
Abstract
Matricaria chamomilla, originally native to the temperate zone of Eurasia, has been one of the most popular herbal plants owing to its pharmacologically active sesquiterpenoid metabolites, such as (-)-α-bisabolol and bisabolol oxide A. Although the sesquiterpenoids of M. chamomilla has been well characterized, information on genes responsible for the biosynthesis of these compounds are scarce, and further investigations on the plant have been hindered by the lack of genomic information. In this study, we performed a de novo assembly of the M. chamomilla transcriptome using Illumina HiSeq 2500 high-throughput sequencing. A single run yielded 21 Gb clean reads that were assembled into 83,741 unigenes. A total of 21,626 and 18,929 unigenes were assigned to Gene Ontology categories and the Cluster of Orthologous Groups, respectively. Differentially expressed genes (DEGs) in flowers, stems, roots, and leaves were screened, and 1115 DEG unigenes were assigned to specific secondary metabolites using the Kyoto Encyclopedia of Genes and Genomes (KEGG). We selected a set of putative genes that were most likely involved in the biosynthesis of sesquiterpenoid in M. chamomilla. The genes in the sesquiterpenoid synthesis pathway displayed significant differential expression in different organs. Out of the 83,741 unigenes, 5555 were identified as having simple sequence repeat motifs, with mono-nucleotide repeats being the most frequently found repeat type. Our data provided a comprehensive sequence resource for M. chamomilla belonging to the Asteraceae family, which contains many medicinal herbs. The present mass sequence data are expected to facilitate further study on the regulatory mechanism of the terpenoid biosynthetic pathway in M. chamomilla.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With more than 55,000 known compounds, terpenoids (or isoprenoids) are regarded as the largest class of secondary metabolites. A number of terpenoids exhibit essential biological functions and processes, including membrane fluidity, electron transport chains, respiration, photosynthesis, plant defense, and regulation of growth and development (Chang et al. 2015; Son et al. 2014). Terpenoids also affect our daily lives as important ingredients in spices, cosmetics, food, and drugs. As such, terpenoids possess significant commercial value (Vranová et al. 2013).
All terpenoids are derived from condensation of two simple common C5 precursors, isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP) (Rodríguez-Concepción and Boronat 2002). Both are synthesized by two separate pathways in plants, namely, the mevalonate (MVA) pathway in cytosol (Buhaescu and Izzedine 2007; Zenoni et al. 2010) and the 2-methyl-d-erythritol-4-phosphate (MEP) pathway in plastid (Heuston et al. 2012). The C5 units from the MEP pathway mostly participate in the synthesis of diterpenes, monoterpenes, hemiterpenes and polyterpenes (Schilmiller et al. 2009), whereas the MVA pathway predominantly provides precursors for sterols, sesquiterpenes, triterpenes, and ubiquinones (Liu et al. 2006; Zhao et al. 2012). Several terpenoid biosynthesis-linked genes have been identified and isolated in recent years (Kalita et al. 2015). The biosynthesis of terpenoids is a complex process that involves a series of chemical reactions such as prenylation, oxidation, reduction, and isomerization. Furthermore, the genes coding the enzymes responsible for these reactions are differentially expressed in different plant organs. Therefore, the sporadic studies on a few genes in the terpene pathway are not sufficient to clarify the control mechanism of terpenoid biosynthesis.
Next-generation sequencing (NGS) has recently been employed to bring about breakthroughs in molecular genetics (Mardis 2008; Shendure and Ji 2008). NGS enables the sequencing of up to 1 million kilobases of DNA in a short time to provide the sequence data for a comprehensive analysis of genomes, transcriptomes, and interactomes (Pop and Salzberg 2008; Shendure and Ji 2008). Transcriptome analysis called RNA sequencing (RNA-Seq) is an NGS application that gives snapshot of changing cellular transcriptome (Libault et al. 2010). RNA-Seq is most powerful in understanding biological processes by identifying genes participating in diverse biological processes, through annotation of DEGs (Benedito et al. 2008; Lange et al. 2000).
Chamomile (Matricaria chamomilla L., synonym M. recutita), also known as German chamomile, is an annual herb of substantial economic value owing to its volatile essential oil (Sayadi et al. 2014). M. chamomilla is one of the most important medicinal plant species widely cultivated in Asia and Europe. The herb accumulates numerous terpenoid secondary metabolites, such as (-)-α-bisabolol and chamazulene as major ingredient (Su et al. 2015). Spiroethers, (-)-α-bisabolone oxide A, anthecotulid, and apigenin are also found in the essential oil from chamomile (Murti et al. 2012; Srivastava et al. 2010). Bioactivities, including antiphlogistic, anti-inflammatory, antiseptic, and spasmolytic properties, have been exploited in biopharmaceuticals, cosmetics, perfume, aromatherapy, and health food industries (Formisano et al. 2015). Despite the importance of M. chamomilla as a medicinal plant, genomics and transcriptome of the plant have yet to be analyzed in detail.
To identify genes involved in the sesquiterpenoids biosynthesis of the chamomile, we adopted high-throughput RNA-Seq method supplemented by quantification of the expression levels of the involved unigenes. In parallel, we determined the content and composition of the essential oil from M. chamomilla samples. We also examined the transcriptional regulation of MVA pathway and downstream thereof in sesquiterpenoids biosynthesis. The putative genes examined were: acetyl-CoA C-acetyltransferase (AACT), mevalonate kinase (MK), 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS), phosphomevalonate kinase (PMK), and 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), isopentenyl diphosphate delta-isomerase (IPPI), (-)-germacrene D synthase (GDS), geranylgeranyl diphosphate synthase (GGPPS), and farnesyl diphosphate synthase (FPPS). The transcriptome data and gene expression profiles are expected to provide invaluable resources for the study of various aspects of M. chamomilla, especially terpenoid biosynthesis in the plant.
Materials and methods
Sample collection and RNA extraction
Matricaria chamomilla L. (Asteraceae) was grown in the botanical garden at Yangtze University in Jingzhou, China (30.35°N, 112.14°E). When the flowers of 4-month-old M. chamomilla opened, roots, stems, leaves, and flowers were collected for transcriptome and sesquiterpenoid anayses. Each sample was cut into small pieces, quick-frozen in liquid nitrogen, and placed at − 80 °C until use. The total RNA of each sample was extracted using the MiniBEST Plant RNA Extraction Kit (Dalian, China) in accordance to the manufacturer’s instructions. Integrity of the total RNA was assessed through electrophoresis on 1% agarose gels, and the RNA was determined using a NanoPhotometer® (Implen, CA).
Library construction, deep sequencing, and de novo assembly
The transcriptome libraries of the plant organs were prepared using a NEBNext®Ultra™ RNA Library Prep Kit for Illumina (NEB, USA) (Li et al. 2016) and were sequenced on an Illumina HiSeq 2500 system at Biomarker Technologies (Beijing, China). The clean reads, generated by removing empty reads, adaptor sequences, and low-quality sequences, were assembled into contigs through Trinity method (Grabherr et al. 2011) to recover extra full-length transcripts across a broad range of expression levels. Trinity method allowed construction of transcripts by connecting the contigs on the basis of the paired-end information of the sequences. Paired-end reads were then applied for gap filling of transcripts, and the longest transcripts thus obtained were defined as unigenes.
Functional annotation and metabolic pathway analysis
For functional annotation, BLASTx (Version 2.2.26) was used with a cut-off E value of 10− 5 to search all of the assembled unigenes of M. chamomilla against the NCBI Nr protein database (Deng et al. 2006), Swiss-Prot (Apweiler et al. 2004), KEGG (Kanehisa et al. 2004), Gene ontology (GO) (Ashburner 2000), and Cluster of Orthologous Groups of proteins (COG) (Tatusov et al. 2000). Hits with E value exceeding 10− 5 were excluded from the analysis, and the optimum comparison results were screened for annotating the assembled unigenes (Altschul et al. 1997). Using the GO program, GO annotation, based on the molecular function, biological process, and cellular component, were obtained. The search of unigenes against the COG database assigned possible functions, and the application of KEGG database allowed analysis of inner-cell KEGG metabolic pathways.
Analysis and validation of the DEGs
To compare the expression characteristics of the genes, the read counts were normalized by calculating the fragments per kilobase of the transcript per million mapped reads and the false discovery rate (FDR) to obtain relative expression value (Trapnell et al. 2010). Differential expression in different organs was assessed using the DESeq R package (version 1.10.1). The package provides a statistical approach for assessing the differential expression by utilizing a negative binomial distribution model (Anders and Huber 2010). FDR was employed to find the threshold of the p values adjusted using Benjamini and Hochberg method (Haynes 2013). An expression was considered significant when |log2 (fold change)| ≥ 1 and the FDR (false discovery rate) was lower than 0.01.
Simple sequence repeats
In this study, frequency and distribution of the simple sequence repeats (SSRs) were analyzed in all obtained unigenes. SSRs were detected using the MISA software (http://pgrc.ipk-gatersleben.de/misa/). The minimum repeat length was set as 12 for di-, tri-, tetra, and hexa-nucleotide repeats, and 15 for penta-nucleotide repeats. FASTA-formatted sequences were the input file (Chen et al. 2016; Reddy et al. 2015).
Gas chromatography–mass spectrometry
For identifying the sesquiterpenes in M. chamomilla, 2 µL of pentane extract of each organ (roots, leaves, stems, and flowers) were directly analyzed through gas chromatography–mass spectrometry (Agilent 5975B GC–MS system with a 6890 N gas chromatograph) with authentic standards under the following temperature program: injection at 250 °C and ramped from 40 to 250 °C at a rate of 10 °C min− 1. The column was DB1-MS (0.25 µm film thickness, 250 µm × 30 m). Helium was carrier gas at 1 mL min− 1. The sesquiterpenes were identified based on comparison of their measured retention times and mass spectra with those of authentic standards. The reference compounds were purchased from Roth (Karlsruhe, Germany), Sigma-Aldrich (Steinheim, Germany), and Herbfine (Jiangxi, China). The mass spectra were searched against the NIST11 database. A previously published GC–MS method and a chiral column were used for chiral analysis (Irmisch et al. 2012).
qRT-PCR validation
The RNAs of the leaves, stems, roots, and flowers were isolated as described above. First-strand cDNA was synthesized from 1 µg high-quality RNA using the PrimeScript™ Reagent Kit (Dalian TaKaRa, China). The 10 × diluted cDNA was used as template for quantitative real-time PCR (qRT-PCR). qRT-PCR was carried out using a SYBR Premix ExTaq™ II Kit (Dalian TaKaRa, China) on the Bio-Rad Mini Opticon™ Real-time PCR system instrument according to the manufacturer’s instructions. The qRT-PCR conditions were the same as those described by Xu et al. (2014). The qRT-PCR data were normalized using 18S rRNA (18SU: 5′-ACCGAGCGTCGAGTGGATTAA-3′ and 18SD: 5′-CTAGTTCGTGCGTCCGTCAAA-3′) as the reference gene (Tao et al. 2016). Gene primers used in qPCR validation are listed in Table S1. The relative expression levels of the target genes in each sample were calculated by the 2− ΔΔCt method (Livak and Schmittgen 2001). The qRT-PCR experiments were repeated using two biological replicates for each organ and three technical replicates for each sample.
Results and discussion
Illumina sequencing and de novo assembly
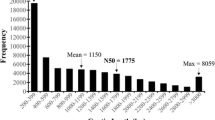
Recent development of NGS technology has made significant progress in gene discovery possible (Reisfilho 2009). In the present study, four cDNA libraries, derived from root, stem, flower, and leaf samples of M. chamomilla, were sequenced on an Illumina HiSeq2500 platform. The sequences were deposited in NCBI Sequence Read Archive under accession numbers SRR3990144 (roots), SRR3990143 (stems), SRR5557958 (flowers), and SRR3961779 (leaves). The libraries produced 21,561,283,776 (21 Gb) clean reads with 90.82% Q30 (sequencing error rate at 0.1%). The reads were de novo assembled using Trinity software after quality control to yield 139,471 transcripts with a mean length of 950 nt. The clean reads were assembled into 83,741 unigenes with N50 of 1277 nt. The number of unigenes exceeding 1000 bp were 20,875 (Fig. 1a, b). The average unigene length of 704 bp was longer than the values from previous reports on medicinal plants, such as Davidia involucrata Baill (Li et al. 2016), Siraitia grosvenorii (Tang et al. 2011), and Panax quinquefolius (Wu et al. 2010).

Statistics of Illumina short read assembly quality
Functional annotation of unigenes
BLASTX of the above-mentioned 83,741 unigenes annotated 42,138 (50.3%) unigenes with a cut-off E value of 10− 5 (Table 1). Among the unigenes annotated against NCBI non-redundant (nr) protein database, 10.11% displayed a close homology to Vitis vinifera (10.11%), followed by Sesamum indicum (6.42%), Coffea canephora (5.83%), Nicotiana sylvestris (4.20%), Nicotiana tomentosiformis (4.13%), Theobroma cacao (3.14%), Nelumbo nucifera (2.66%), Solanum tuberosum (2.34%), Citrus sinensis (2.25%), and Erythranthe guttata (2.15%) (Fig. 2).

No. of M. chamomilla transcripts homologous to genes from other species
Gene ontology classification
To describe the functions of the predicted genes in M. chamomilla, we performed GO assignments in the three main domains, cellular component, biological process, and molecular function, and the results were plotted using WEGO (Ashburner 2000; Ye et al. 2006). A total of 21,626 sequences were categorized into 52 functional groups (Fig. 3). “Cell”, followed by “cell part” was the most highly represented GO term under cellular component. In the case of molecular function and biological process categories, catalytic activity and metabolic process were most highly represented, respectively. The same trend in maximum GO categories as the present study was previously reported for the Cassia angustifolia Vahl transcriptome (Li et al. 2013). We also observed high percentages of genes under the categories of “organelle”, “binding”, and “cellular process”, as well as several genes under the terms of “extracellular matrix part”, “protein tag”, “translation regulator activity” and “cell killing”. Under biological process category, the maximum number of unigenes was associated with “metabolic process”, suggesting possibility that novel genes involved in important metabolic activities in M. chamomilla could be identified in the present study.

Gene Ontology categories of the M. chamomilla unigenes. The unigenes are summarized in three categories: cellular component, biological process, and molecular function
Clusters of orthologous group classification
To further validate the transcriptome library and the annotation process, 18,929 sequences out of the 42,138 nr hits were classified into a cluster of orthologous group (COG). The COG annotation distributed the sequences into a minimum of 25 categories according to their biological function (Fig. 4). The largest group was the cluster “general function prediction only” (3433, 18.14%), followed by “replication, recombination, and repair” (1806, 9.54%) and “transcription” (1695, 8.95%). In contrast, the unigenes classified under extracellular and nuclear structures were few. Under secondary metabolite biosynthesis, transport, and catabolism categories, a total of 725 unigenes (3.83%) were assigned and several of these were involved in sesquiterpene biosynthesis (vide infra).

Clusters of orthologous groups (COG) classification of the M. chamomilla unigenes. Out of 36,021 nr hits, 16,008 sequences have a COG classification among the 25 categories
Functional classification by the Kyoto encyclopedia of genes and genomes
Sesquiterpenes have been regarded as pharmacologically active constituents of M. chamomilla. Insight into sesquiterpene biosynthesis in this plant thus can accelerate the future engineering of the pathway that can impart high sesquiterpene content. We adopted KEGG, which has been widely used to identify unigenes involved in the biological pathways (Reddy et al. 2015), to analyze the unigenes for terpenoid pathway. All of these unigenes would be important resources for genetic manipulation of M. chamomilla.
The metabolic pathways of sesquiterpene biosynthesis in plants are under active study (Lange et al. 2000; Son et al. 2014) because of myriad variations in structure and modification. To construct the biological pathways in M. chamomilla, 15,746 sequences were assigned to 128 KEGG pathways. The most predominant among these pathways was ribosome (867, 5.51%), followed by carbon metabolism (658, 4.18%), biosynthesis of amino acids (545, 3.46%), protein processing in the endoplasmic reticulum (525, 3.33%), and spliceosome (430, 2.73%) (Table S2). Regarding the biosynthesis of secondary metabolites, 1115 unigenes were found to be involved (Table 2). Among these unigenes, the cluster of “phenylpropanoid biosynthesis [PATH: ko00940]” represented the largest group (348, 31.21%), followed by “flavonoid biosynthesis [PATH: ko00941]” (90, 8.07%) and “terpenoid backbone biosynthesis [PATH: ko00900]” (81, 7.26%). Most terpene-related enzymes were mapped to the terpenoid backbone biosynthesis [PATH: ko00900] and sesqui- and triterpenoid biosyntheses [PATH: ko00909] groups by KEGG.
In plants, the building blocks for isoprenoids are synthesized via MVA and MEP pathways (Seemann et al. 2002; Vranová et al. 2013). The BLASTX search identified and classified 61 enzymes as terpenoid synthase in ko00900 metabolic pathways (Table 3). All of these unigenes were important genetic resources for studying the MVA and MEP pathways in M. chamomilla. These results again confirmed the usefulness of high-throughput sequencing in identifying metabolic pathways genes.
Identification of differentially expressed genes
To estimate the expression difference of genes in different M. chamomilla organs, the FDR and absolute log2 ratio value were used as thresholds for assessing the significance of differential gene expression (Gao et al. 2015). A total of 29,975 genes had substantial expression differences between the flower, stem, root, and leaf libraries (Fig. 5).

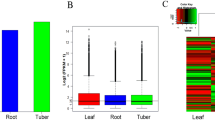
Differentially expressed genes (DEGs) in different M. chamomilla organs. a Hierarchical cluster analysis of common DEGs. A scale indicating the color assigned to log2 FPKM is shown to the right of the cluster. Yellow colors indicate high expression, blue colors indicate low expression, and each horizontal bar represents a single gene. b The number of upregulated and downregulated genes between flowers and stems, flowers and roots, flowers and leaves, stems and roots, stems and leaves, and roots and leaves
Figure 5 depicts summary of DEGs between different M. chamomilla organs. Tables S3 through S8 list detail of the DEGs between flower and stem (Table S3), flower and root (Table S4), flower and leaf (Table S5), stem and root (Table S6), stem and leaf (Table S7), and root and leaf (Table S8). As shown in Fig. 5 5355 DEGs were detected between the flowers and the stem with 1004 genes upregulated and 4351 downregulated (Table S3). Between flowers and roots, a total of 5793 DEGs, 1912 upregulated and 3881 downregulated, was detected (Table S4). Exactly 3000 DEGs, which included 896 upregulated and 2104 downregulated genes, were found between flowers and leaves (Table S5). Then, 5342 DEGs, including 3084 upregulated and 2258 downregulated genes, were observed between stems and roots (Table S6). A total of 4552 DEGs, which included 2707 upregulated and 1845 downregulated genes, were identified between stems and the leaves (Table S7). Finally, 5933 DEGs, which included 3245 upregulated and 2688 downregulated genes, were observed between roots and leaves (Table S8). The results shows that the number of DEGs was highest between flower and root pair, whereas the lowest number was shown between flower and leaf pair. The findings suggested that these DEGs were differentially expressed in different organs of M. chamomilla.
DEGs in terpenoid pathway among four organs
To clarify differential expression of terpenoid biosynthetic genes in the four of M. chamomilla organs, the DEGs involved in the terpenoid metabolism were further analyzed. Gene candidates that may correlate with the terpenoid pathway were examined by cluster analysis of the gene expression pattern with HemI software. One gene cluster for sesquiterpene biosynthesis was identified in the cluster analysis of the intersection of DEGs (Fig. 6a). Comparison of stems with roots revealed that the expression levels of 20 unigenes were significantly higher in stems than in roots, and eight significantly lower in stems compared with roots. Similarly, the expression levels of 19 unigenes were considerably higher in leaves in comparison with stems, and five unigenes showed a substantially lower expression in leaves relative to stems. The expression patterns of 28 unigenes in roots and leaves were comparable, and seven unigenes presented considerably higher expression in roots than in leaves. Figure 6a shows that 24 unigenes exhibited a lower expression in flowers than in leaves; 23 unigenes lower expression in flowers than in stems; and 21 unigenes lower expression in flowers than in roots. These results suggested that the unigenes involved in the MVA pathway were expressed higher in leaves and stems than in roots and flowers. These differential expression patterns of terpene genes may give rise to the differences in the types and contents of sesquiterpenes in various organs.

Expression patterns of genes related to terpenoids. a Expression patterns of MVA pathway-related genes. b Expression patterns of MEP pathway-related genes. Each column shows a pair of organs used in comparison, and the names are shown at the bottom. Each row shows a unigene, and expression differences are observed in different colors. Expression undetected is in black
Another cluster, containing 31 unigenes related to the MEP pathway, was obtained as the union of DEGs (Fig. 6b). Comparison of the expression levels of these genes showed similar spatial expression patterns of the genes in MVA pathway-higher expression in leaves and stems than in roots and flowers. Among the 31 unigenes, 22 were expressed higher in stems than in flowers, 15 higher in leaves than in flowers, 18 higher in stems than in roots, and 15 higher in leaves than in roots.
Putative sesquiterpene biosynthesis genes in different organs
Because sesquiterpenoid profiles in various organs of this chamomile cultivar is not available, flowers, roots, stems, and leaves were analyzed for their terpene composition and content. Flowers had the highest sesquiterpene content, which was nearly 2.5 times of that in roots (Table 4). The flowers accumulated bisabolol oxide A (27.9%), α-bisabolol (22.8%), bisabolol oxide B (17.3%), and β-farnesene (22.5%) as the major sesquiterpenoid components. In addition, small amounts of isocomene and β-caryophyllene were detected, accounting for 0.02 and 0.27% of the total sesquiterpenes, respectively. By contrast, the sesquiterpene blend of the leaves was dominated by α-farnesene (55.4%) and germacrene D (27.5%), whereas α-bisabolol and bisabolol oxide A and B were not detected. Stems and roots displayed comparable bouquet of sesquiterpenes, with β-farnesene as the major compound (stems, 88.6%; roots, 77.2%). Our analysis confirmed the organ-selective production of essential oils in chamomile as described in the previous studies (Presibella et al. 2006).
In plants, sesquiterpene synthases is localized in cytosol (Degenhardt et al. 2009), and MVA pathway supplies their precursors (Vranová et al. 2013). The entry reaction of the MVA pathway is catalyzed by AACT to produce acetoacetyl-CoA, which is then converted into 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by HMGS. HMG-CoA is consequently reduced to MVA by HMGR. Successive phosphorylation reactions catalyzed by MK and PMK converts MVA into MVA 5-diphosphate. ATP-dependent decarboxylation of MVA 5-diphosphate by MVD yields IPP. Ultimately, reversible conversion of IPP by IPPI into DMAPP completes MVA pathway. IPP and DMAPP are then used for synthesizing isoprenoids in the cytosol and the mitochondria (Vranová et al. 2013). Condensation of DMAPP and IPP by FPPS produces FPP. Subsequently, sesquiterpene skeletons are generated from the precursor FPP by sesquiterpene synthases (Fig. 7a).

Spatial expression analysis of DEGs related to sesquiterpene biosynthesis. a MVA pathway for sesquiterpene biosynthesis in M. chamomilla. Heat-Map of the hierarchical clustering of 12 gene expression profiles in four different organs F flower, R root, S stem, L leaf. Expression ratios are expressed as log2 values. b Expression analysis of the 12 genes in various organs. Relative expression level in flowers was set to 1.0
Economic and pharmacological importance of chamomile justifies detailed study on terpene-related genes in M. chamomilla. We thus selected 10 genes that are known to be responsible for the sesquiterpenoid biosynthesis (AACT, HMGS, HMGR1-3, MK, PMK, IPPI, FPPS, and GDS) and, in addition, two for diterpenoid synthesis (GGPPS1 and GGPPS2) in this plant. Homologs of these terpenoid biosynthesis-linked genes have been isolated and identified from various plant species. In Ginkgo biloba, AACT and MVK genes are suggested to act in terpene trilactone biosynthesis. On salicylic acid and methyl jasmonate treatment, the expression levels of GbAACT and GbMVK correlate positively with terpene trilactone (TTL) production in G. biloba seedlings (Chen et al. 2017). TmHMGS was expressed in needles and the stems at similar levels in Taxus × media (Kai et al. 2006). In M. chamomilla, the highest and lowest expression levels of McHMGS were observed in flowers and stems (Tao et al. 2016). CaHMGS is strongly expressed in hypocotyls and cotyledons, exhibiting good correlation with camptothecin content in the tested tissues (Kai et al. 2013). The HMGR gene from numerous plants has been isolated and its function has been studied. CmHMGR is involved in determining the fruit size of melons (Kobayashi et al. 2002). In Artemisia annua L., the co-overexpression of HMGR and FPPS genes enhances artemisinin content (Wang et al. 2011). When HMGR is overexpressed, the production level of β-sesquiphellandrene in Lactococcus lactis increases by 1.25 to 1.60-fold (Song et al. 2012). Two enantioselective GDSs involved in the biosynthesis of (+)- and (−)-germacrene D were known in Solidago canadensis (Schmidt et al. 1998). In German chamomile, four sesquiterpene synthases (TPS) and one monoterpene synthase were identified, and the expression patterns of TPSs in different organs of chamomile positively correlate with the content of terpenoid products in the corresponding organs (Irmisch et al. 2012).
In the present study, qRT-PCR demonstrated the organ-specific expression of above-mentioned genes, reflecting the composition and content of sesquiterpenoids in the respective plant organs. The expression of seven unigenes operating in MVA pathway, namely, AACT, HMGR1, HMGR2, HMGR3, MK, PMK, and IPPI, was significantly higher in flowers compared to other organs (Fig. 7b) explaining the highest sesquiterpene content in flowers (Table 4). Previous study of Litsea cubeba also showed the highest expression levels of six genes (AACT, HMGR, HMGS, PMVK, MVK, and MVD) in flowers (Han et al. 2013). Among the MVA pathway genes, HMGR is the rate-limiting enzyme in isoprenoid biosynthesis, catalyzing the conversion of HMG-CoA into MVA (Liu et al. 2014). HMGR thus could serve as a marker representing overall MVA pathway flux. The enzyme is highly expressed in flowers and abundantly expressed in leaves and roots in the present study, displaying a similar pattern to that of Panax quinquefolius HMGR (Wu et al. 2012). AACT, MK, PMK, and IPPI are also involved in the MVA pathway to provide building block to form sesquiterpene skeleton. Therefore, the expression levels of these genes positively correlated with the total sesquiterpenoid content in the present study. These results suggested that flowers are crucial to total sesquiterpenoid biosynthesis in M. chamomilla. In practice, only the flowers of chamomile have been harvested for essential oil extraction (Raal et al. 2012). However, the present study demonstrated that the leaves are promising sources of α-farnesene and germacrene D.
FPPS catalyzes the condensation of DMAPP with two molecules of IPP to form FPP that is the precursor of all sesquiterpenes (Zhao et al. 2015). FPPS and GGPPS showed identical expression pattern in M. chamomilla (Fig. 7). The highest expression of FPPS and GGPPS appeared in the leaves, followed by flowers, roots, and stems. These data suggested that the organ-specific expression of FPPS and GGPPS regulated the biosynthesis of α-farnesene and germacrene D. This assumption confirms the previous studies that organ-specific expression of terpene synthases parallels the organ-specific accumulation of essential oils in chamomile (Irmisch et al. 2012).
As the second enzyme involved in the MVA pathway, HMGS combines acetyl-CoA with acetoacetyl-CoA to form HMG-CoA (Liu et al. 2014). In our study, HMGS was predominantly expressed in stems, where β-farnesene is the major sesquiterpene compound. These results alluded that HMGS played an important role in the accumulation of β-farnesene by regulating the flux of MVA pathway. GDS was detected in all the tested M. chamomilla organs at a comparable level. Functional diversity of terpenoids calls for highly complex regulation in the synthesis and metabolism. Combining transcriptome analysis and metabolomics makeup of sesquiterpenoids in M. chamomilla, we concluded that flowers, leaves, stems, and roots respectively synthesizes and accumulates particular composition of sesquiterpenoids. This study confirmed that the organ-specific expression of these genes correlated with the dominant sesquiterpenoid compounds in the respective organs in M. chamomilla. Mining and identification of the sesquiterpenoid biosynthesis genes in M. chamomilla would be useful not only in understanding regulation of terpenoid biosynthesis but also in providing molecular information for genetic improvement of this important medicinal plant.
Identification of unigene-derived microsatellite markers
Microsatellites or SSRs, as important genetic markers, have been extensively employed in assessment of genome organization and phenotypic diversity (Murat et al. 2011). A total of 5555 SSRs were identified out of the 82,946 unigenes generated in the present study (Table S9). The proportion of SSRs were not even among the unigens. The proportion of mono-nucleotide SSR was the largest (2872 or 51.70%), followed by the tri- (1482 or 26.68%), di- (771 or 13.88%), tetra- (75 or 1.35%), penta- (13 or 0.23%), and hexa-nucleotide (9 or 0.16%) (Fig. 8). (GT/TG)n, (CA/AC)n, and (TA/AT)n were the other three major motif types prevailing among the di-nucleotide SSRs, displaying frequencies of 4.2, 3.4, and 2.9%, respectively. Among the 20 types of tri-nucleotide SSRs, TGA (1.4%) was most common, followed by TCA (1.2%), GAA (1.1%), and ATC (1.1%). The unigene-derived markers identified in the present study can provide a useful genetic approach on M. chamomilla and other Asteraceae species.

Distribution of different types of SSRs identified in the Matricaria chamomilla unigenes. The scale at the bottom is the amount of repeated nucleotides
Conclusions
Although M. chamomilla, a non-model plant, has not been accessible for total genome analyses, transcriptome analyses as shown in the present study offers an efficient and cost-effective approach to gain access to genetic data for genomic study. This is the first report of such study to identify and assess genes with emphasis on terpene metabolism in M. chamomilla using NGS technology. A total of 1115 DEG unigenes were assigned to specific secondary metabolites using KEGG. Genes in the sesquiterpenoid synthesis pathway exhibited significant differential expression in different organs. The obtained transcriptome resources would provide foundation for identifying functional genes involved in sesquiterpenoid biosynthesis in M. chamomilla. Several genes of agronomical and medicinal importance as well as numerous microsatellite markers were also found. Our data provide useful information for identifying the genes involved in the secondary metabolism in Asteraceae species, especially M. chamomilla.
Author contribution statement
FX and JC designed and conceived the experiments. WWZ, TTT and XML finished the experiments. WWZ, TTT, and FX analyzed the data. WWZ, TTT, XML, FX, and YLL contributed reagents/materials/analysis tools. WWZ, TTT and FX prepared the manuscript. All authors reviewed and approved the manuscript.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11:R106
Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res 32:115–119
Ashburner M (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25:25–29
Benedito VA et al (2008) A gene expression atlas of the model legume Medicago truncatula. Plant J 55:504–513
Buhaescu I, Izzedine H (2007) Mevalonate pathway: a review of clinical and therapeutical implications. Clin Biochem 40:575–584
Chang J, Ning Y, Xu F, Cheng S, Li X (2015) Research advance of 3-hydroxy-3-methylglutaryl-coenzyme a synthase in plant isoprenoid biosynthesis. J Anim Plant Sci 25:1441–1450
Chen H, Chen X, Tian J, Yang Y, Liu Z, Hao X, Wang L, Wang S, Liang J, Zhang L, Yin F (2016) Development of gene-based SSR markers in Rice Bean (Vigna umbellata L.) based on transcriptome data. PLoS One 11:e0151040
Chen Q, Yan J, Meng X, Xu F, Zhang W, Liao Y, Qu J (2017) Molecular cloning, characterization, and functional analysis of acetyl-CoA C-acetyltransferase and mevalonate kinase genes involved in terpene trilactone biosynthesis from Ginkgo biloba. Molecules 22:74
Degenhardt J, Köllner TG, Gershenzon J (2009) Monoterpene and sesquiterpene synthases and the origin of terpene skeletal diversity in plants. Phytochemistry 70:1621–1637
Deng Y, Jianqi LI, Songfeng WU, Zhu Y, Chen Y, Fuchu HE (2006) Integrated nr database in protein annotation system and its localization. Comput Eng 32:71–72
Formisano C, Delfine S, Oliviero F, Tenore GC, Rigano D, Senatore F (2015) Correlation among environmental factors, chemical composition and antioxidative properties of essential oil and extracts of chamomile (Matricaria chamomilla L.) collected in Molise (South-central Italy). Ind Crops Prod 63:256–263
Gao B, Zhang D, Li X, Yang H, Zhang Y, Wood AJ (2015) De novo transcriptome characterization and gene expression profiling of the desiccation tolerant moss Bryum argenteum following rehydration. BMC Genom 16:416
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Han XJ, Wang YD, Chen YC, Lin LY, Wu QK (2013) Transcriptome sequencing and expression analysis of terpenoid biosynthesis genes in Litsea cubeba. PLoS One 8:e76890
Haynes W (2013) Benjamini-Hochberg method. In: Encyclopedia of systems biology. Springer, New York
Heuston S, Begley M, Davey MS, Eberl M, Casey PG, Hill C, Gahan CGM (2012) HmgR, a key enzyme in the mevalonate pathway for isoprenoid biosynthesis, is essential for growth of Listeria monocytogenes EGDe. Microbiology 158:1684–1693
Irmisch S, Krause ST, Kunert G, Gershenzon J, Degenhardt J, Köllner TG (2012) The organ-specific expression of terpene synthase genes contributes to the terpene hydrocarbon composition of chamomile essential oils. BMC Plant Biol 12:84
Kai G, Miao Z, Zhang L, Zhao D, Liao Z, Sun X, Zhao L, Tang K (2006) Molecular cloning and expression analyses of a new gene encoding 3-hydroxy-3-methylglutaryl-CoA synthase from Taxus × media. Biol Plant 50:359–366
Kai GY, Li SS, Wang W, Lu Y, Wang J, Liao P, Cui LJ (2013) Molecular cloning and expression analysis of a gene encoding 3-hydroxy-3-methylglutaryl-CoA synthase from Camptotheca acuminata. Russ J Plant Physiol 60:131–138
Kalita R, Patar L, Shasany AK, Modi MK, Sen P (2015) Molecular cloning, characterization and expression analysis of 3-hydroxy-3-methylglutaryl coenzyme A reductase gene from Centella asiatica L. Mol Biol Rep 42:1431–1439
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:277–280
Kobayashi T, Kato-Emori S, Tomita K, Ezura H (2002) Detection of 3-hydroxy-3-methylglutaryl-coenzyme A reductase protein Cm-HMGR during fruit development in melon (Cucumis melo L.). Theor Appl Genet 104:779–785
Lange BM, Rujan T, Martin W, Croteau R (2000) Isoprenoid biosynthesis: the evolution of two ancient and distinct pathways across genomes. Proc Natl Acad Sci USA 97:13172–13177
Li ZQ, Zhang S, Ma Y, Luo JY, Wang CY, Lv LM, Dong SL, Cui JJ (2013) First transcriptome and digital gene expression analysis in neuroptera with an emphasis on chemoreception genes in Chrysopa pallens (Rambur). PLoS One 8:e67151
Li M, Dong X, Peng J, Xu W, Ren R, Liu J, Cao F, Liu Z (2016) De novo transcriptome sequencing and gene expression analysis reveal potential mechanisms of seed abortion in dove tree (Davidia involucrata Baill.). BMC Plant Biol 16:82
Libault M, Farmer A, Brechenmacher L, Drnevich J, Langley RJ, Bilgin DD, Radwan O, Neece DJ, Clough SJ, May GD, Stacey G (2010) Complete transcriptome of the soybean root hair cell, a single-cell model, and its alteration in response to Bradyrhizobium japonicum infection. Plant Physiol 152:541–552
Liu Z, Li X, Simoneau AR, Jafari M, Zi X (2006) Isoprenoid biosynthesis in plants: pathways, genes, regulation and metabolic engineering. J Biol Sci 351:371–374
Liu YJ, Zhao YJ, Zhang M, Su P, Wang XJ, Zhang XN, Gao W, Huang LQ (2014) Cloning and characterisation of the gene encoding 3-hydroxy-3-methylglutaryl-CoA synthase in Tripterygium wilfordii. Molecules 19:19696–19707
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25:402–408
Mardis ER (2008) The impact of next-generation sequencing technology on genetics. Trends Genet 24:133–141
Murat C, Riccioni C, Belfiori B, Cichocki N, Labbé J, Morin E, Tisserant E, Paolocci F, Rubini A, Martin F (2011) Distribution and localization of microsatellites in the Perigord black truffle genome and identification of new molecular markers. Fungal Genet Biol 48:592–601
Murti K, Panchal MA, Gajera V, Solanki J (2012) Pharmacological properties of Matricaria recutita: a review. Pharmacologia 3:348–351
Pop M, Salzberg SL (2008) Bioinformatics challenges of new sequencing technology. Trends Genet 24:142–149
Presibella M, Villas-Bôas L, Belletti KM, Santos C, Weffort-Santos A (2006) Comparison of chemical constituents of Chamomilla recutita (L.) rauschert essential oil and its anti-chemotactic activity. Braz Arch Biol Technol 49:717–724
Raal A, Orav A, Püssa T, Valner C, Malmiste B, Arak E (2012) Content of essential oil, terpenoids and polyphenols in commercial chamomile (Chamomilla recutita L. Rauschert) teas from different countries. Food Chem 131:632–638
Reddy NRR, Mehta RH, Soni PH, Makasana J, Gajbhiye NA, Ponnuchamy M, Kumar J (2015) Next generation sequencing and transcriptome analysis predicts biosynthetic pathway of sennosides from senna (Cassia angustifolia vahl.), a non-model plant with potent laxative properties. PLoS One 10:e0129422
Reisfilho JS (2009) Next-generation sequencing. Breast Cancer Res 11:2033–2033
Rodríguez-Concepción M, Boronat A (2002) Elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis in bacteria and plastids. A metabolic milestone achieved through genomics. Plant Physiol 130:1079–1089
Sayadi V, Mehrabi AA, Saidi M, Nourollahi K (2014) In vitro culture and callus induction of chamomile (Matricaria chamomilla L.) explants under different concentrations of plant growth regulators. Int J Biosci 4:206–2011
Schilmiller AL, Schauvinhold I, Larson M, Xu R, Charbonneau AL, Schmidt A, Wilkerson C, Last RL, Pichersky E (2009) Monoterpenes in the glandular trichomes of tomato are synthesized from a neryl diphosphate precursor rather than geranyl diphosphate. Proc Natl Acad Sci USA 106:10865–10870
Schmidt CO, Bouwmeester HJ, de Kraker JW, König WA (1998) Biosynthesis of (+)- and (–)-Germacrene D in Solidago canadensis: isolation and characterization of two enantioselective germacrene D synthases. Angew Chem Int Edit 37:1400–1402
Seemann M, Campos N, Hoeffler JF, Grosdemange-Billiard C, Boronat A, Rohmer M (2002) Isoprenoid biosynthesis via the methylerythritol phosphate pathway: accumulation of 2-C-methyl-d-erythritol 2,4-cyclodiphosphate in a gcpE deficient mutant of Escherichia coli. Tetrahedron Lett 43:775–778
Shendure J, Ji H (2008) Next-generation DNA sequencing. Nat Biotechnol 26:1135–1145
Son YJ, Kwon M, Ro DK, Kim SU (2014) Enantioselective microbial synthesis of the indigenous natural product (-)-α-bisabolol by a sesquiterpene synthase from chamomile (Matricaria recutita). Biochem J 463:239–248
Song AA, Abdullah JO, Abdullah MP, Shafee N, Othman R, Tan EF, Noor NM, Raha AR (2012) Overexpressing 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) in the lactococcal mevalonate pathway for heterologous plant sesquiterpene production. PLoS One 7:e52444
Srivastava JK, Shankar E, Gupta S (2010) Chamomile: a herbal medicine of the past with bright future. Mol Med Rep 3:895–901
Su SS, Zhang HM, Liu XY, Pan GF, Ling SP, Zhang XS, Yang XM, Tai YL, Yuan Y (2015) Cloning and characterization of a farnesyl pyrophosphate synthase from Matricaria recutita L. and its upregulation by methyl jasmonate. Genet Mol Res 14:349–361
Tang Q, Ma X, Mo C, Wilson IW, Song C, Zhao H, Yang Y, Fu W, Qiu D (2011) An efficient approach to finding Siraitia grosvenorii triterpene biosynthetic genes by RNA-seq and digital gene expression analysis. BMC Genom 12:343
Tao T, Chen Q, Meng X, Yan J, Xu F, Chang J (2016) Molecular cloning, characterization, and functional analysis of a gene encoding 3-hydroxy-3-methylglutaryl-coenzyme A synthase from Matricaria chamomilla. Genes Genom 38:1179–1187
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annu Rev Plant Biol 64:463–476
Wang Y, Jing F, Yu S, Chen Y, Wang T, Liu P, Wang G, Sun X, Tang K (2011) Co-overexpression of the HMGR and FPS genes enhances artemisinin content in Artemisia annua L. J Med Plants Res 5:3396–3403
Wu Q, Song J, Sun Y, Suo F, Li C, Luo H, Liu Y, Li Y, Zhang X, Yao H, Li X (2010) Transcript profiles of Panax quinquefolius from flower, leaf and root bring new insights into genes related to ginsenosides biosynthesis and transcriptional regulation. Physiol Plant 138:134–149
Wu Q, Chao S, Chen SL (2012) Identification and expression analysis of a 3-hydroxy-3-methylglutaryl coenzyme A reductase gene from American ginseng. Plant Omics 5:414–420
Xu F, Ning Y, Zhang W, Liao Y, Li L, Cheng H, Cheng S (2014) An R2R3-MYB transcription factor as a negative regulator of the flavonoid biosynthesis pathway in Ginkgo biloba. Funct Integr Genom 14:177–189
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297
Zenoni S, Ferrarini A, Giacomelli E, Xumerle L, Fasoli M, Malerba G, Bellin D, Pezzotti M, Delledonne M (2010) Characterization of transcriptional complexity during berry development in Vitis vinifera using RNA-SEq. Plant Physiol 152:1787–1795
Zhao L, Chang W, Xiao Y, Liu H, Liu P (2012) Methylerythritol phosphate pathway of isoprenoid biosynthesis. Annu Rev Biochem 82:497–530
Zhao YJ, Chen X, Zhang M, Su P, Liu YJ, Tong YR, Wang XJ, Huang LQ, Gao W (2015) Molecular cloning and characterisation of farnesyl pyrophosphate synthase from Tripterygium wilfordii. PLos One 10:e0125415
Acknowledgements
This study was supported by the National Natural Science Foundation of China (31400603).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by M. Stobiecki.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11738_2018_2706_MOESM1_ESM.rar
Table S1 Primers sequences used in qRT-PCR. qRT-PCR primers of fourteen candidate unigenes involved in sesquiterpene biosynthesis were designed using the Primer premier 5.0. Table S2 List of Unigenes with KEGG Pathway ID and their categorization into various functional classes. Table S3 DEGs between flower and stem. Table S4 DEGs between flower and root. Table S5 DEGs between flower and leaf. Table S6 DEGs between stem and root. Table S7 DEGs between stem and leaf. Table S8 DEGs between root and leaf. Table S9 The frequency of SSR structure and the number of repeat units. Six types of SSRs were identified from the M. chamomilla unigenes using the MISA. The gene ID, length, SSR type, SSR, SSR start, SSR end, and sequence were listed in this table (RAR 1372 KB)
Rights and permissions
About this article

Cite this article
Zhang, W., Tao, T., Liu, X. et al. De novo assembly and comparative transcriptome analysis: novel insights into sesquiterpenoid biosynthesis in Matricaria chamomilla L.. Acta Physiol Plant 40, 129 (2018). https://doi.org/10.1007/s11738-018-2706-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-018-2706-8