
Abstract
Key message
Analysis of terpenoids content, transcriptome from Chamaemelum nobile showed that the content of the terpenoids in the roots was the highest and key genes involved in the terpenoids synthesis pathway were identified.
Abstract
Chamaemelum nobile is a widely used herbaceous medicinal plant rich in volatile oils, mainly composed of terpenoids. It is widely used in food, cosmetics, medicine, and other fields. In this study, we analyzed the transcriptome and the content and chemical composition of the terpenoids in different organs of C. nobile. Gas chromatography–mass spectrometry analysis showed that the total content of the terpenoids among C. nobile organs was highest in the roots, followed by the flowers. Illumina HiSeq 2500 high-throughput sequencing technology was used to sequence the transcripts of roots, stems, leaves, and flowers of C. nobile. We obtained 139,757 unigenes using the Trinity software assembly. A total of 887 unigenes were annotated to secondary metabolism. In total, 55,711 differentially expressed genes were screened among different organs of C. nobile. We identified 16 candidate genes that may be involved in the terpenoid biosynthesis from C. nobile and analyzed their expression patterns using real-time PCR. Results showed that the expression pattern of these genes was tissue-specific and had significant differential expression levels in different organs of C. nobile. Among these genes, 13 were expressed in roots with the highest levels. Furthermore, the transcript levels of these 13 genes were positively correlated with the content of α-pinene, β-phellandrene, 1,8-cineole, camphor, α-terpineol, carvacrol, (E,E)-farnesol and chamazulene, suggesting that these 13 genes may be involved in the regulation of the synthesis of the volatile terpenoids. These results laid the foundation for the subsequent improvement of C. nobile quality through genetic engineering.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Roman chamomile (Chamaemelum nobile L.) is a perennial herb that belongs to the Asteraceae family and is native to southwestern Europe and now is distributed throughout Europe, South Africa and Southwest Asia. (Ma et al. 2007). Chamaemelum nobile is rich in volatile aromatic oil, and its active components mainly include ester volatile oil, flavonoids, and terpenoids (Farhoudi 2013). Among these active components, terpenoids, especially sesquiterpenoids, are the main pharmacological components. At present, various terpenoids, such as β-pinene, germacrene D, and chamazulene (Newall et al. 1996), have been isolated and identified. These terpenoids possess anti-inflammatory, bacteriostatic, antioxidant, and anticancer properties and are widely used in pharmaceuticals, cosmetics, spices, and other fields (Yu and Utsumi 2009). However, the essential oil content of C. nobile is immensely low, and the medicinal active ingredients extracted from the plant are far from meeting the market demand. Therefore, improving the terpenoid content in C. nobile is one of the goals in the research field of C. nobile cultivation. With the progress in isolation and identification of genes related to the synthesis of compounds in metabolic pathways, the use of genetic engineering to alter the transcript level of key genes to increase the yield and species of compounds has become a means of improving plant quality (Banyai et al. 2010). However, the detailed analysis of transcriptome and genomic information of C. nobile has not yet been reported.
Terpenoids are a class of natural organic compounds composed of isoprene (C5) structural units widely found in nature. At present, more than 60,000 terpenoids, including their derivatives, are known (Bohlmann and Keeling 2008). These compounds can be classified into monoterpene (C10), sesquiterpene (C15), diterpene (C20), and triterpene (C30), according to the number of isoprene units, and can also be categorized into chain, monocyclic, bicyclic, and tricyclic terpenes, according to their carbocycle number. In plants, the terpene compounds have two synthetic routes: the mevalonate (MVA) pathway (Buhaescu and Izzedine 2007; Zenoni and Delledonne 2010) and the 2-methyl-D-erythritol 4-phosphate (MEP) pathway (Heuston et al. 2012). The MVA pathway mainly occurs in the w of eukaryotes to provide isoprene units that eventually form sesquiterpenes and triterpenes (Liu et al. 2006; Zhao et al. 2013); the MEP pathway exists in some prokaryotes and plant plastid and leads to synthesis of monoterpene, diterpenoids, and some polyterpenes (Schilmiller et al. 2009). In recent years, several genes involved in biosynthetic pathway of terpenoid genes have been identified from C. nobile (Yan et al. 2017; Meng et al. 2016; Cheng et al. 2016). However, terpenoid biosynthetic pathway is a complex process that involves various genes specifically expressed in different organs. Thus, the regulation mechanism of terpenoid biosynthesis in a given organism cannot be fully understood just by identifying and characterizing a few genes.
In this study, we used Illumina HiSeq 2500 high-throughput sequencing technology to sequence the transcriptomes in roots, stems, leaves, and flowers of C. nobile. The transcriptome database, namely the unigene library, thus constructed was functionally annotated. In addition, the composition and content of terpenoid in C. nobile were analyzed by gas chromatography–mass spectrometry (GC–MS), and several genes involved in the biosynthesis of terpenoids were identified by correlation analysis between transcript level of genes and terpenoid content. Our transcriptome data and related findings would provide valuable genetic resources for studying various biochemical processes of C. nobile, particularly the biosynthesis of terpenoids.
Materials and methods
Plant materials
We planted the C. nobile plants in the botanical garden at Yangtze University, located in Jingzhou, Hubei province, China (around N30.35, E112.14). Then, we collected the roots, stems, leaves, and flowers of C. nobile after one week of flowering for terpenoid content and transcriptome analysis.
Gas chromatography–mass spectrometry (GC–MS)
The essential oil of each organ (roots, stems, leaves, and flowers) from C. nobile was extracted using the method described in Chinese Pharmacopoeia Appendix XD and Soxhlet extraction (Misra et al. 2003). The terpenoid composition and content of the pentane extract were directly analyzed on GC–MS system (Agilent 5975B with 6890N gas chromatograph) with authentic standards under the following temperature program: injection at 250 °C and ramped from 40 to 250 °C at a rate of 10°C·min−1 (Irmisch et al. 2012). We used the DB1-MS (0.25 µm film thickness, 250 µm × 30 m) as the column with helium at 1 ml·min−1 as carrier gas. Injection volume was 2 µL. The terpenoids were identified based on comparison of their measured retention times and mass spectra with those of authentic standards. The reference compounds were purchased from Roth (Karlsruhe, Germany), Sigma-Aldrich (Steinheim, Germany), and Herbfine (Nanchang, China). In addition, we used the NIST11 database to search the fragmentation pattern to identify the corresponding terpenoid compounds.
RNA extraction
After cleaning and cutting the bulk materials into small pieces, we collected the roots, stems, leaves, and flowers separately and immediately frozen in liquid nitrogen and stored at − 80 °C for later use. According to the manufacturer’s instructions, we isolated the total RNA from each sample using TaKaRa MiniBEST plant RNA extraction kit (Dalian, China). We then assessed the quality and quantity of the total RNA using 1% agarose gels and a NanoPhotometer® spectrophotometer (Implen, CA, USA).
Library construction, deep sequencing, and de novo assembly
We conducted library construction and RNA-Seq by Biomarker Biotechnology Corporation (Beijing, China). We then prepared the transcriptome libraries of roots, stems, leaves and flowers using the NEBNext® Ultra™ RNA library prep kit for Illumina (NEB, USA) (Li et al. 2016). And the libraries were sequenced using the Illumina HiSeq 2500 high-throughput sequencing platform to obtain a large number reads referred to as raw data. We pooled and assembled all the clean reads using the Trinity de novo assembly program (Grabherr et al. 2011). First, we broke the sequenced reads into smaller segments (k-mer), and then we extended these small segments into contigs. Next, we used the overlapping portions of these fragments to obtain a collection of fragments. Finally, we relied on genome alignments to construct transcripts, and we obtained the UniGene database by aggregating the assembled sequences.
Functional annotation and metabolic pathway analysis
We used the BLAST (Version 2.2.26) (Kent 2002) software to obtain information on protein function annotation to compare the sequence of the UniGene databases and the National Center for Biotechnology Information non-redundant (NCBI nr) (Deng et al. 2006), Swiss-Prot (Apweiler et al. 2004), Gene ontology (GO) (Ashburner 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al. 2004), and Cluster of Orthologous Groups of proteins (COG) (Tatusov et al. 2000) databases. In addition, we set the E value ≤ 10−5 to obtain the protein sequences that were highly similar to the C. nobile unigene sequences and finally, to acquire function annotations. We utilized the GO program to obtain GO annotation, according to molecular function, biological process, and cellular component. Meanwhile, we searched the assembled unigenes against the COG and KEGG databases to predict possible functional classifications and molecular pathways and to classify possible COG functions and KEGG pathways, respectively.
Identification of differentially expressed genes (DEGs)
For all the comparisons, we normalize the read counts by calculating the fragments per kilobase of transcript, per million mapped read (FPKM) (Trapnell et al. 2010) and the false discovery rate (FDR) to obtain relative expression levels. The DESeq R package (1.10.1) for different expression analyses in different organs provided statistical routines that determine the differential expression in digital gene expression data using a model based on negative binomial distribution (Anders and Huber 2010). P value corresponds to differential gene expression test. The threshold of P value in multiple tests was determined through manipulating the FDR value (Benjamini and Yekutieli 2001). An FDR < 0.01 and fold change ≥ 2 were used as the threshold to determine the significance of gene expression differences.
Simple sequence repeats (SSRs)
All the unigenes obtained were searched to determine the frequency and distribution of SSRs. We detected the SSRs using an SSR identification tool, called the Perl script MISA (microsatellite searching tool) (http://pgrc.ipk-gatersleben.de/misa/). This tool accepts FASTA-formatted sequence files and reports the GenBank ID, microsatellite motifs (dimers to hexamers), number of repeats, and sequence coordinates for each microsatellite (Chen et al. 2016; Rama et al. 2015). We set the search parameters for the maximum motif-length group to hexamer, and the minimum number of repeats was set at five.
Co-expression analysis
To study the interaction between genes, we analyzed the correlation coefficient between each two genes and used correlation coefficients to determine the correlation between gene expressions. We set the thresholds (> 0.9 or < − 0.9) to screen certain genes for network analysis to ensure a strong gene co-expression relationship. We constructed the gene co-expression network using the Cytoscape software because network elements represent the ways in which genes might regulate other genes; gene networks can be divided into subgraphs, called k-core networks, where all genes are linked to at least k in other genes in the subgraph (Huber et al. 2007).
qRT-PCR validation
We prepared the samples using the same method mentioned above and isolated the total RNA from the plant in different stages of development. Experiments were performed on two independent biological replicates, each containing three technical replicates for each sample. We synthesized first-strand cDNA from total RNA using PrimeScriptTM One Step RT-PCR kit ver. 2 (Takara, Japan) and diluted tenfold as template. We normalized the data of qPCR using 18S gene as the reference gene. Since we were mainly concerned with the key genes involved in the synthesis of sesquiterpenes, we selected 16 relevant genes excluding the monoterpene synthase genes for RT-PCR and designed specific primers based on the open reading frame of the sequence database. Primers used in qPCR are shown in Supplementary Table S1. We also performed real-time assays by SYBR Premix ExTaq™ II kit (Dalian TaKaRa) and Bio-Rad Mini OpticonTM Real-time PCR Mini Cycler (BioRad, Hercules, CA, USA), with the following cycle conditions: 95 °C for 5 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 30 s; 95 °C for 15 s, 60 °C for 60 s, and 95 °C for 15 s (Xu et al. 2014). Furthermore, we calculated the relative expression fold of each sample by its Ct value normalized to the Ct value of the reference gene using the 2−ΔΔCt method (Livak and Schmittgen 2001).
Results and discussion
Composition and content of terpenoid in different organs of C. nobile
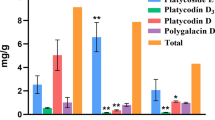
As shown in Table 1, the total terpenoid content in C. nobile was significantly highest in roots, followed by flowers, being significantly low in stems and leaves. The total terpenoids in the stems and leaves were less than 5% and 10% of the roots and the flowers, respectively. The main terpenoids in the roots were chamazulene (71.31%), 1,8-cineole (9.33%), (E,E)-farnesol (4.00%), and germacrene D (3.91%). Similarly, the main terpenoids in flowers were chamazulene (65.93%), germacrene D (19.87%), and terpinen-4-ol (3.17%). In stems and leaves, the main components were germacrene D (26.36%) and carvacrol (25.39%), respectively. Recently, Farhoudi (2013) reported the composition and content of terpenoids using the whole C. nobile seedlings as sample, but the composition and content of the terpenoids were not separately measure in different organs of C. nobile. In this study, we compared the composition and content of terpenoids among different organs. Compared with the results of Farhoudi (2013), the composition of terpenoids was similar, and the main component of total terpenoids was chamazulene. Furthermore, we found that the content of chamazulene, germacrene D and (E,E)-Farnesol were higher in roots and flowers compared to stems and leaves.
Sequencing analysis and de novo assembly
We constructed separate cDNA libraries from C. nobile root, stem, leaf, and flower samples each. In total, the libraries produced 23,572,568,706 clean reads. Each sample produced no less than 4 Gb data, the percentage of Q30 reached more than 88.03%, and the GC content was 42.88–43.90% (Table 2). We brought all the clean reads together and assembled them de novo using Trinity (Grabherr et al. 2011). The sequencing reads deposited with the National Center for Biotechnology Information (NCBI) under the SRA accession numbers SRS1610818 (T01, roots), SRS1610819 (T02, stems), SRS1610820 (T03, leaves), and SRS1610821 (T04, flowers). We obtained a total of 40,201 transcripts with an average length of 856 nt and an N50 length of 1280 nt. A total of 139,757 unigenes, of which 22,198 (15.88%) were longer than 1000 bp (Supplementary Fig. S1), were obtained with a total length of 83,444,804 nt with an average length of 597 nt and an N50 length of 924 nt. These data illustrated that the results of the assembly were favorable and applicable for subsequent studies. We compared the unigene sequence with the Nr, Swiss-Prot, GO, COGs of proteins, KOG, Pfam, and KEGG databases using BLAST. As shown in Supplementary Table S2, we obtained 37,644 unigenes with annotated information, of which 10,642, 17,427 and 14,383 were annotated against COG, GO, and KEGG databases, respectively.
Nr annotation analysis
By comparing the Nr database with non-redundant proteins, the species distribution of the best match results in Nr was obtained (Fig. 1). The C. nobile unigenes showed the closest matches with Vitis vinifera (3054, 11.10%), followed by Sesamum indicum (2046, 7.43%), Coffea canephora (1817, 6.60%), Nicotiana sylvestris (1363, 4.95%), Nicotiana tomentosiformis (1291, 4.69%), Theobroma cacao (1063, 3.86%), Nelumbo nucifera (774, 2.81%), Solanum tuberosum (744, 2.70%), Erythranthe guttata (739, 2.69%), and Citrus sinensis (701, 2.55%).

Species distribution of the BLASTX results of C. nobile transcriptome. The numbers in the pies indicate the percentage of difference in each category
Functional classification
GO is a functional classification system of genes and have three ontologies that describe the molecular function of a gene, its cellular components, and biological processes in which the gene is involved. To understand macroscopically the functional distribution of unigenes, we used WEGO to categorize the GO annotation results of unigenes into GO functions and to draw a histogram (Ashburner 2000; Ye et al. 2006). A total of 83,658 unigenes were noted in the 49 GO functional categories (Fig. 2), among which 16 (25%), 13 (25.1%) and 20 (49.9%), respectively, belonged to the molecular function, cellular components, and biological processes. Catalytic activity, cell part, and metabolic process were most highly represented, respectively, in these three subclasses.

Histogram of GO classification. The results are summarized in three main categories: cellular component, molecular function, and biological process. The left y-axis indicates the number of genes in a category. The right y-axis indicates the percentage of a specific category of genes in that main category
The unigenes of C. nobile were compared with those in the COG database. We predicted the possible unigene functions, which were classified statistically. A total of 10,642 unigenes were annotated to 25 protein families, accounting for 7.61% of the total unigenes (Fig. 3). In the 25 COG categories, the “general function prediction only” accounted for the largest proportion (3002 unigenes, 28.21%), followed by “replication, recombination, and repair” (1787 unigenes, 16.79%) and “transcription” (1644 unigenes, 15.45%). Only three sequences were annotated to the “nuclear structure”, and no sequences were annotated into the “extracellular structures” category. Notably, 599 (5.63%) of the functional unigenes were annotated to “secondary metabolite biosynthesis, transport, and catabolism” category.

COG classification. In total, 10,642 unigenes were grouped into 25 COG classification
To identify unigenes involved in metabolic pathway of C. nobile, we compared all unigene sequences with the reference canonical pathways in KEGG (Kanehisa et al. 2004). The results showed that 29,056 unigene sequences were successfully assigned to 335 KEGG pathways (Supplementary Table S3). The number of unigenes involved in the carbon metabolism pathway was the largest (473, 1.63%), followed by protein processing in the endoplasmic reticulum (460, 1.58%), spliceosome (439, 1.51%), and biosynthesis of amino acids (436, 1.50%). In addition, 19 KEGG pathways included 887 unigenes associated with secondary metabolism (Table 3). Among them, the cluster of “Phenylpropanoid biosynthesis (PATH: ko00940)” was predominant (294, 33.15%), followed by “Terpenoid backbone biosynthesis (PATH: ko00900)” (118, 13.30%) and “Flavonoid biosynthesis (PATH: ko00941)” (91, 10.26%). These annotations could help further study in the metabolic processes and offer the possibility of identifying genes involved in the secondary metabolism in C. nobile. We screened several genes in the terpene biosynthetic pathway by RNA-seq technique to identify 24, 27, and 27 genes involved in the MVA pathway, the MEP pathway, and the branch point, respectively. At the same time, we identified 12 monoterpene synthases by local blast based on the protein sequence of the previously known monoterpene synthases (Dudareva et al. 1996, 2003; Landmann et al. 2007; Nagegowda et al. 2008) and our transcriptome data (Table 4). These genes will be important resources for subsequent molecular breeding of C. nobile to improve terpenoid accumulation.
Identification of DEGs
To analyze the specificity of unigene expression in various organs of C. nobile, we mapped the reads from the roots, stems leaves and flowers libraries to the assembled transcriptome. Most of the unigenes (11,244, 18.6%) were commonly expressed in all four organs of C. nobile (Fig. 4). We detected 18,273 tissue-specific unigenes (30.2%) from different tissues of C. nobile. Among these unigenes, 5324 (8.8%), 3012 (5.0%), 4129 (6.8%), and 5808 (9.6%) were expressed exclusively in roots, stems, leaves, and flowers of C. nobile, respectively.

Venn diagram illustrating the number of unigenes expressed in roots, stems, leaves, and flowers of C. nobile
To assess differential expression level of genes in various C. nobile organs, we used a FDR and a fold change (FC) in the log10 ratio (Gao et al. 2015). According to the relative expression level between the two samples, the DEGs can be divided into up-regulated and down-regulated genes (Fig. 5a, b). In total, 55,711 DEGs were discovered among different organs of C. nobile (Fig. 6a), and we compared the two combinations between the four tissue samples of C. nobile (Fig. 6b), respectively, to obtain the up-regulated and down-regulated genes in the latter relative to the former. Tables S4 through S9 list detail of the DEGs between root and stem (Supplementary Table S4), root and leaf (Supplementary Table S5), root and flower (Supplementary Table S6), stem and leaf (Supplementary Table S7), stem and flower (Supplementary Table S8), and leaf and flower (Supplementary Table S9). The results show that the number of DEGs between flower and root pairs is the highest, and interestingly, the number of annotated DEGs is also the most, with a total of 9125 DEGs (Table 5), indicating that relatively larger number of gene plays an important role in the process of growth and development of the roots or flowers of C. nobile.

Differentially expressed unigenes of four organs of roots, stems, leaves, and flowers of C. nobile. a Distribution of FPKM values from each sample. b Pearson correlation of the samples. Each grid denotes the corresponding Pearson correlation coefficient between the two samples

DEGs in different C. nobile organs. a Hierarchical cluster analysis of common DEGs. A scale indicating the color assigned to log2FPKM is shown to the right of the cluster. Red represents high expression, blue represents low expression, and each horizontal bar represents a single gene. b The number of up-regulated and down-regulated genes between roots and stems, roots and leaves, roots and flowers, stems and leaves, stems and flowers, and leaves and flowers
DEGs in terpenoid pathway among four organs
Terpenoids are diverse in type and structure and are widely found in higher plants. To elucidate the expression patterns of terpenoid biosynthetic genes in four organs, we screened out all unigenes using RNA-seq data. We identified 78 unigenes that were involved in terpenoid synthesis, and their expression level was presented as a heat map (Fig. 7). Furthermore, 28 DEGs were present in the biosynthetic pathway of terpenoids. The number of DEGs between organs is shown in Table 6. The gene ID and gene name of the DEGs between the organs were shown in Supplementary Table S10. In detail, the number of DEGs between roots and stems was the highest, with 11 up-regulated and 5 down-regulated genes in the stem, followed by the roots and leaves with 13 DEGs. The number of DEGs in leaves and flowers was the least, with only two down-regulated genes.

Expression patterns of genes involved in terpenoids. Each column represents a pair of organs used in comparison. Each column represents a tissue organ of C. nobile, whereas each row represents a gene. Expression differences are observed in different colors. The sample names are shown at the top. Gray blocks indicate that the expression is undetected
Construction and analysis of DEG co-expression network for terpenoid synthesis in various organs of C. nobile
Most of the gene co-expression networks have been constructed on the basis of the correlation of gene expression data (Nikiforova and Willmitzer 2007) in an undirected graph. The nodes in the network represent genes or gene products, and genes with similar expression profiles are linked to form networks. The edge represents a significant association between a pair of nodes in gene expression, that is, co-expression between genes. The construction of a co-expression network is conceptually simple and intuitive. Using RNA-seq data, we obtained the gene expression data and analyzed the relationship, including both correlation coefficients and correlations, between each two genes. The correlation coefficient represents the degree of similarity between the expression profiles of the two genes, with higher values indicating greater similarity (Carlson et al. 2006). The relationship indicates whether the expression of the two genes is positively or negatively correlated. The similarity of gene expression enabled us to analyze possible interactions between genes or gene products to understand the interactions between genes and find core genes (Barabasi and Oltvai 2004), which are important hubs and play a key role in the network. In the present study, 28 DEGs involved in terpenoid biosynthesis were screened based on RNA-seq data of four C. nobile organs. We constructed the co-expression network based on the FPKM values of these genes (Fig. 8). The octagon represents the gene, and the straight line represents the regulatory relationship of the gene. The rank of k-core values describes the complexity of gene-related relationships (Supplementary Table S11). The co-expression network of 28 DEGs was divided into three subnetworks (Fig. 8). The k-core value of DXR2, DXR3, MDS, GPPS1, and GPPS2 genes was 7; 6 for HDR, HMGR3, FPPS1, and FPPS3 genes; and 5 for HMGR2, HMGR4, and FPPS6. These genes might play a key role in terpenoid biosynthesis of C. nobile.

Gene co-expression network. Nodes in the network represent genes, and edges represent a significant correlation between gene expression levels in a pair of nodes
Putative terpenoid biosynthesis genes in different organs
There are two pathways to provide isoprene units in terpene biosynthesis: the MVA and the MEP pathways (Fig. 9a). We further profiled the expression of 16 selected genes that are known to participate in the terpenoid biosynthesis of C. nobile using qRT-PCR. We then generated the differential expression heat maps of these genes based on the qRT-PCR data (Fig. 9a). Overall, RNA-seq data did not significantly correlated with qRT-PCR data from most of the genes (Fig. 9b). The consistency between the FPKM value and the qRT-PCR value was not desirable possibly because we did not repeat the sequencing of the C. nobile transcriptome. Therefore, we chose the qRT-PCR data for assessing the expression level of selected tissue-specific genes. Most of the tested genes, including AACT, HMGS, HMGR1, HMGR2, MDPS, MDS, MK1, MK2, PMK, IPPI, FPPS1, DXS2, and GPPS, had the highest expression levels in roots. The expression levels of these genes positively correlated with the total terpenoid content, indicating that these genes may play an important regulatory role in terpenoid synthesis. We attempted to confirm the relationship between the transcript level of these genes and the accumulation of various components of terpenoids in different organs of C. nobile. We edited the script using the software EditPlus and RStudio to calculate the correlation coefficient (Supplementary Table S12). As indicated in Fig. 9c, the expression levels of most terpenoid genes (AACT, HMGS, HMGR1, HMGR2, MDPS, MDS, MK1, MK2, PMK, IPPI, FPPS1, DXS2, and GPPS) positively correlated with the content of most of the monoterpenes (α-pinene, β-phellandrene, 1,8-cineole, camphor, α-terpineol, and carvacrol) and the sesquiterpenes ((E,E)-farnesol and chamazulene). Therefore, these 13 genes extensively contributed in regulating the biosynthesis of terpenoids in C. nobile. Moreover, these 13 genes are involved in MEP, MVA and common downstream pathways of terpenoid biosynthesis, respectively, suggesting cross-talk between MEP and MVA pathways in C. nobile. Interestingly, GDS1 was mainly expressed in flowers, and germacrene D was higher in flowers than other organs. Therefore, GDS1 was likely to be one of the key genes in the accumulation of germacrene D. Several genes involved in the synthesis of terpenoids have been isolated and identified from C. nobile. For example, CnHMGR showed different expression patterns in roots, stems, leaves, and flowers, with higher expression in flowers and the lowest expression in stems (Meng et al. 2016). HMGS gene of C. nobile was also cloned and identified from C. nobile, CnHMGS had the highest expression level in flowers, followed by roots, and low expression levels in stems and leaves (Cheng et al. 2016). In the present study, CnHMGR1 had the highest expression level in roots, followed by flowers and stems, and low expression levels in leaves. CnHMGR2 also had the highest expression level in roots, followed by flowers, and low expression levels in leaves and stems. Similarly, CnHMGS had the highest expression level in roots, followed by stems and leaves, and with the lowest level of expression in flowers. These findings were different from the tissue expression patterns of the genes in our previous studied, which may be related to the different functions of different members of the HMGS and HMGR gene families in the synthesis of terpenoids and growth in C. nobile.

a MVA and MEP pathways for terpenoid biosynthesis in C. nobile. Heat map of the hierarchical clustering of 16 gene expression profiles in four different organs; the heat map data were the results of qRT-PCR. F, flowers; R, roots; S, stems; and L, leaves. b Expression analysis of 16 genes involved in the terpenoid biosynthesis of C. nobile. 18S gene was used as the internal control. The error bars represent the standard error of three biological replicates. Bar and line charts represent the qRT-PCR and FPKM values of the genes, respectively. c Correlation analysis of 16 gene expression levels and terpenoid content in different components. Each column represents a gene used in comparison, whereas each row represents a terpenoid. Different colors indicate the size of the correlation coefficient
Overall, the accumulation levels of terpenoids significantly varied among the four organs of C. nobile. The differential expression of the terpene-related genes in different organs may be one of the reasons for this differential accumulation. Therefore, analysis of the biosynthetic genes involved in terpenoids provides deeper understanding into terpene synthesis as well as molecular basis for improvement in the quality of this medicinal plant through biotechnology.
Identification of unigene-derived microsatellite markers
SSRs are a series of simple, repetitive short-sequence composition, widely distributed in the eukaryotic genome. SSR, as a molecular marker, is widely used in crossbreeding, population genetic diversity, genetic linkage map construction, and other research fields. SSR plays an important role in genomic and phenotypic diversity and is broadly applied in molecular marker technology (Murat et al. 2003). At present, molecular markers of C. nobile are considerably limited. The present study identified 4614 SSRs from all unigenes (Supplementary Table S13), and the mononucleotide repeats were the most abundant (2241), accounting for 48.57% of the total SSRs, followed by tri- (1416, 30.69%), di- (657, 14.24%), tetra- (57, 1.24%), penta- (10, 0.02%), and hexa-nucleotide, with 8 SSRs (Fig. 10). In the di-nucleotide repeats, AC/CA, GT/TG, AT/TA type distribution ratios were predominant, accounting for 27.25%, 25.11%, and 27.25%, respectively; in the tri-nucleotide, GAT was the most widely distributed, accounting for 5.72%. The study of C. nobile SSRs will provide important resource in genetic marker research of C. nobile.

Distribution of different types of SSRs identified in the C. nobile unigenes. The scale at the bottom is the amount of repeated nucleotides
Conclusion
In this study, we determined the composition and content of terpenoids in different organs of C. nobile and found that the roots were the main synthesis site of terpenoids with chamazulene being the main component among the terpenoids. Transcriptome datasets, established from roots, stems, leaves, and flowers of C. nobile, generated basic molecular information for identifying genes involved in terpenoid synthesis through the MVA and MEP pathways. We screened 78 unigenes, of which 28 DEGs were identified, putatively involved in terpenoid biosynthesis. In addition, we found that 13 genes (AACT, HMGS, HMGR1, HMGR2, MDPS, MDS, MK1, MK2, PMK, IPPI, FPPS1, DXS2, and GPPS) among them had the highest expression levels in roots. The expression levels of these 13 genes positively correlated with the content of most terpenoid compounds in different organs. These findings would be helpful for understanding of the anabolic pathways of terpenoids, which are the active ingredients of many medicinal plants, and provide diverse genetic resources for further study.
Author contribution statement
FX and JC conceived the idea and designed the experiment. XL, XW, JY, ZC, YL and WZ carried out various experiments. XL, XW, JY and FX analyzed the data. XL and FX wrote the manuscript.
References
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:1–12
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:115–119
Ashburner M (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29
Banyai W, Kirdmanee C, Mii M, Supaibulwatana K (2010) Overexpression of farnesyl pyrophosphate synthase (FPS) gene affected artemisinin content and growth of Artemisia annua L. Plant Cell Tiss Org 103:255–265
Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell’s functional organization. Nat Rev Genet 5:101–113
Benjamini Y, Yekutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Bohlmann J, Keeling CI (2008) Terpenoid biomaterials. Plant J 54:656–669
Buhaescu I, Izzedine H (2007) Mevalonate pathway: A review of clinical and therapeutical implications. Clin Biochem 40:575–584
Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF (2006) Gene connectivity, function, and sequence conservation: predictions from modular yeast co-expression networks. BMC Genom 7:40
Chen H, Xin C, Jing T, Yong Y, Liu Z, Hao X, Wang L, Wang S, Liang J, Zhang L, Yin F, Cheng X (2016) Development of gene-based SSR markers in rice bean (Vigna umbellata L.) based on transcriptome data. PLoS One 11:e0151040
Cheng S, Wang X, Xu F, Chen Q, Tao T, Lei J, Zhang W, Liao Y, Chang J, Li X (2016) Cloning, expression profiling and functional analysis of CnHMGS, a gene encoding 3-hydroxy-3-methylglutaryl coenzyme a synthase from Chamaemelum nobile. Molecules 21:316
Deng YY, Li JQ, Wu SF, Zhu YP, Chen YW, He FC (2006) Integrated nr database in protein annotation system and its localization. Comput En 32:71–74
Dudareva N, Cseke L, Blanc VM, Pichersky E (1996) Evolution of floral scent in Clarkia: novel patterns of S-linalool synthase gene expression in the C. breweri flower. Plant Cell 8:1137–1148
Dudareva N, Martin D, Kish CM, Kolosova N, Gorenstein N, Faldt J, Miller B, Bohlmann J (2003) (E)-beta-ocimene and myrcene synthase genes of floral scent biosynthesis in snapdragon: function and expression of three terpene synthase genes of a new terpene synthase subfamily. Plant Cell 15:1227–1241
Farhoudi R (2013) Chemical constituents and antioxidant properties of Matricaria recutita and Chamaemelum nobile essential oil growing wild in the south west of Iran. J Essent Oil Bear Plant 16:531–537
Gao B, Zhang D, Li X, Yang H, Zhang Y, Wood AJ (2015) De novo transcriptome characterization and gene expression profiling of the desiccation tolerant moss Bryum argenteum following rehydration. BMC Genom 16:1–14
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Heuston S, Begley M, Davey MS, Eberl M, Casey PG, Hill C, Gahan CGM (2012) HmgR, a key enzyme in the mevalonate pathway for isoprenoid biosynthesis, is essential for growth of Listeria monocytogenes EGDe. Microbiology 158:1684–1693
Huber W, Carey VJ, Long L, Falcon S, Gentleman R (2007) Gentleman R: Graphs in molecular biology. BMC Bioinformatics 8:S8
Irmisch S, Krause ST, Kunert G, Gershenzon J, Degenhardt J, Köllner TG (2012) The organ-specific expression of terpene synthase genes contributes to the terpene hydrocarbon composition of chamomile essential oils. BMC Plant Biol 12:1–13
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:277–280
Kent WJ (2002) BLAT-the BLAST-like alignment tool. Genome Res 12:656–664
Landmann C, Fink B, Festner M, Dregus M, Engel KH, Schwab W (2007) Cloning and functional characterization of three terpene synthases from lavender (Lavandula angustifolia). Arch Biochem Biophys 465:417–429
Liu Z, Li X, Simoneau AR, Jafari M, Zi X (2006) Isoprenoid biosynthesis in plants: pathways, genes, regulation and metabolic engineering. J Biol Sci 351:371–374
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2–∆∆C T method. Methods 25:402–408
Ma CM, Winsor L, Daneshtalab M (2007) Quantification of spiroether isomers and Herniarin of different parts of Matricaria matricarioides and flowers of Chamaemelum nobile. Phytochem Anal 18:42–49
Meng XX, Yan JP, Liu XM, Liao YL, Chang J, Xu F (2016) Cloning and expression analysis of HMGR gene from Chamaemelum nobile. Acta Agric Boreali Sin 31:68–75
Misra I, Wang CZ, Miziorko HM (2003) The influence of conserved aromatic residues in 3-hydroxy-3-methylglutaryl-CoA synthase. J Biol Chem 278:26443–26449
Murat C, Riccioni C, Belfiori B, Cichocki N, Labbé J, Morin E, Tisserant E, Paolocci F, Rubini A, Martin F (2011) Distribution and localization of microsatellites in the Perigord black truffle genome and identification of new molecular markers. Fungal Genet Biol 48:592–601
Nagegowda DA, Gutensohn M, Wilkerson CG, Dudareva N (2008) Two nearly identical terpene synthases catalyze the formation of nerolidol and linalool in snapdragon flowers. Plant J 55:224–239
Newall CA, Anderson LA, Phillipson JD (1996) Herbal medicines. A guide for health-care professionals. Pharm Press 296
Nikiforova VJ, Willmitzer L (2007) Network visualization and network analysis. Plant Syst Biol 97:245–275
Rama Reddy NR, Mehta RH, Soni PH, Makasana J, Gajbhiye NA, Ponnuchamy M, Kumar J (2015) Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from Senna (Cassia angustifolia Vahl.), a non-model plant with potent laxative properties. PLoS One 10:e0129422
Schilmiller AL, Schauvinhold I, Larson M, Xu R, Charbonneau AL, Schmidt A, Wilkerson C, Last RL, Pichersky E (2009) Monoterpenes in the glandular trichomes of tomato are synthesized from a neryl diphosphate precursor rather than geranyl diphosphate. Pro Natl Acad Sci USA 106:10865–10870
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg LS, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Xu F, Ning Y, Zhang W, Liao Y, Li L, Cheng H, Cheng S (2014) An R2R3-MYB transcription factor as a negative regulator of the flavonoid biosynthesis pathway in Ginkgo biloba. Funct Integr Genomic 14:177–189
Yan JP, Meng XX, Li Z, Zhang WW, Jie C, Xu F (2017) Molecular cloning and expression analysis of germacrene A synthase gene in Chamaemelum nobile. Chinese Tradit Herb Drugs 48:1851–1859
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297
Yu F, Utsumi R (2009) Deversity, regulation, and genetic manipulation of plant mono-and sesquiterpenoid biosynthesis. Cell Mol Life Sci 66:3043–3052
Zenoni S, Delledonne M (2010) Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-SEq. Plant Physiol 152:1787–1795
Zhao L, Chang W, Xiao Y, Liu H, Liu P (2013) Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu Rev Biochem 82:497–530
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant no. 31400603).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Salim Al-Babili.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, X., Wang, X., Chen, Z. et al. De novo assembly and comparative transcriptome analysis: novel insights into terpenoid biosynthesis in Chamaemelum nobile L.. Plant Cell Rep 38, 101–116 (2019). https://doi.org/10.1007/s00299-018-2352-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-018-2352-z