tool to engineer multidomain proteins of important food crops to cope with global climate changes for sustainable agriculture and future food security.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) cleavage usually produces blunt-end double-stranded breaks (DSBs) at a position three base pairs upstream of the NGG PAM sequence (Jinek et al. 2012; Van Vu et al. 2019). The DSB blunt ends are efficiently religated via nonhomologous end-joining (NHEJ), generating precise repair products with relatively rare indel products (Chiruvella et al. 2013; Davis and Chen 2013).
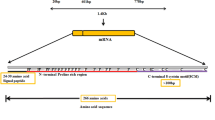
It is possible that when we introduce two or more DSB sites in a genome, the 3′-end of the downstream DSB site could be ligated to the 5′-end of the upstream DSB site, generating precise removal of the DNA sequence located between the DSB sites (Betermier et al. 2014; Cong et al. 2013). The approach was validated in mammals and plants for precision genome editing (Zheng et al. 2014; Guo et al. 2018; Zhao et al. 2016). A recent paper characterized DSB repair by canonical NHEJ and showed high efficacy of precise ligation of compatible DSB ends (blunt or complementary cohesive ends) (Stinson et al. 2019), thereby supporting the CRISPR/Cas9-based domain deletion strategies (Fig. 1a) proposed in this work.

System construction and expected outcomes of the approach. a A multiplexed CRISPR/Cas9 editing system was used with four guide RNAs (gRNA1, gRNA2, gRNA3, and gRNA4) or in pairs (gRNA1 and gRNA2; gRNA3 and gRNA4) for the targeted double-stranded break (DSB) formation and religation by NHEJ, thereby potentially producing expected products (PR1, PR2, and PR3). The peptide lengths of each part have been denoted above its representative block (aa, amino acids). SP = signal peptide. SlHyPRP1-Fwd1 and SlHyPRP1-Rev1 were the primer pair for PCR. b All four gRNAs or pairs of gRNAs were cloned and arranged in the pDR1 binary plasmid together with a plant selection marker (pNOS-NptII-tOCS) and SpCas9 (p35S-SpCas9-tNOS). The gRNA transcription was driven by the Arabidopsis U6 promoter (AtU6) core element (75 bp). The gRNA1-gRNA2 expressing plasmids are pDR2 and pDR3 for gRNA3-gRNA4
Hybrid proline-rich proteins (HyPRPs), a subgroup of putative plant cell wall glycoproteins, consist of a repetitive proline-rich N-terminal domain (PRD) and a conserved eight-cysteine motif (8CM) C-terminal domain. HyPRPs were shown to have differential roles in biotic and abiotic stress responses of different plant species. In pepper (Capsicum annuum) and tobacco (Nicotiana benthamiana), HyPRPs played roles as both positive regulators of cell death and negative regulators of biotic stress (Yeom et al. 2012). Overexpression of a pigeon pea HyPRP (CcHyPRP) showed multiple abiotic stress tolerance in yeast, Arabidopsis (Priyanka et al. 2010), and rice (Mellacheruvu et al. 2015). Similarly, EARLI1 in Arabidopsis was found to play roles as a positive regulator under cold and salt stresses (Xu et al. 2011). In contrast, SlHyPRP1 was shown to be a negative regulator of multistress responses in tomato. Suppression of SlHyPRP1 led to multistress (e.g., oxidative stress, dehydration, and salinity) tolerance without obvious developmental phenotype changes (Li et al. 2016).
Considering the conservation and diversity of HyPRP functions, the PRD and 8CM domains of SlHyPRP1 may play different roles under different stresses. Therefore, we hypothesize that the generation of precisely edited alleles retaining only one of the two domains or complete removal of both domains provides a variable range of agronomical traits. These divergences of agronomical traits and a selection process may offer an opportunity to obtain improved stress tolerance plants relative to simple knockout mutant plants, with minimal agronomical trait penalties. Our data revealed variable roles of the SlHyPRP1 domains in salt stress responses and indicated that precise removal of each of the domains generated salt stress-tolerant events in tomato.
Materials and methods
Construction for cloning multiplex gRNAs using the CRISPR/Cas9 system
HyPRP1 was shown to be involved in multistress responses (Li et al. 2016). The functions of HyPRP1 could be separately accounted for by the PRD and 8CM domains. In this study, using the multiplexed CRISPR/Cas9 editing system, we sought to generate three types of precise removal of SlHyPRP1 (Solyc12g009650) domains: PRD, 8CM or PRD-8CM removal. The multiplexed editing tools were designed with pairs of guide RNA (gRNA1 and gRNA2 for PRD removal; and gRNA3 and gRNA4 for 8CM removal) (Figs. 1a, S1, S2) and all four gRNAs (gRNA1, gRNA2, gRNA3, and gRNA4) together in one construct to evaluate multiplexed editing efficiencies. The gRNAs were designed using the most updated criteria for their efficient activities: sequence contents, secondary structure prediction, and targeted genome sequence contexts connected with a nucleosome map. Guide RNA expression cassettes, plant selection markers, and CRISPR/Cas9 expression cassettes were cloned and assembled into binary vectors for tomato transformation using the Golden Gate/MoClo system (Weber et al. 2011; Engler et al. 2014) (Fig. 1b). The AtU6 promoter (Addgene #46968) was used to drive transcription of the gRNAs, and the Cas9 expression cassette (humanized SpCas9, Addgene #49771) was driven with the 2xCaMV 35S promoter. The plant selection marker was kanamycin (NptII) with pNOS as a promoter (Addgene #51144). The CRISPR/Cas9-gRNAs were based on the T-DNA system as used with the Agro-mediated plant transformation approach (Fig. 1b).
Agrobacterium-mediated transformation into the tomato and transgenic plant analysis
Agrobacterium tumefaciens GV3101 (pMP90) was used to deliver the editing tools into tomato cotyledon fragments (explants). Tomato transformation was conducted following the protocol of Vu and coworkers (Vu et al. 2020) with minor modifications (Fig. S2). Briefly, agrobacteria were cultured overnight (16 h, 180 r.p.m., 28 °C) in 3 mL of liquid LB medium containing 20 mg/L rifampicin, 25 mg/L gentamycin, and 50 mg/L kanamycin (LB + RGK). The overnight culture was diluted at a 1:10 ratio by 30 mL of fresh LB + RGK medium and incubated for an additional 4–6 h in a shaker (180 r.p.m) at 28 °C. The Agrobacterium cells (OD600nm ~ 0.6–0.8) were then collected by centrifuging at 3500 r.p.m. for 15 min and subsequently resuspended in 30 mL of liquid ABM-MS medium (pH 5.2) (Wu et al. 2014) containing acetosyringone (AS, 200 µM). The agrobacterial suspension was activated in shaking conditions (180 r.p.m) at 28 °C for 1 h before plant transformation.
Our study used Hongkwang and 15T01 local tomato varieties for transformation. The explants (between 0.2 and 0.3 cm from the cotyledon) for transformation were produced by cross-sectioned 7-day-old cotyledons germinated on 1/2 MSO medium (half-strength MS medium containing 30 g/L sucrose and 7.5 g/L agar, pH 5.8) at 25 ± 2 °C under 16-h/8-h light/dark conditions. The explants were transferred onto PREMC medium [MS basal salts, Gamborg B5 vitamins, 2.0 mg/L zeatin trans isomer, 0.2 mg/L indolyl acetic acid (IAA), 1 mM putrescine, 30 g/L glucose, 100 µM AS and no antibiotics, pH 5.7] for 1 day and then incubated in agrobacterium cell solution for 20 min. The explants were blotted onto sterilized filter papers and transferred to cocultivation medium plates containing all of the components in the ABM-MS medium (pH 5.8), 7.5 g/L agar, and 200 µM AS. After incubation for 2 days in dark conditions at 25 °C, the explants were washed with 500 mg/L timetin and moved to the selection medium SEL4-60 that contained MS basal salts, Gamborg B5 vitamins, 0.5 mg/L zeatin ribose trans-isomer, 0.05 mg/L of IAA, 1 mM putrescine, 30 g/L glucose and 60 mg/L kanamycin, pH 5.7, for 14 days and subcultured in the same medium until the shoots were adequately long (1.5–3 cm). The shoots were placed in the root induction medium (RIM) containing 0.1 mg/L IBA, 0.3 mg/L NAA and all of the elongation medium components except for the zeatin trans isomer. The intact plants from the rooting medium were transferred to vermiculite pots to allow them to harden before being transferred to soil pots in a greenhouse with a temperature of 25 ± 2 °C under a 16-h/8-h photoperiod.
Genomic DNA preparation
Plant tissue (~ 200 mg) was crushed in liquid nitrogen using a ceramic mortar and pestle, and 0.5 mL of sol I (NaCl 1 M + Sarcosyl 2% + 50 µg of RNase) was added to the powder. The mixture was transferred into an Eppendorf tube (1.5 mL), mixed by inverting, and incubated at 37 °C for 30 min. The tubes were centrifuged at 13,000 rpm for 10 min at 4 °C, and then 250 µL of the upper phase was transferred to new tubes (15-mL Falcon tubes). Two volumes of freshly prepared extraction buffer (Tris–Cl 100 mM [pH 8.0] + EDTA 20 mM + NaCl 1.4 M + CTAB (Hexadecyltrimethylammonium bromide) 2%) were added into the tubes. The tubes were then incubated at 60 °C in a shaking water bath at 100 rpm for 35 min. Subsequently, an equal volume of chloroform:isoamyl alcohol (24:1) was added into the tubes and mixed gently. The tubes were centrifuged at 13,000 rpm at room temperature for 15 min, and the upper phase was transferred to new tubes. Amounts equal to 1/30 volume of sodium acetate 3 M (pH 5.2) and 0.6 volume of isopropanol were added into the tubes. Then, the DNA was pelleted down by centrifuging at 13,000 rpm for 10 min at 4 °C. The supernatant was poured off, the pellets were washed by adding 80% ethanol into the tubes, and the tubes were centrifuged at 13,000 rpm for 5 min at 4 °C. Ethanol was poured off, and the pellets were air-dried. The pellets were dissolved in 100–200 µL of TE (Tris–Cl 10 mM (pH 8.0) + EDTA 1 mM). The concentration of the gDNA samples was quantified by NanoDrop 1000 UV/Vis Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE, USA).
Screening and validating editing events by sequencing
Assessment of editing events was conducted by conventional PCR, followed by Sanger sequencing. Sequences flanking the targeted sites were amplified by PCR using the primer pair SlHyPRP1-Fwd1 (5′-GCATTCAACAATCATCAAGTACTCA-3′) and SlHyPRP1-Rev1 (5′-CAGTAGTACGACGGGTGTTTAAT-3′) (Fig. 1a) and sequenced to identify the modification type post-NHEJ repair of the DSBs formed by the designed CRISPR/SpCas9 complexes by comparison with the WT sequence. The decomposition of sequencing data for allele retrieval was conducted by bioinformatics tools (TIDE or ICE Synthego) (Hsiau et al. 2019) or pJET1.2/blunt cloning and sequencing. Similar analyses were performed with genome-edited generation 1 (GE1) and GE2 to confirm the inheritance of the indel mutated alleles.
Identification of off‑target loci
To identify potential off-target loci in the tomato genome, the 20-nt sequence of each gRNA was used as a query sequence in https://www.rgenome.net/cas-offinder. PAM types of SpCas9 (5′NGG3′) and Solanum lycopersicum (SL2.4)-Tomato were selected. For experimental validation, offtarget-specific PCR primers were designed by PrimerQuest Tool and used for PCR amplification and Sanger sequencing (Table S3). Data from the off-target analysis are presented in Table S4.
Salinity test
Plants carrying homozygous edited alleles and WT control were challenged with stresses to assess tolerance/susceptibility levels. A comparison of tested data (tri-replicates) of WT and edited plants was performed to determine the performance of the edited alleles.
Salinity tolerance test in the germination stage
GE1, GE2, and WT-Hongkwang control seeds were sterilized and grown on 1/2 MSO medium supplemented with various concentrations of NaCl including 0, 50, 100, 150, and 200 mM, pH 5.8. The seed germination containers were kept in the dark for 3 days and then placed under a 16 h/8 h light/dark photoperiod at 25 ± 2 °C. The germination rate was counted daily, and the data were collected including fresh weight, stem length, and root length recorded on the 19th day after sowing.
Salinity tolerance tests in the growth stage
Salinity tolerance tests at the growth stage were conducted following the protocol published by Renau-morata and coworkers (Renau-Morata et al. 2014) with minor modifications. Homozygous GE1 and GE2 edited lines and WT-Hongkwang and WT-like were sterilized before sowing on wetted Whatman paper and kept in the dark at 25 ± 2 °C for 2 to 3 days. On the 3rd day, all seeds were transferred into Eppendorf 1.5 mL tubes containing 0.6% agar (pH 5.8) and kept in a tray with water to maintain moisture, and the tray was kept under 16-h/8-h light/dark conditions at 25 ± 2 °C with a lid. At the fully expanded cotyledon stage, one drop of ¼ MS nutrient salts was added, and the lips were progressively opened for hardening. All seedlings were screened by the direct PCR method for the presence of respective edited alleles, and 3–5 plants of each of the edited lines were moved to a hydroponic system supplemented with Hoagland nutrient solution (Hoagland and Arnon 1950) containing various NaCl concentrations (0, 50, 100 and 150 mM) and placed under a 16-h light/8-h dark photoperiod. The hydroponic solution was changed every 4 days. Data were recorded at 0, 4, 8, 12, and 16 days after salt exposure including stem and root lengths.
qRT-PCR procedure and analysis of data
All of the qPCR and qRT-PCR analyses were performed following the MIQE guideline (Bustin et al. 2009) and protocol published by Vu and coworkers (Vu et al. 2020). Briefly, total RNA was isolated from plant tissues using RNeasy mini Qiagen kits (cat. no. 74104, Qiagen, USA) and subjected to reverse transcription for synthesizing 1st cDNA strands using the QuantiTect Reverse Transcription Kit and protocol (cat. no. 205311, Qiagen, USA). At least two pairs of primers (Table S5) were tested for each target gene/cDNA to evaluate their efficiencies, and primer pairs with ~ 100% efficiency were ultimately used for qPCR/qRT-PCR. A similar assessment was also applied for the internal genes to normalize the amplicon levels. qPCR/qRT-PCRs were performed using intercalating dyes (KAPA SYBR FAST Universal, cat. No. KK4601, Sigma, USA) to detect products. Thermocycling was conducted with the Illumina Eco Real-Time PCR System (Illumina, USA). Analyses of amplicon levels were performed using the delta-delta Cq method (Livak and Schmittgen 2001) with internal gene/transcript of SlPDS and were plotted using Excel software (Microsoft, USA).
Statistical analysis
The salinity stress tolerance data were collected as tri-replicates and analyzed by pairwise comparison using the t test function of MS Excel (Tables S6, S7, S8, S9, S10).
Agronomical trait assessment
Morphology, growth, yields, and fruit quality of well-performing lines in stress tolerance tests were evaluated in comparison with WT.
Results
System construction for precise elimination of SlHyPRP1 domain(s) by the CRISPR/Cas9-based multiplexed editing system
The tomato HyPRP1 showed widely conserved sequences of its functional domains (PRD and 8CM) among various plant species (Fig. S1a) (Li et al. 2016). We sought to engineer the SlHyPRP1 gene by precise removal of its PRD or 8CM domain or both, retaining only signal peptide (SP) (knockout) (Fig. 1) and taking advantage of blunt-end double-stranded break (DSB) formation by CRISPR/Cas9-based gene editing complexes. Based on the boundaries of the domains and genomic sequence of HyPRP1 (Fig. S1b, c), four gRNA target sites were selected for the work, namely, gRNA1, gRNA2, gRNA3, and gRNA4, that correspond to the start and end of each of the domains (Fig. 1a; Table S1). A precise editing (PR) strategy was designed to obtain the DNA sequences that were cut and precisely ligated at the two cutting sites of two gRNAs. Likewise, PR1 is the event that was perfectly cut and ligated at the gRNA1 and gRNA2 cutting sites, thereby eliminating the PRD domain from the edited chromosome. PR2 was formed by targeted cutting and perfect ligation at gRNA3 and gRNA4, thus deleting the 8CM domain. Moreover, PR3 is a knockout event (PRD and 8 CM domain removal) that could be obtained from the activity of any of the gRNAs with frameshift and early stop codons (Fig. 1a). In the beginning, we simultaneously designed CRISPR/Cas9 systems using all four gRNA (gRNA1, gRNA2, gRNA3, and gRNA4) expression cassettes in a single T-DNA vector for plant transformation (Fig. 1b). In many events, we found gRNA1, gRNA2, and gRNA3 working together; thus, we subsequently designed each pair of gRNAs (gRNA1 and gRNA2) to obtain PR1 and/or PR3 events and (gRNA3 and gRNA4) PR2 and/or PR3 transformants in separate T-DNAs for tomato transformation (Fig. 1b).
Multiplexed CRISPR/SpCas9-based genome editing tools could generate precise deletion of SlHyPRP1 domain(s) to obtain expected alleles
To obtain expected editing events, CRISPR/SpCas9 editing tools (Fig. 1b) were introduced into tomato cotyledon explants via the Agrobacterium-mediated transformation method (Fig. S2). Hardened plants were analyzed by PCR using primers flanking the targeted sites (Fig. 1a; Materials and Methods), and Sanger sequencing of the PCR products was conducted for tracing the presence of indel mutations at the targeted sites. A total of 12 edited events (Table 1) including two (PR1 and PR3) out of the three precise editing events and their variants, PR1 (PR1v1), PR2 (PR2v1, PR2v2, and PR2v3) and PR3 (PR3v1) (i.e., editing more base pairs than expected) (Fig. 2b, c), in either Hongkwang (8 events, Fig. 2c) or 15T01 (4 events, Fig. S3) cultivars, were obtained from the transformation.

Analysis of SlHyPRP1 edited events. a PCR products amplified from targeted deletion sites of the SlHyPRP1. L: 1 kb plus BIOFACT ladder; WT-HK and WT-15T01: wild-type Hongkwang and 15T01 cultivars, and 9 independently regenerated events. b Sanger sequencing results of HyPRP1 (Hongkwang) T0 edited lines. The positive clones in a and others were sequenced to confirm the indel patterns and are shown in b including the precise PRD removal events PR1 and PR1v1; precise 8CM removal variants (PR2v1, PR2v2, and PR2v3), and PRD-8CM removal (event #PR3) and its variants (PR3v1 and PR3v4). Green letters: gRNA sequence, red-letter: inserted nucleotide, red star: stop codon, blue letters: sequence outside genome after being produced frameshift. The guide RNA name and cutting sites (arrows) are shown above each WT sequence. c SlHyPRP1 indel alleles were obtained from the multiplexed editing. The major alleles obtained from the study are illustrated as the editing layout. Overall, 12 major alleles were obtained including the precise elimination of PRD or 8CM or both at high frequency (Table 1). Several allele variants were also shown as the ligation occurred with damaged ends or with single nucleotide insertion. Detailed information for the events carrying the alleles, frequencies, and segregation rates in GE0 and GE1 offspring are shown in Table 1. SP signal peptide, PRD proline-rich domain, 8CM 8-Cysteine Motif
The PCR products showed potential precise deletions of sequences intervening in the gRNA1 and gRNA2 and between the gRNA3 and gRNA4 cutting sites (Fig. 2a). Sanger sequencing of the PCR products revealed not only precise edited events but also their variants with additional modifications (Figs. 2b, S1). For instance, in the case of the PR1 event, 423 bp from the PRD domain was precisely removed via the activities of gRNA1 and gRNA2 (Table S1). Event PR1v1 (PR1 variant 1) was formed by the precise elimination of 423 bp from the PRD domain by gRNA1 and gRNA2 but also contained a 1 bp insertion at the gRNA4 cutting site, resulting in a frame-shift and the addition of 96 bp before reaching a premature stop codon (Fig. 2b, c). Only the transformation using the construct containing gRNA1 and gRNA2 expression cassettes generated a perfect PR1 event (Fig. 2b, c; Table 1). None of the transformed events showed a precise PR2 event. However, we obtained three PR2 variants, namely, PR2v1, PR2v2, and PR2v3 (Fig. 2a-c), with the Hongkwang variety. The PR2v1 variants were obtained due to the long DNA deletion (228 bp in a total of 255 bp of the 8CM domain) that started from the gRNA4 cutting site and prolonged up to 2 bp downstream of the gRNA3 cutting site (Fig. 2b, c). Event PR2v3 showed 246 bp removal from its 8CM domain and 6 aa from its PRD domain thanks to the activities of gRNA2 and gRNA4 (Fig. 2b). Finally, three PR3 variants (PR3v1, PR3v4, and PR3v5)-containing knockout lines of Hongkwang cultivar were revealed (Fig. 2b, c). Among them, the event PR3v1 was obtained through activities of gRNA1 and gRNA2. However, a sequence 281 bp downstream of the gRNA1 cutting site was inverted and incorrectly ligated at the 6 bp end of the signal peptide (SP) domain. In addition, a 1 bp deletion at the gRNA1 cutting site and a long deletion up to 363 bp extending 154 bp downstream of the PRD domain and 167 bp in the first half of the 8CM domain were shown in the event PR3v4 (Fig. 2a, b). There was a 10 bp deletion around the gRNA1 cutting site, which then produced a premature stop codon after 28 bp, as indicated in event PR3v5 (Fig. 2c). Moreover, we also obtained several edited events in the 15T01 cultivar including PR1, PR3, PR3v2, and PR3v3 that were characterized in detail in Fig. S3. In general, the precise ligation frequencies were identified at ~ 10% among total analyzed plants, and the allelic frequency varied from 12 to 45.07% in both the studied varieties (Table 1).
Off-target analysis of the genome-edited events
To determine the off-target incidence of the multiplexed gRNAs, GE2 plants of the two events (#PR1v1 and #PR2v3) were selected for the assessment of 12 potential off-target loci that contain less than 4 mismatches outside the seed sequences (Table S2). The potential off-target loci were identified using Cas-OFFinder (Bae et al. 2014) with the Solanum lycopersicum (SL2.4) chromosome database. PCRs using primers flanking the potential off-target sites (Fig. S4; Table S3) were performed, and the PCR products were sent for sequencing. The off-target activity was found at only one out of the 12 potential off-target sites and was only significant in the lines of event PR2v3 (Table S4). The line with 37% indel showed a normal leaf color, but the other line with a more drastic change at the off-target site showed a reduced level of chlorophyll but no effect on other growth parameters (Fig. S5).
Salinity tolerance performance of the edited lines
One of the important traits of crop plants is salinity tolerance. HyPRP1 was shown to be a negative regulator in salt stress responses (Li et al. 2016). Therefore, a test to assay salinity tolerance was carried out on HyPRP1 domain deletion lines carrying homozygous edited alleles. We reasoned that the behaviors of the edited lines may be different in the various growth stages. Similarly, salinity tolerance evaluation of the PRD, 8CM, and PRD-8CM eliminated lines was carried out at both the germination and growth stages. We used G1′s or G2′s seeds carrying homozygous alleles of PR1 (precise PRD elimination line); PR1v1 (precise PRD elimination variant 1); PR2v2 and PR2v3 (precise 8CM elimination variant 2 and 3, respectively); and PR3v1 and PR3v4 (PRD and 8CM elimination variant 1 and 4, respectively) (Fig. S6; Table 1) for the salinity test. Some of the lines were free of T-DNA (Fig. S6).
Salinity tolerance at the germination stage
At the germination stage under various salinity levels, some of the tested lines (PR1 and PR2v2) showed lower germination performance than WT, but the others out-performed the WT (Figs. 3, 4; Table S6). The PR1 showed a low germination rate even in the mock medium. The germination of WT control was at 100% in the 0 and 50 mM NaCl media, even better than some edited lines (i.e., PR1 and PR2v2 at 50 mM). However, at the higher salt concentrations (100 and 150 mM), some edited lines (PR1v1, PR3v1, and PR3v4) germinated much better than the WT control. In particular, the precisely edited variants of PR1 (PR1v1) and PR3 (PR3v1 and PR3v4) were germinated and survived on the medium containing up to 150 mM NaCl (Fig. 3). By contrast, the edited lines PR1, PR2v2, PR2v3, and WT control strongly reduced germination ability on the medium containing 100 mM and could not germinate under 150 mM NaCl (Fig. 4; Table S6). All the lines could not germinate on the medium containing 200 mM NaCl (Table S6).

Performance of the edited lines at the germination stage under salinity stress (150 mM NaCl). The seeds of WT, as well as edited lines, were sterilized and germinated on the medium containing 0 mM or 150 mM NaCl. Only some plants of the edited lines could germinate and grow on the medium containing 150 mM NaCl (upper panels). The germinated seedlings from the 150 mM NaCl-containing medium were further characterized by photographing in comparison with the WT in the lower panels. WT: Hongkwang wild-type; WT-HK_F3: PR1v1-2, PR3v1-1, and PR3v4-1: edited lines. Scale bars: 1 cm

Salinity tolerance at the germination stage of the HyPRP1 edited lines. WT: wild-type Hongkwang cultivar; PR1: precise edited event PR1; PR1v1: PR1 variant; PR2v2: variant 2 of precise edited event PR2; PR2v3: variant 3 of precise edited event PR2; PR3v1: variant 1 of precise edited event PR3; PR3v4: variant 4 of precise edited event PR3
Statistical analysis of the fresh weight, stem, and root lengths collected at day 19 post salinity exposure (100 mM NaCl) (Table S7) revealed a significant difference (P < 0.05) in fresh weight of the PR3 variants (0.12 ± 0.03 g/plant of PR3v1-1 and 0.11 ± 0.01 g/plant of PR3v4) but not the PR1v1 line (0.07 ± 0.01 g/plant) compared to that of the WT (0.04 ± 0.02 g/plant). The stem and root lengths of the lines that grew on the salty medium (100 mM NaCl) were also nearly significantly different (P ~ 0.05) compared to the WT control (Table S7). There was a trend of better performance regarding the fresh weight, root, and stem lengths measured from plants of the three lines (PR1v1, PR3v1, and PR3v4) compared to the WT (Table S7), and extremely better salinity tolerance was shown in the medium containing 150 mM NaCl.
Salinity tolerance at the vegetative growth stage
Due to the differences in the physiological stages of plant growth that may affect salinity tolerance of the tomato plants, we sought to test the salinity tolerance of the edited lines at the vegetative growth stage. In this set of experiments, most of the edited lines and WT appeared to be equally healthy on the control medium and the medium containing 50 mM NaCl. Different survival rates of the edited lines were recorded at higher salinity levels (Table S8). At 150 mM NaCl, except PR1v1 line, most of the plants of the other edited lines showed better growth compared to WT control and WT siblings (WT-like) (Figs. 5, 6). Furthermore, all the edited lines exhibited higher survival rates that varied from 71.43 to 93.33%, compared to WT (33.33%) and WT-like plants (Fig. 5, Table S8).

Salinity tolerance at the growth stage of the HyPRP1 edited lines. The data were collected at day 16 post-exposing the plants to NaCl treatments. WT: wild-type Hongkwang cultivar; PR1: precise edited event PR1; PR1v1: PR1 variant; PR2v2: variant 2 of precise edited event PR2; PR2v3: variant 3 of precise edited event PR2; PR3v1: variant 1 of precise edited event PR3; PR3v4: variant 4 of precise edited event PR3

Performance of the edited lines compared to the WT and WT-like plants at the vegetative growth stage under salinity stress (150 mM NaCl). Seeds of the WT and edited lines were germinated, and seedlings with 2 fully expanded true leaves were transferred into a salinity test system in hydroponic conditions. Photographs were taken with plants treated with 150 mM NaCl at 16 days post salt challenge. The name of plants/lines are denoted at the bottom of each panel. The horizontal lines indicate plants from the same edited lines. WT: Hongkwang wild-type; WT-like: segregated WT siblings of the edited plants; PR1, PR1v1; PR2v2; PR2v3; PR3v1 and PR3v4: edited lines. Vertical scale bars: 2 cm
However, when comparing the growth parameters, such as the stem and root lengths, using a t-test, only plants from the PR1 lines but not the other lines showed significantly better stem growth compared to that of the WT control (Table S9). Stem and root lengths measured at the 16th-day post salt challenge of the PR1v1 line were even significantly lower than those of WT control in 50 mM NaCl. On the 16th-day post 50 mM NaCl salt exposure, plants from the line PR2v2 also showed significantly higher stem lengths.
Assessment of transcript levels of the edited alleles under stress conditions
We wondered whether the expression of the edited alleles under the salt stress conditions would be affected or not. Therefore, leaf samples from three plants were collected from each edited and WT control line grown at 0 and 150 mM NaCl for total RNA isolation and quantitative RT-PCR (qPCR) analysis in a real-time manner at day 8 (Table S10) and day 16 post salt exposure. RT-PCR reactions were also conducted to indicate the expected size of mRNAs transcribed from the precisely edited lines (Fig. S7). On day 8, compared to the WT control that showed no significant difference in relative expression between the mock and salty conditions, both of the knockout (PR3) lines showed similar expression levels of the edited transcripts in both conditions (Fig. 7). In contrast, all of the PR1, PR1v1, PR2v2, and PR2v3 lines showed higher RNA levels compared to WT control in the mock condition but appeared as opposite trends under salt stress. The true PR1 line appeared to be upregulated under the stress of 150 mM NaCl, while the PR1v1 and PR2v3 variants were dramatically downregulated to lower levels than that of the WT (Fig. 7). The RNA levels of the PR2v2 line were similar in both salt conditions. On day 16, all the transcripts of the edited lines grown on a medium containing 150 mM NaCl showed lower levels than that of the mock control (Table S11).

Relative expression of mRNAs transcribed from the edited alleles in comparison with that of the WT. qRT-PCRs were conducted using primers specific for the SlHyPRP1 allele and the edited alleles. Relative expression of the alleles at the RNA level was calculated using the delta Cq method with SlPDS as an internal control. Data were analyzed and plotted by the Excel 2016 program. WT: wild-type Hongkwang cultivar; PR1: precise edited event PR1; PR1v1: PR1 variant; PR2v2: variant 2 of precise edited event PR2; PR2v3: variant 3 of precise edited event PR2; PR3v1: variant 1 of precise edited event PR3; PR3v4: variant 4 of precise edited event PR3
Discussion
DSB repair via the canonical NHEJ (cNHEJ) and/or noncanonical NHEJ pathways, such as microhomology-mediated end joining (MMEJ), are well-studied in plants (Puchta 2005; Lieber 2010) and widely applied in the generation of CRISPR/Cas-based targeted gene mutagenesis (Van Vu et al. 2019; Schindele et al. 2020). The repaired outcomes mediated by the cNHEJ are predominantly identical to the WT sequence if the two broken ends are compatible, thus maintaining the stability of the genome (Waters et al. 2014; Guo et al. 2018). Therefore, introducing two or more DSBs for the generation of DNA blunt ends at targeted loci by CRISPR/SpCas9 complexes (Jinek et al. 2012) in quite a proximity to each other may trigger precise ligations among the ends. In our study, multiplexed CRISPR/SpCas9 complexes with four gRNAs simultaneously targeting four sites on the SlHyPRP1 within less than a 1 kb zone produced two out of three possible precision deletion outcomes (PR1 and PR3) (Fig. 1a) in a single transformation, which has never been reported before. Our data showed various DSB repair patterns including precise ligations, small insertions and deletions at the junctions of DSB ends leading to unintended imprecise product variants (Fig. 2). The data successfully confirmed the precise ligations of the broken ends from the two different DSB sites in tomato to generate the PR1 and PR3 but not the PR2 event. This finding was observed because of the lack of gRNA3 activity. The result indicates that for efficient precise ligation of two DSB sites, the similar activity of the paired gRNAs is also important. The precise ligation frequencies were at approximately 10%, which was slightly low compared to that in animals (Guo et al. 2018). However, the precise editing frequencies revealed in our study appeared to be sufficient for obtaining several events (Fig. 2, Table 1).
Salinity stress is among the most important threats to global cropping (van Zelm et al. 2020). Knocking down the expression of tomato HyPRP1 by an RNAi approach enhanced various abiotic stress tolerances including salinity stress (Li et al. 2016). Therefore, it is interesting to examine the performance of the edited lines under salty conditions. There was a mild difference in salinity responses of the tested lines between the germination and growth stages. On the germination media, the PR1v1 and PR3 variants (knockout lines) performed the best and survived under the 150 mM NaCl concentration, indicating that they are salt-tolerant (Fig. 4, Table S6). The 200 mM NaCl killed all tested plants and WT control. The data suggest that either the PRD domain (eliminated in the PR1v1 lines) or the entire HyPRP1 protein may function as a negative regulator of the salt-tolerant responses at the germination stage. Another reasonable explanation is that the PRD majorly contributes as a negative regulator of stress responses; thus, its removal strongly enhanced salinity tolerance in both the PR1 and PR3 variants (Fig. 4, Table S6).
It is interesting that all the edited lines showed higher survival rates than the WT control in the medium containing 150 mM of NaCl, indicating an overall effect of truncation of the HyPRP1 protein (Figs. 5, 6, Table S8, S9). That also indicated the complex involvement of the PRD and/or 8CM domains in salinity stress responses during the vegetative stage of growth compared to the germination stage. Statistically, only the surviving plants from the PR1 line showed significantly higher stem lengths, suggesting that the deletion of the PRD domain significantly contributed to the salt tolerance of the edited line and that the presence of 8CM in the alleles but not the other lines somehow was strongly involved in the salt stress responses that were not observed in the earlier stage (Table S9). The 8CM domain was shown to be a determinant of programmed cell death in stressed tobacco plants, which could fulfill the functions of the HyPRP1 protein in the same plants (Yeom et al. 2012). It is not known if the 8CM motif contributed to salt tolerance. Another possibility is that due to the removal of the PRD domain, free prolines in the cellular space increased, leading to enhancement of stress tolerance.
In agreement with the statistical analysis of the phenotypic data collected from the surviving plants under saline conditions, only the PRD-eliminated PR1 plants showed significantly higher RNA transcript levels at ~ ninefold than that of the WT in 150 mM NaCl (Fig. 7) at 8 days post-exposure. The removal of the PRD domain in the PR1, PR1v1, PR3v1, and PR3v4 plants might help them to tolerate salt conditions, resulting in higher survival rates than the WT control, and keeping the 8CM might synergistically enhance the stem growth of the PR1 plants (Table S8). The PR1v1 is a variant of PR1 with the addition of 32 nonsense aa at its C-terminus due to the gRNA4 activity that results in a G insertion at the cutting site (Fig. 2c). The added G might destabilize the PR1v1 transcripts under the salinity stress conditions to a similar level as that of the knockout lines, leading to even significantly lower root and stem lengths than that of the WT control under 50 mM NaCl (Fig. 7; Table S9, S10). Between the two PR2 variants 2 and 3, the stable RNA level in the case of the PR2v2 plants might help them perform better under 50 mM NaCl exposure compared to WT, but not the PR2v3. However, at a higher salt concentration, the 8CM removal lines did not exhibit synergistic effects on plant growth (Table S9 and S10).
In some of the tested lines, there might be low levels of off-target modifications; therefore, we cannot exclude the possible involvement of the off-target alleles in the salinity tolerance of the line PR2v3. The potential off-target sites annexed in the tomato genome SL2.40 database (location SL2.40ch09-42199794.0.42201794) are located within an uncharacterized open reading frame (protein LOC101257680, accession no. XP_004243133.1). This protein may link to the chlorophyll reduction phenotype (Fig. S5) since a segregated line of the PR2v2 event showed a pale yellow color but did not affect the plant growth under normal conditions in our lab.
Conclusion
Random mutagenesis using radiation or chemicals to produce nonspecific DNA damage has played important roles in the improvement of crops to address increasing demands for food production. The drawback of random mutagenic approaches includes high costs as they require extensive labor costs, large-scale laboratory stations, and time to test for useful traits and to "clean up" unwanted mutations that potentially occur throughout the genomes subjected to radiation/chemical mutagenesis. While widely accepted and applied in plant breeding due to the abovementioned barriers, the rate of increase in yield upon crop improvement lags very far behind the rate of population expansion. The recently emerged CRISPR/Cas-based targeted mutagenesis is a revolutionary approach to plant improvement and potentially is expected to meet food requirements. An excellent example of the application of CRISPR/Cas is engineering negative stress response regulators to obtain improved crops capable of dealing with changes in growing conditions. In this study, we show the precise editing of HyPRP1, a negative regulator of multistress responses. Precise elimination of SlHyPRP1 negative-response domain(s) has led to high salinity tolerance (up to 150 mM NaCl) in our experimental conditions.
The flexible engineering of SlHyPRP domains to track their multi-function and performance in stress responses is a smart and novel approach for genome editing-mediated precision breeding. The risky points of this approach are the limited understanding of SlHyPRP1 functions in tomato. It seems that each member of the HyPRP family may play counter roles in different plant species and at different growth stages. Although SlHyPRP1 likely plays roles as a negative regulator(s) of multistress responses (Li et al. 2016), the tested RNAi-based approach might also reflect some levels of requirement for either of its domain functions or both and hence may introduce some minor unexpected penalties to agronomical traits of the obtained editing events. Therefore, our work needs to be further extended to test other abiotic and biotic stresses that could help to reveal the importance of the edited alleles of SlHyPRP1.
Author contributions statement
TVV and JYK conceived and designed the research. MTT, TVV, DTHD, JK, YJS, YWS, SD, EJK, GHS, and conducted experiments. TVV, MTT, SHK, and JYK analyzed data. MTT and TVV wrote the manuscript. TVV and JYK finalized the manuscript. All authors read and approved the manuscript.
References
Bae S, Park J, Kim JS (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30(10):1473–1475. https://doi.org/10.1093/bioinformatics/btu048
Betermier M, Bertrand P, Lopez BS (2014) Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10(1):e1004086. https://doi.org/10.1371/journal.pgen.1004086
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622. https://doi.org/10.1373/clinchem.2008.112797
Chiruvella KK, Liang Z, Wilson TE (2013) Repair of double-strand breaks by end joining. Cold Spring Harb Perspect Biol 5(5):a012757. https://doi.org/10.1101/cshperspect.a012757
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. https://doi.org/10.1126/science.1231143
Davis AJ, Chen DJ (2013) DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2(3):130–143. https://doi.org/10.3978/j.issn.2218-676X.2013.04.02
Engler C, Youles M, Gruetzner R, Ehnert TM, Werner S, Jones JD, Patron NJ, Marillonnet S (2014) A golden gate modular cloning toolbox for plants. ACS Synth Biol 3(11):839–843. https://doi.org/10.1021/sb4001504
Guo T, Feng YL, Xiao JJ, Liu Q, Sun XN, Xiang JF, Kong N, Liu SC, Chen GQ, Wang Y, Dong MM, Cai Z, Lin H, Cai XJ, Xie AY (2018) Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol 19(1):170. https://doi.org/10.1186/s13059-018-1518-x
Hoagland DR, and Arnon DI (1950) The Water-Culture Method for Growing Plants without Soil. California Agricultural Experiment Station Circular-347
Hsiau T, Conant D, Rossi N, Maures T, Waite K, Yang J, Joshi S, Kelso R, Holden K, Enzmann BL, Stoner R (2019) Inference of CRISPR Edits from Sanger Trace Data.251082. doi:https://doi.org/10.1101/251082
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821. https://doi.org/10.1126/science.1225829
Li J, Ouyang B, Wang T, Luo Z, Yang C, Li H, Sima W, Zhang J, Ye Z (2016) HyPRP1 gene suppressed by multiple stresses plays a negative role in abiotic stress tolerance in tomato. Front Plant Sci 7:967. https://doi.org/10.3389/fpls.2016.00967
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211. https://doi.org/10.1146/annurev.biochem.052308.093131
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Mellacheruvu S, Tamirisa S, Vudem DR, Khareedu VR (2015) Pigeonpea hybrid-proline-rich protein (CcHyPRP) confers biotic and abiotic stress tolerance in transgenic rice. Front Plant Sci 6:1167. https://doi.org/10.3389/fpls.2015.01167
Priyanka B, Sekhar K, Reddy VD, Rao KV (2010) Expression of pigeonpea hybrid-proline-rich protein encoding gene (CcHyPRP) in yeast and Arabidopsis affords multiple abiotic stress tolerance. Plant Biotechnol J 8(1):76–87. https://doi.org/10.1111/j.1467-7652.2009.00467.x
Puchta H (2005) The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot 56(409):1–14. https://doi.org/10.1093/jxb/eri025
Renau-Morata B, Sánchez-Perales M, Medina J, Molina RV, Carrillo L, Fernández-Nohales P, Pollmann S, Vicente-Carbajosa J, Granell A, Nebauer SG (2014) Salinity assay in tomato. Bio-protocol 4(16):e1215. https://doi.org/10.21769/BioProtoc.1215
Schindele A, Dorn A, Puchta H (2020) CRISPR/Cas brings plant biology and breeding into the fast lane. Curr Opin Biotechnol 61:7–14. https://doi.org/10.1016/j.copbio.2019.08.006
Stinson BM, Moreno AT, Walter JC, Loparo JJ (2019) A mechanism to minimize errors during non-homologous end joining. Mol Cell. https://doi.org/10.1016/j.molcel.2019.11.018
Van Vu T, Sung YW, Kim J, Doan DTH, Tran MT, Kim JY (2019) Challenges and perspectives in homology-directed gene targeting in monocot plants. Rice (N Y) 12(1):95. https://doi.org/10.1186/s12284-019-0355-1
van Zelm E, Zhang Y, Testerink C (2020) Salt tolerance mechanisms of plants. Annu Rev Plant Biol 71:403–433. https://doi.org/10.1146/annurev-arplant-050718-100005
Vu TV, Sivankalyani V, Kim EJ, Doan DTH, Tran MT, Kim J, Sung YW, Park M, Kang YJ, Kim JY (2020) Highly efficient homology-directed repair using CRISPR/Cpf1-geminiviral replicon in tomato. Plant Biotechnol J. https://doi.org/10.1111/pbi.13373
Waters CA, Strande NT, Pryor JM, Strom CN, Mieczkowski P, Burkhalter MD, Oh S, Qaqish BF, Moore DT, Hendrickson EA, Ramsden DA (2014) The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat Commun 5:4286. https://doi.org/10.1038/ncomms5286
Weber E, Engler C, Gruetzner R, Werner S, Marillonnet S (2011) A modular cloning system for standardized assembly of multigene constructs. PLoS ONE 6(2):e16765. https://doi.org/10.1371/journal.pone.0016765
Wu HY, Liu KH, Wang YC, Wu JF, Chiu WL, Chen CY, Wu SH, Sheen J, Lai EM (2014) AGROBEST: an efficient Agrobacterium-mediated transient expression method for versatile gene function analyses in Arabidopsis seedlings. Plant Methods 10:19. https://doi.org/10.1186/1746-4811-10-19
Xu D, Huang X, Xu ZQ, Schlappi M (2011) The HyPRP gene EARLI1 has an auxiliary role for germinability and early seedling development under low temperature and salt stress conditions in Arabidopsis thaliana. Planta 234(3):565–577. https://doi.org/10.1007/s00425-011-1425-9
Yeom SI, Seo E, Oh SK, Kim KW, Choi D (2012) A common plant cell-wall protein HyPRP1 has dual roles as a positive regulator of cell death and a negative regulator of basal defense against pathogens. Plant J 69(5):755–768. https://doi.org/10.1111/j.1365-313X.2011.04828.x
Zhao Y, Zhang C, Liu W, Gao W, Liu C, Song G, Li WX, Mao L, Chen B, Xu Y, Li X, Xie C (2016) An alternative strategy for targeted gene replacement in plants using a dual-sgRNA/Cas9 design. Sci Rep 6:23890. https://doi.org/10.1038/srep23890
Zheng Q, Cai X, Tan MH, Schaffert S, Arnold CP, Gong X, Chen CZ, Huang S (2014) Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 57(3):115–124. https://doi.org/10.2144/000114196
Acknowledgments
We wish to thank Mrs. Jeong Se Jeong for her valuable technical support in this study. This work was supported by the National Research Foundation of Korea (Grant NRF 2020R1I1A1A01072130, 2020M3A9I4038352, 2020R1A6A1A03044344), the Next-Generation BioGreen 21 Program (SSAC, Grant PJ01322601) and the Program for New Plant Breeding Techniques (NBT, Grant PJ01478401), Rural Development Administration (RDA), Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Günther Hahne.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tran, M.T., Doan, D.T.H., Kim, J. et al. CRISPR/Cas9-based precise excision of SlHyPRP1 domain(s) to obtain salt stress-tolerant tomato. Plant Cell Rep 40, 999–1011 (2021). https://doi.org/10.1007/s00299-020-02622-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-020-02622-z