
Abstract
Eukaryotic gene expression is regulated at least by two processes, RNA interference at the post-transcriptional level and chromatin modification at the transcriptional level. Distinct small RNAs (approximately 21–24 nucleotides; sRNAs) were demonstrated to play vital roles in facilitating gene silencing. In plants, the generation of these sRNAs mainly depends on some proteins encoded by respective Dicer-like (DCL), Argonaute (AGO) and RNA-dependent RNA polymerases (RDR) gene families. Here, we analyzed the DCL, AGO and RDR gene families in maize, including gene structure, phylogenetic relationships, protein conserved motifs and genomic localization among gene family members. A total of 5 Zmdcl, 18 Zmago and 5 Zmrdr genes were identified in maize. Phylogenetic analyses clustered each of these genes families into four subfamilies. In addition, gene chromosomal localization revealed that five pairs of Zmago genes resulted from tandem or segmental duplication, respectively. EST expression data mining revealed that these newly identified genes had temporal and spatial expression pattern. Furthermore, the transcripts of these genes were detected in the leaves by two different abiotic stress treatments using semi-quantitative RT-PCR. The data demonstrated that these genes exhibited different expression levels in stress treatments. The results of this study provided basic genomic information for these gene families and insights into the probable roles of these genes in plant growth and development. This will further provide a solid foundation for future functional genomics studies of Dicer-like, Argonaute and RDR gene families in maize.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At least two general types of small RNA molecules (approximately 21–24 nucleotides) are produced by multicellular eukaryotes, most notably microRNA (miRNA) and short interfering RNA (siRNA). These RNA molecules direct a range of biological processes, including developmental timing and patterning, formation of heterochromatin, genome rearrangement and antiviral defense (Carrington and Ambros 2003; Finnegan and Matzke 2003; Lai 2003). These small RNA molecules are primarily associated with both post-transcriptional forms of RNA interference (RNAi) and transcriptional silencing involving chromatin modification (Finnegan and Matzke 2003). In plants, the generation of small RNAs mainly depends on some proteins encoded by respective Dicer-like (DCL), Argonaute (AGO) and RNA-dependent RNA polymerases (RDR) gene families. DCLs undergo RNaseIII-type activities to process complementary dsRNAs into small RNAs, 21–24 nucleotides in length (siRNA or miRNA). These siRNAs or miRNAs provide specificity to the endonuclease-containing, RNA-induced silencing complex (RISC). The specificity is facilitated via AGO proteins with RNaseH-type activities, which target homologous RNAs for degradation with sequence complementary to the small RNAs. Alternatively, transcriptional gene silencing occurs by implementation of RNA-directed DNA methylation or chromatin remodeling to regulatory sequences of the target genes (Wassenegger et al. 1994; Xie et al. 2004).
Among these RNAi machinery components, the plant DCL proteins mainly process long double-stranded RNAs into mature small RNAs (Bernstein et al. 2001; Hammond et al. 2001; Millar and Waterhouse 2005; Chapman and Carrington 2007; Großhans and Filipowicz 2008). Based on sequence comparisons, these proteins have been characterized including the DExD, Helicase-C, DUF283, PAZ, RNaseIII and the double-stranded RNA-binding (dsRB) domain (Margis et al. 2006). The PAZ domain functions to bind the double-stranded 5′ end of the siRNA precursor. Subsequently, the two catalytic RNaseIII domains cleave the dsRNA. The distance between the PAZ domain and the two Dicer RNaseIII domains determines the siRNA length (Zhang et al. 2004). The AGO proteins belong to the core components of RNAi effector complexes and play central roles in RNA silencing (Moazed 2009). The domain structure shared by all AGO proteins includes an N-terminal, PAZ, MID and C-terminal PIWI domain (Kapoor et al. 2008). The PAZ domain contains a specific binding pocket that anchors a characteristic two-nucleotide 3′ overhang that results from RNA digestion by RNase III. A highly basic pocket characteristic of the MID domain specifically binds the 5′ phosphate of the small RNAs and therefore anchors the small RNA onto AGO proteins (Peters and Meister 2007). The PIWI domain, which binds the siRNA 5′ end to the target RNA, exhibits extensive homology to RNase H (Höck and Meister 2008). AGO proteins are highly conserved evolutionarily in eukaryotes and can be phylogenetically subdivided into three groups: Ago-like, Piwi-like, and C. elegans-specific group 3 AGO proteins (Yigit et al. 2006; Hutvagner and Simard 2008). The Ago-like proteins are present in many organisms such as plants, animals, fungi, yeasts and bacteria, while the Piwi-like proteins have only been detected in animals (Girard et al. 2006; Yigit et al. 2006). Expression studies revealed that the Ago-like group was ubiquitously expressed throughout plants, animals and yeasts, but Piwi-like group expression was restricted to germ cells in some mammals such as human and rat (Girard et al. 2006). The C. elegans-specific group 3 AGO proteins usually lose some important catalytic residues in their Piwi domains, which are usually conserved in the Ago-like and Piwi-like proteins (Yigit et al. 2006). A third major type of RNAi protein is the RDR proteins. They are present and required for RNAi in fungi, nematodes and plants, but have not been identified in insects or vertebrates (Djupedal and Ekwall 2009). These enzymes catalyze the formation of phosphodiester bonds between ribonucleotides in an RNA template-dependent fashion and are necessary for initiation and amplification of silencing signals (Kapoor et al. 2008).
At present, some reports reveal that the plant DCL, AGO and RDR gene families usually possess multiple members and are involved in different RNAi pathways, respectively. For example, in Arabidopsis, 4 DCL, 10 AGO and 6 RDR genes have been identified. Furthermore, 8 DCL, 19 AGO and 5 RDR genes have been detected in rice (Kapoor et al. 2008). However, to date, few gene functions have been characterized for these DCLs, AGOs and RDRs in plants. For example, in Arabidopsis, Henderson et al. (2006) reported that miRNA biogenesis is influenced by AtDCL1. AtDCL3 and AtAGO4 are required for RNA-directed DNA methylation of the FWA transgene, which is linked to histone H3 lysine 9 (H3K9) methylation (Zilberman et al. 2003; Henderson et al. 2006). Also, AtDCL2 generates siRNAs associated with virus defense and production of siRNAs from natural cis-acting antisense transcripts, and AtDCL4 generates trans-acting siRNAs that regulate vegetative phase change (Margis et al. 2006). Furthermore, RDRs are proposed to be involved in several types of gene silencing, including co-suppression in plants (Dalmay et al. 2000; Mourrain et al. 2000), RNA interference in Caenorhabditis elegans (Smardon et al. 2000) and gene quelling in Neurospora crassa (Cogoni and Macino 1999).
In maize, few RNAi machinery components have been characterized and reported to date. The present study was performed to obtain a comprehensive understanding of all members of maize DCL, AGO and RDR gene families. The results presented in this study will provide basic genomic information for these gene families and insights into the probable roles of these genes in plant growth and development.
Materials and methods
Identification of DCL, AGO and RDR genes
Maize genome sequences were downloaded from http://www.maizesequence.org/index.html. Hidden Markov Model (HMM) analysis was used to search for DCL, AGO and RDR genes encoded in the maize genome. The HMM profiles of DCL, AGO and RDR families were extracted from the Pfam (http://www.sanger.ac.uk/), respectively. Based on the HMM profiles, the corresponding conserved sequences of DCL, AGO and RDR proteins were obtained with the aid of HMMEMIT utility (Eddy 2008). The above conserved sequences were adopted to search for all predicted DCL, AGO and RDR genes in the B73 maize sequencing database (http://www.maizesequence.org/index.html) by BLASTP program (P value = 0.001). Significant hits were then used as query sequences to search against the National Centre for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/BLAST) using the TBLASTN program (P value = 0.001). The Pfam database (http://www.sanger.ac.uk/Software/Pfam/search.shtml) was finally used to confirm each predicted ZmDCL, ZmAGO or ZmRDR protein sequence as a DCL, AGO or RDR protein, respectively. These genes in this study were designated on the basis of their phylogenetic relationship to other members of the same gene family in Arabidopsis and rice. Some basic physical and chemical parameters of these genes were calculated by online Protparam tool (http://www.expasy.org/tools/protparam.html).
Sequence alignment and phylogenetic analysis
Predicted gene coding sequences were determined using tBlastn and manual comparisons of Clustal-W-aligned genomic sequences, cDNA sequences and predicted coding sequences. All protein sequence alignments were made using Clustal-W (Thompson et al. 1994). Phylogenetic analysis was performed with the MEGA v4.0 program (Kumar et al. 2004) by the neighbor-joining method (Saitou and Nei 1987) and 1,000 bootstrap replicates were performed. Protein domains were analyzed by scanning protein sequences against the InterPro protein signature database (http://www.ebi.ac.uk/InterProScan) with the InterProScan program. Unless otherwise stated, domains were defined according to Pfam predictions (http://www.sanger.ac.uk/Software/Pfam/).
Analysis of conserved motifs and domains
The conserved motif divergence among DCL, AGO and RDR genes in maize was further assessed by a complete amino acid online sequence analysis using Multiple Expectation Maximization for Motif Elicitation (MEME) (Bailey and Elkan 1995) with the following parameters: (1) optimum motif width was set to ≥6 and ≤50; (2) maximum number of motifs were designated to identify 20 motifs. SMART (http://smart.embl-heidelberg.de) program and Pfam database were adopted to annotate the MEME motifs.
DCL, AGO and RDR gene duplication events in maize were also investigated. All of the confirmed DCL, AGO and RDR genes from the maize genome were aligned using Clustal-W and calculated using MEGA v4.0 (Yang et al. 2008). Gene duplication was defined according to the following criteria (Gu et al. 2002; Yang et al. 2008): (1) the length of alignable sequence cover >80% of the longer gene; and (2) the similarity of the aligned regions >70%.
Chromosomal localization of Zmdcl, Zmago and Zmrdr genes
The physical locations of DCL, AGO and RDR genes were determined by initially confirming the starting positions of these candidate genes from the maize genome. The positions of maize DCL, AGO and RDR genes were subject to online analysis using the TBLASTN program (P value = 0.001) (http://www.maizesequence.org/blast) using predicted coding sequences as query sequences. Through this method, the physical locations of all candidate DCL, AGO and RDR genes were confirmed and the redundant sequences with the same chromosome location were rejected from the DCL, AGO and RDR candidate list. Genome Pixelizer software was subsequently used to draw the location images of maize DCL, AGO and RDR genes (http://www.niblrrs.ucdavis.edu/GenomePixelizer/GenomePixelizer_Welcome.html).
EST expression profile analysis of Zmdcl, Zmago and Zmrdr genes in silico
The analysis of Zmdcls, Zmagos and Zmrdrs expression profiles was accomplished by searching the maize dbEST database (http://www.ncbi.nlm.nih.gov/dbEST/) and finding expression information provided at the Web sites. Maize expression data were first obtained through blast searches against the maize dbEST database downloaded from NCBI by conducting the DNATOOLS Blast program. Searching parameters were as follows: maximum identity >95%, length >200 bp and Evalue <10−10. In addition to the maize EST database, maize expression data were also extracted from the Maize Assembled Genomic Island (MAGI) (http://magi.plantgenomics.iastate.edu/) and the Plant Genomic Database (Plant GBD) (http://www.plantgdb.org/) including EST, cDNA and PUTs (PlantGDB unique transcripts).
Plant materials and stress treatment
Maize (Zea mays L. inbred line B73) plants were grown in a greenhouse at 28°C with a photoperiod of 15-h light and 9-h dark. Three-week-old seedlings were subjected to two abiotic stress treatments. Drought stress was induced by 15% PEG-6000 (polyethylene glycerol), and seedling leaves were sampled at 24 h after the treatment. Seedling roots were submerged in 0.15 M NaCl solution for salt stress, and seedling leaves were sampled at 8 h after the treatment. The controls were treated with fresh water.
Semi-quantitative RT-PCR analysis
Total RNA was isolated from the seedlings using Trizol reagent according to manufacturer’s directions (Invitrogen, USA), followed by DNase I treatment to remove any genomic DNA contamination. First-strand cDNA was synthesized using oligo (dT) primer and Superscript II reverse transcriptase (Invitrogen). As a control, reactions were run in parallel that excluded reverse transcriptase.
To examine the expression patterns of these predicted genes in maize and to further confirm their stress responsiveness to abiotic stresses such as drought and salt, all 28 genes were subjected to semi-quantitative RT-PCR using specific primers designed using Primer 5.0 software (Table S2). To adjust for RNA quality and differences in cDNA concentration, we amplified actin as an internal control with the following primers: ZmActin-F (5′-ATGGCTGACGGTGAG-3′) and ZmActin-R (5′-TTAGAA GCACTTCCG-3′). These genes were amplified from first-strand cDNA using Taq polymerase (Promega) on a thermal cycler (Tpersonal 48; Biometra, Germany),with the following profile: initial denaturation at 94°C for 5 min, followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 61.5–69°C (Table S2) for 45 s, polymerization at 72°C for 45 s, and final elongation at 72°C for 5 min. Each PCR pattern was verified by triple replicate experiments; mixture without template was used as negative control and maize actin DNA fragment as positive control for each gene amplified. A 3-μL aliquot of the reaction was separated on 1% agarose gel.
Results
Isolation and characterization of Zmdcl, Zmago and Zmrdr genes
HMM analysis identified 5 DCL genes encoding ZmDCL proteins, 18 AGO genes (ZmAGO) and 5 RDR genes (ZmRDR) in the maize genome. Five DCL loci were confirmed as Zmdcl genes in the maize genome on the basis of analysis of all six type of conserved domains of DExD, Helicase-C, DUF283, PAZ, RNaseIII and dsRB from the putative polypeptide sequence. The length of the Zmdcl open reading frames (ORFs) varied from 4,335 bp for Zmdcl2 to 5,544 bp for Zmdcl1, with the respective coding potential of 1,444 and 1,847 amino acids (Table 1). By SMART analysis and NCBI databases, search for the conserved domains revealed the ubiquitous presence of the conserved DExD, Helicase-C, DUF283, PAZ, RNaseIII and dsRB domains in most proteins, characteristic of all plant DCL proteins from the DCL family (class 3 RNase III family) (MacRae and Doudna 2007; Nicholson 2003). In addition, ZmDCL2 lacked one of the dsRB domains, while all of the ZmDCL1, ZmDCL3a, ZmDCL3b and ZmDCL4 proteins in maize had a second dsRB (dsRBb) domain, which completely lacked in non-plant DCLs, as reported previously (Margis et al. 2006). Interestingly, compared to other ZmDCL proteins, the SMART and Pfam analysis revealed that the N-terminal DExD domain in ZmDCL1 might consist of two isolated segments (57 and 79 amino acids) of amino acid polypeptides, encoded by two different portions of the coding region in maize genome, respectively. According to genomic sequence analysis, these two portions of the coding region were separated by an insertion segment of about 100 kb nucleotide sequence, such as a transposon that probably results in the loss of function of this gene. Additionally, the newly identified DCL locus, Zmdcl4 (GRMZM2G160473), with coding potential of 1,447 amino acid polypeptides, was rearranged and encoded all DCL domains in two different orientations. The third terminal of the gene was inverted and inserted in the first portion of the gene, which was likely a pseudo gene. The SMART and Pfam analysis of the predicted protein sequence revealed that the second RNase III (RNase IIIb) and dsRB domains were inverted and inserted in between the N-terminal DExD and Helicase-C domains.
Based on HMM analysis of PAZ and PIWI conserved domains from the putative polypeptide sequence, a total of 18 AGOs were identified in the maize genome. The length of the Zmago open reading frames (ORFs) varied from 1,665 bp for Zmago1e to 3,309 bp for Zmago1a, with the respective coding potential of 554 and 1,102 amino acids (Table 1). By SMART analysis and NCBI databases, search for the conserved domains revealed that all ZmAGOs shared an N-terminus PAZ domain and a C-terminus PIWI domain, characteristic of the plant AGO proteins. Furthermore, previous studies revealed that the PIWI domain exhibiting extensive homology to RNase H binds the siRNA 5′ end to the target RNA (Höck and Meister 2008) and cleaves target RNAs that exhibit sequence complementary to small RNAs (Rivas et al. 2005; Baumberger and Baulcombe 2005). This is related to three conserved metal-chelating residues in the PIWI domain, aspartate, aspartate and histidine (DDH) (Kapoor et al. 2008). This catalytic triad was firstly revealed in Arabidopsis AGO1, and a conserved histidine at position 798 (H798) was also found to be critical for AGO1 for in vitro endonuclease activity (Baumberger and Baulcombe 2005). In this study, we aligned the PIWI domains of all ZmAGOs with the paralogs, OsAGOs and AtAGOs, in rice and Arabidopsis using CLUSTALX as described in Kapoor et al. (2008) (Fig. 1). The results revealed that there were 11 ZmAGO proteins possessing the conserved DDH/H798 residues. Among the other seven ZmAGOs with missing PIWI domain catalytic residue(s) in the third histidine of ZmAGO1c, ZmAGO2 and ZmAGO5a at the 986th position in AGO1, it remained missing or was replaced by a lysine or aspartate. In ZmAGO4a and ZmAGO4d, the second and/or third catalytic residue (aspartate and/or histidine) was not only missing, but also the AGO1 histidine at the 798th position was replaced by an alanine or proline. In ZmAGO5c, only one catalytic residue (aspartate) was missing, while in ZmAGO18a two catalytic residues (aspartate and histidine) were replaced by histidine and glutamine, respectively (Table 2).

Alignment profile of piwi domain amino acids of maize, rice and Arabidopsis AGO proteins with the ClustalX (1.83) program. The beginning and end positions of the Piwi domains in each protein are marked. The conserved DDH triad residues corresponding to D760, D845 and H986 of Arabidopsis AGO1 are highlighted with downward arrows, while the conserved H residue corresponding to H798 of Arabidopsis AGO1 are boxed
Consistent with the previous reports, the newly identified five ZmRDR proteins share a common sequence motif corresponding to the catalytic β′ subunit of DNA-dependent RNA polymerases (Iyer et al. 2003). The length of the Zmrdr ORFs varied from 2,802 bp for Zmrdr1 to 3,726 bp for Zmrdr5, with a coding potential of 933 and 1,241 amino acids, respectively.
Phylogenetic analysis of DCL, AGO and RDR proteins in maize, rice and Arabidopsis
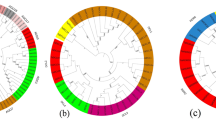
To determine the phylogenetic relationships between DCL, AGO and RDR proteins and to assess the evolutionary history of these families among maize, rice and Arabidopsis, full-length protein sequences from these plants were used to construct an unrooted neighbor-joining phylogenetic tree (Fig. 2).

Phylogenetic relationship among DCL, AGO and RDR proteins of maize, rice and Arabidopsis. a AGO. Unrooted neighbor-joining (NJ) phylogenetic tree of maize, rice and Arabidopsis AGO proteins with bootstrap values shown for each clade. The maize AGOs have been highlighted for each group. Four clades are marked: AGO1, MEL1/AGO5, ZIPPY/AGO7 and AGO4 as reviewed in Kapoor et al. (2008). Protein sequences were downloaded from National Center for Biotechnology Information (NCBI). Accession numbers and abbreviations are as follows: OsAGO1a(Os02g45070), OsAGO1b(Os04g47870), OsAGO1c(Os02g58490), OsAGO 1d(Os06g51310), OsAGO2(Os04g52540), OsAGO3(Os04g52550), OsAGO4a(Os01g16870), OsAGO4b(Os04g06770), OsAGO14(Os07g09020), OsMEL1(Os03g58600), OsAGO13(Os03g57560), OsAGO16(Os07g16224), SHL4(Os03g33650), OsPNH1(Os06g39640), OsAGO17(Os02g07310), OsAGO12(Os03g47820), OsAGO11(Os03g47830), OsAGO18(Os07g28850), OsAGO15(Os01g16850), AtAGO1(At1g48410), AtAGO2(At1g31280), AtAGO3(At1g31290), AtAGO4(At2g27040), AtAGO5(At2g27880), AtAGO6(At2g32940), AtAGO7(At1g69440), AtAGO8(At5g21030), AtAGO9(At5g21150) and AtAGO10 (At5g43810). b DCL. Unrooted NJ phylogenetic tree of maize, rice and Arabidopsis DCL proteins with bootstrap values shown for each clade. The maize DCLs have been highlighted for each group. Four clades are marked: DCL1, DCL2, DCL3 and DCL4 as reviewed in Kapoor et al. (2008). Protein sequences were downloaded from National Center for Biotechnology Information (NCBI). Accession numbers and abbreviations are as follows: OsDCL1a(Os03g02970), OsDCL2a(Os 03g38740), OsDCL2b(Os09g14610), OsDCL3a(Os01g68120), OsDCL3b(Os10g34430), SHO1(Os04g43050), OsDCL1c(Os05g18850), OsDCL1b(Os06g25250); AtDCL1(At1g01040), AtDCL2(At3g03300), AtDCL3 (At3g43920) and AtDCL4(At5g20320). c RDR. Unrooted NJ phylogenetic tree of maize, rice and Arabidopsis RDR proteins with bootstrap values shown for each clade. The maize RDRs have been highlighted for each group. Four clades are marked: RDR1, RDR2, RDR3 and RDR4. Protein sequences were downloaded from National Center for Biotechnology Information (NCBI). Accession numbers and abbreviations are as follows: OsRDR1(Os02g50330), OsRDR2(Os04g39160), OsRDR3(Os01g 10130), OsRDR4(Os01g10140) and SHL2(Os01g34350); AtRDR1(At1g14790), AtRDR2(At4g11 130), AtRDR3(At2g19910), AtRDR4(At2g19920), AtRDR5(At2g19930), and AtRDR6(At3g49500)
The unrooted phylogenetic tree, generated from aligned full-length protein sequences of all 18 ZmAGOs, 19 OsAGOs and 10 AtAGOs, grouped maize, rice and Arabidopsis AGO proteins into four subfamilies, AGO1, MEL1/AGO5, ZIPPY/AGO7 and AGO4 (Fig. 2a), with well-supported bootstrap values. The AGO1 subfamily comprised five maize proteins clustered with the single Arabidopsis protein AtAGO1 and four rice OsAGOs (OsAGO1a, OsAGO1b, OsAGO1c, OsAGO1d), which were designated ZmAGO1a, ZmAGO1b, ZmAGO1c, ZmAGO1d and ZmAGO1e based on the high sequence similarity to AtAGO1 and OsAGO1a–1d. Three additional maize proteins clustered with a single Arabidopsis protein (AtAGO10/PNH) and a rice protein (OsPNH1), closely related to the AtAGO1 and OsAGO1 clade. These proteins were designated ZmAGO10a, ZmAGO10b and ZmAGO10c on the basis of high sequence similarity to AtAGO10 and OsPNH1. The MEL1/AGO5 subfamily contained three maize proteins clustered with one Arabidopsis protein (AtAGO5) and five rice proteins (OsMEL1 and OsAGO11–14). Based on high sequence similarity, the maize proteins were designated ZmAGO5a, ZmAGO5b and ZmAGO5c. The ZIPPY/AGO7 subfamily comprised two maize members designated ZmAGO2 and ZmAGO7, in addition to Arabidopsis and rice. Based on sequence comparisons, ZmAGO2 shared higher similarity with two Arabidopsis proteins (AtAGO2, AtAGO3) and two rice proteins (OsAGO2, OsAGO3), while ZmAGO7 exhibited the highest similarity to Arabidopsis protein AtAGO7 and rice protein SHL4. The AGO4 subfamily shared two highly similar maize members, ZmAGO4a and ZmAGO4d, which exhibited greater similarity with OsAGO4b and OsAGO4a, respectively. These two proteins clustered with four Arabidopsis proteins (AtAGO4, AtAGO6, AtAGO8 and AtAGO9) and four rice members (OsAGO4a, OsAGO4b, OsAGO15 and OsAGO16). Moreover, exclusive of the four subfamilies mentioned above, three maize members displayed high similarity to OsAGO18 and were named ZmAGO18a, ZmAGO18b and ZmAGO18c.
A monophyletic family comprised plant DCL and ZmDCL proteins exhibiting high sequence conservation with their counterparts in Arabidopsis and rice (Fig. 2b). In the DCL1 subfamily, one newly identified ZmDCL protein, designated ZmDCL1, was closely allied with AtDCL1 and OsDCL1a with high similarity. In addition, the other three subfamilies each contained one, two and one newly identified ZmDCL protein. They have been named after their counterparts in Arabidopsis and rice on the basis of increased sequence similarity.
Based on phylogenetic analysis of maize, rice and Arabidopsis RDR proteins, four major classes of RDR proteins were revealed as shown in Fig. 2c. Patterns of monophyletic origin can be hypothesized for members of each class on the basis of high sequence conservation with their counterparts in Arabidopsis and rice. With the exception of the RDR4 subfamily that shares two newly identified ZmRDR proteins, designated ZmRDR4 and ZmRDR5, the other three subfamilies each contained only one member of five newly identified ZmRDR proteins.
Analysis of conserved motifs and domains in DCL, AGO and RDR proteins
The results from the preliminary Pfam analyses of the entire predicted proteins were applied to the MEME motif search tool. This analysis was used to further identify conserved motifs in the corresponding conserved domains of all 17 DCL, 47 AGO and 16 RDR proteins encoded by the three maize, rice and Arabidopsis gene families. The search was performed separately for each family of proteins and 20 conserved motifs were identified (Fig. 3). Moreover, SMART and Pfam were further used to annotate the motifs identified by MEME. The majority of the motifs were well conserved in the DCL family and found in order in almost all four subfamilies of the DCL family from rice and Arabidopsis. However, in the maize DCL4 subfamily, ZmDCL4 was shown having a few C-terminal motifs rearranged, compared to the DCL4 paralogs, OsDCL4 and AtDCL4 from rice and Arabidopsis. This is coincident with the previous results of SMART and Pfam analysis, which revealed that the second RNase III (RNase IIIb) and dsRB domains were inverted and inserted between the N-terminal DExD and Helicase-C domains in the predicted ZmDCL4 protein. Individual and sequential manual motif analyses using SMART and Pfam programs were performed to further elucidate the major functional roles of these conserved motifs in the DCL protein family. Figure 3a shows the annotated conserved motifs 6, 9, 3 and 16 specified for the DExD domain in the N-terminal of the DCL protein; motif 4 for the Helicase-C domain; motifs 10 and 11 for the DUF283 domain; motifs 18, 14 and 13 for the PAZ domain; motifs 5, 8, 7, 1, 15 and 2 for the RNase III domain, and motif 20 for the last dsRB domain in the C-terminal of the DCL protein. These analyses clearly demonstrated that these motifs were well conserved and shared major functional roles in the DCL family of proteins.


Distribution of conserved motifs in maize DCL, AGO and RDR proteins identified using the MEME search tool. Schematic representation of motifs identified in maize DCL, AGO and RDR proteins using MEME motif search tool for each group. Each motif is represented by a number in the box. Box length does not correspond to length of motif. Order of the motifs corresponds to position of motifs in individual protein sequence. For motif detail refer to supplementary material (Table S1)
Manual and MEME analyses of the AGO family identified 16 of 20 conserved motifs in common among all the AGO proteins from maize, rice and Arabidopsis. The MEME motifs matched four common domains in the AGO protein family, which were indicated as DUF1785 (N-terminal), PAZ, MID and PIWI (C-terminal) domains by SMART and Pfam analyses. A detailed scheme is depicted in Fig. 3b: namely, conserved motifs 6 and 20 annotated to specify the first DUF1785 domain in the AGO protein N-terminal; motifs 17, 14 and 9 for the PAZ domain; motifs 15 and 10 for the MID domain; and motifs 8, 13, 11, 7, 3, 5, 2, 1 and 4 for the PIWI domain in the AGO protein C-terminal. The protein motif schemes of the individual AGO family members clearly demonstrated structural similarities among the proteins within the three species examined in this study. Furthermore, these data suggested that the motifs might share major functional roles in these proteins. Although the motif configurations identified by MEME reflected conservation and specificity within the AGO families of maize, rice and Arabidopsis species, we detected some variability distributed between different subfamilies in the individual members of the AGO family. For example, the absence of motif 6 was only found in three AGO proteins, ZmAGO1a and ZmAGO1e from the AGO1 subfamily in maize and OsAGO11 from the MEL1 subgroup in rice, which resulted in a deformity of the DUF1785 domain detected by SMART and Pfam analyses. Motif 6 was also absent in MEL 1 and ZIPPY subfamilies. Furthermore, one and three AGO members were found separately with the respective absence of motif 17 and 14, but the two motifs were well conserved in all other AGO members of the four subfamilies (i.e., representing all the AGO family). Furthermore, for each AGO protein member, at least seven conserved motifs were detected in the PIWI domain by MEME analysis. However, in MEL1 and AGO4 subfamilies, a subset of AGO members shared the absence or duplication of one or two motifs from the PIWI domain. For example, in the AGO4 subgroup, a distinct copy of the newly duplicated motif 2 was located between motifs 7 and 3 in each of the PIWI domains from OsAGO4b, AtAGO4, OsAGO16 and ZmAGO4a.
In the RDR protein family, 15 of 20 motifs were characterized as conserved by MEME analysis (Fig. 3c). Among them, eight distinct MEME motifs were identified as major motifs of the RDR domain that spanned ~400 amino acids among the RDR family members. These annotated motifs included motifs 10, 12, 5, 3, 4, 1, 9 and 2 (Fig. 3c). Although the motifs were well conserved, the protein motif schemes of the individual RDR family members did not always follow the same rules in all subgroups. For example, motifs 12, 3 and 9 shared distinct diversification in the RDR family class III subfamily, different from all other RDR family subfamily members. Motifs 12 and 9 were completely absent in class III subfamily rice and Arabidopsis species. Parallel to the same location, motif 12 could be replaced by a new motif 18 in rice and Arabidopsis, because a similar motif was not detected at the same location in other subfamily members. Moreover, motifs 10, 5, 4, 1 and 2 were well conserved in all members of RDR families from maize, rice and Arabidopsis species. The functional roles of these motifs remain unclear; however, the observed conservation and widespread distribution throughout the subfamily suggest that the motifs are important in protein function.
Chromosomal localization of Zmdcls, Zmagos and Zmrdrs
The physical locations of DCL, AGO and RDR genes in maize were investigated by analysis of genomic distribution on chromosomes (Fig. 4, Table 1). A total of five Zmdcl genes were distributed on four chromosomes. All chromosomes (3, 5 and 10) exhibited single representative of Zmdcl, with the exception of chromosome 1, which contained Zmdcl1 and Zmdcl3b. Of these, a pair of maize orthologs, Zmdcl3a and Zmdcl3b, which share significant homology were found localized on duplicated regions of chromosomes 1 and 3; however, the orthologs of ZmDCL3a and ZmDCL3b in maize are highly divergent showing about 51% similarity at the amino acid level, similar to the DCL3 paralogs, OsDCL3a and OsDCL3b, in rice (Margis et al. 2006; Kapoor et al. 2008). This result revealed that the maize DCL3 orthologs might play a vital role in the evolution of the ZmDCL family in maize.

Chromosomal localization of maize DCL, AGO and RDR genes. A total of 5 Zmdcl, 18 Zmago and 5 Zmrdr genes have been mapped on maize chromosomes according to 5′ and 3′ coordinates mentioned in Maize Genome Database. The respective chromosome numbers are written on the left. Segmentally duplicated genes have been joined using dashed lines, while tandem duplications are indicated by filled triangles
Localization of Zmago on maize chromosomes indicated that the 18 Zmagos families were distributed on eight of the ten chromosomes (Fig. 4). Zmagos were not detected on chromosomes 3 and 4. Four Zmagos were detected on chromosome 2; two each on chromosomes 1, 5, 8, 9 and 10; three on chromosome 6; and one on chromosome 7 (Fig. 4, Table 1). Three pairs of maize Argonautes, Zmago1a/Zmago1e, Zmago1c/Zmago1d and Zmago4a/Zmago4d, were found located in duplicated segments of the genome, while another two gene pairs, Zmago10a/Zmago10c and Zmago18b/Zmago18c, appeared to have undergone tandem duplication on the basis of more than 99% similarity at the amino acid level.
The small family of five Zmrdr genes was distributed on chromosomes 2, 3, 5 and 9. Zmrdr3 and Zmrdr5 were located on chromosome 9, and each of the other Zmrdrs was detected on chromosomes 2, 3 and 5. The Zmrdr genes were not found located in duplicated segments of the genome.
EST expression profiles of Zmdcl, Zmago and Zmrdr gene families in silico
The NCBI EST database provides a large number of ESTs generated from the maize FLcDNA project. These EST data mainly consist of mixed or individual tissue and organ types released by the maize FLcDNA project. In this study, a MEGABLAST search in EST database available at NCBI was performed and resulted in the identification of ESTs for 21 of the total 28 Zmdcl, Zmago and Zmrdr genes (Table 3). Further, on the basis of tissue and organ types, obtained EST data were classified into only five groups including the mixed type. Most of these genes were shown to obtain expression evidence from mixed tissues and organs, but few from other tissues or organs except embryo and endosperm (Table 3). Additionally, some expression evidences of these genes were supported by transcript data in the MAGI and PlantGDB databases (Table 3).
The expression pattern assay of Zmdcl, Zmago and Zmrdr gene families under stress treatment
To confirm these predicted genes and understand their expression profiles under various stress conditions, two abiotic stress treatments (PEG for drought stress and NaCl for salt stress) were investigated. Semi-quantitative RT-PCR analyses on RNA isolated from maize leaves was performed. The results revealed that these genes were differentially expressed in the leaves under either normal condition (control) or stress conditions (15% PEG or 0.15 M NaCl treatment) (Fig. 5, Table 4). On the basis of the brightness of the bands, most of the expressed genes showed increased expression levels in maize leaves by PEG or NaCl treatment, whereas eight members including Zmdcl1, Zmdcl4, Zmago1a, Zmago1c, Zmago5c, Zmago10a, Zmrdr3 and Zmrdr5 showed no obvious increase or even decrease in the leaves of maize under stress conditions. Moreover, four genes, Zmago1e, Zmago2, Zmago5b and Zmago18a, exhibited differential expression with decreased expression levels in maize leaves by PEG treatment and increased expression levels by NaCl treatment. These results demonstrated that these predicted genes exhibit different expression levels in stress treatments.

Semi-quantitative RT-PCR analysis of maize DCL, AGO and RDR genes under stress treatments. Total RNA was extracted from the 21-day-old seedlings germinated on wet filter paper soaked with distilled water and grown in a growth chamber. Either fresh water (1), 15% PEG (2) or 0.15 M NaCl (3) was applied 24 h before harvest. An amplified maize actin gene was used as an internal control
Discussion
Gene expression regulation is associated with RNA interference (RNAi) at the post-transcriptional level and chromatin modification in transcriptional silencing during plant vegetative and reproductive development (Finnegan and Matzke 2003). DCLs, AGOs and RDRs play integral roles in these processes. In this study, a total of 5 Zmdcls, 18 Zmagos and 5 Zmrdrs genes encoding these protein families were identified in maize. Phylogenetic analysis provided insights into the evolution of gene family members and gene multiplicity in maize. EST expression data mining revealed that these newly identified genes had temporal and spatial expression pattern. Furthermore, the transcripts of these genes were detected in the leaves by two different abiotic stress treatments using semi-quantitative RT-PCR. The data demonstrated that these genes exhibited different expression levels in stress treatments. These results provided basic genomic information for these gene families and insights into the probable roles of these genes in plant growth and development.
Maize Dicer-like genes
Dicer or Dicer-like (DCL) proteins are key components in miRNA and siRNA biogenesis pathways and serve to process long double-stranded RNAs into mature small RNAs. Relative to animals and fungi, the notable expansion of DCL family members in monocots and dicots may reflect the deployment of RNA silencing in antiviral defense (Deleris et al. 2006; Margis et al. 2006). For example, A. thaliana encodes four DCL proteins and eight putative DCL proteins that have also been identified in rice (Oryza sativa). Genetic analysis in Arabidopsis has revealed both specialized and overlapping functions of DCL proteins (Fahlgren et al. 2006; Henderson et al. 2006). AtDCL1 and AtDCL3 functions overlap to promote Arabidopsis flowering (Schmitz et al. 2007). It has been revealed that AtDCL2 and AtDCL4 share functional overlap in antiviral defense (Deleris et al. 2006), and AtDCL2, AtDCL3 and AtDCL4 also exhibit overlapping functions in siRNA and tasiRNA production, establishment and maintenance of DNA methylation (Henderson et al. 2006). Unlike the well-characterized DCL proteins in Arabidopsis, little is known about their action in other plants, including rice and maize. The knockdown of OsDCL1 caused pleiotropic phenotypes in rice due to a failure of miRNA metabolism (Schauer et al. 2002; Liu et al. 2005). However, phylogenetic analysis suggested that the functional diversification of DCLs occurred before the divergence of monocots and dicots ~200 million years ago (Henderson et al. 2006; Margis et al. 2006). Therefore, it is possible that during the course of evolution, rice and Arabidopsis DCLs acquired distinct functions in small RNA biogenesis and/or plant development for each species. However, in maize, few reports regarding the identification and functional diversification of the DCL gene family are available.
In the present study, we identified all four subfamilies of DCL genes in maize. Based on phylogenetic analysis of DCL proteins from maize, rice and Arabidopsis, four subfamilies comprising DCL1, DCL2, DCL3 and DCL4 homologs were distinguished. One DCL protein most similar to Arabidopsis AtDCL1 and rice OsDCL1a–1c was identified, as well as two most similar to Arabidopsis DCL3 and rice OsDCL3a–3b and two most similar to DCL2 and DCL4. MEME analyses from conserved motifs in DCL protein families further confirmed similar functional diversification in DCL protein families from maize, rice and Arabidopsis. In view of these similarities, we suggest a similar evolutionary alliance in the functional diversification of the maize DCL gene family and rice/Arabidopsis. However, few reports of the biochemical or genetic analysis of DCL genes in maize are available. Therefore, these data provide a basis for continued research of the maize DCL protein family.
Maize Argonaute genes
The Argonaute protein family was first identified in plants, and members are defined by the presence of PAZ and PIWI domains. AGO proteins are highly conserved among species and many organisms encode multiple family members. In eukaryotes, with the notable exception of fission yeast, most exhibit AGO multigene families, the members of which have specialized biological function, as revealed by a variety of mutant phenotypes (Carmell et al. 2002). In Arabidopsis, the AGO family comprises ten members (Carmell et al. 2002; Fagard et al. 2000), two of which have been unambiguously associated with different forms of RNA silencing. It is therefore likely that functional diversification of RNA silencing is linked to variation between AGO family members, similar to that of animals. AtAGO1 is associated with the miRNA and transgene-silencing pathways (Fagard et al. 2000; Vaucheret et al. 2004), and AtAGO4 with endogenous siRNAs, which affect epigenetic silencing (Zilberman et al. 2004, 2003). In addition, AtAGO7 and AtZLL/AtAGO10 function in the transition from juvenile to adult plant growth phases (Hunter et al. 2003) and meristem maintenance (Moussian et al. 1998; Lynn et al. 1999), respectively. Although a role in sRNA-mediated regulation seems likely, the evidence to support this role is not yet available. Yigit et al. (2006) reported that rice possesses the largest number of AGOs among plants, nearly doubling the number reported in Arabidopsis. However, little is known regarding the functional diversification of monocot AGO family members. In the present study, we investigated all four subfamilies of AGO genes in the maize AGO family. Interestingly, almost the same number of AGO members in rice was found in the maize AGO family, which is equivalent to almost double the number reported in Arabidopsis. We propose that the functional diversification of AGOs occurred after the divergence of monocots and dicots ~200 million years ago. Furthermore, based on the phylogenetic and MEME analyses of conserved motifs of AGO protein families from maize, rice and Arabidopsis, these gene families share major similarities between the species investigated. These results should provide further insights into the functional diversification of the maize AGO gene family.
Argonautes belong to highly basic RNA-binding proteins that include PAZ and PIWI domains. AGO proteins undergo endonuclease activity that is primarily associated with the PIWI domain, which contains three conserved metal-chelating amino acids (DDH). Interestingly, many AGO proteins are endonucleolytically inactive, although the catalytic residues are conserved. For instance, in Arabidopsis and rice, there are 5 and 11 AGO genes, which do not code for the conserved catalytic residues, respectively. Also, in maize PIWI domains, we identified seven genes lacking the conserved catalytic residues. The absence of conserved catalytic residues could lead to loss of function of target RNA processing by endonucleolytic cleavage in these proteins (Kapoor et al. 2008).
Maize RNA-dependent RNA polymerase genes
RNA-dependent RNA polymerases usually amplify RNAi silencing signals by generating more aberrant RNA population (Sijen et al. 2001). The proteins can catalyze the formation of phosphodiester bonds between ribonucleotides in an RNA template-dependent fashion. Astier-Manifacier and Cornuet (1971) first reported the activity of RDR in Chinese cabbage. Since then, several RDR gene paralogs have been identified in many other plant species, including Arabidopsis and rice. In Arabidopsis, at least three RDR types serve in distinct and overlapping biological processes such as viral defense, chromatin silencing and PTGS (Kapoor et al. 2008). Of these, AtRDR1s were elicited by salicylic acid (SA) or viral infection and were reported to be involved in antiviral defense in several other plants (Yu et al. 2003; Chan et al. 2004; Xie et al. 2001; Jovel et al. 2007). Furthermore, AtRDR2 plays a critical role in RNA-directed DNA methylation and repressive chromatin modifications on certain transgenes, endogenous genes and centromeric repeats that correlate with the production of 24-nt interfering RNAs (Matzke et al. 2007; Zaratiegui et al. 2007). In addition, AtRDR6 amplifies some aberrant RNAs generated from transgenes or inverted repeats to trigger degradation of complementary RNA species (Luo and Chen 2007). Moreover, the Arabidopsis RDR2 homolog in maize, MOP1 (mediator of paramutation1), was essential for paramutation at the b1, pl1 and r1 loci (Dorweiler et al. 2000) and was involved in the maintenance of Mutator transposon silencing and the silencing of certain transgenes (Lisch et al. 2002; McGinnis et al. 2006). In a recently study, MOP1 was found to play a significant role in regulating the expression of not only transposons, but also of genes (Jia et al. 2009). In this study, we identified five maize RDR gene family members. Similar to rice and Arabidopsis, these genes were clustered into four distinct subgroups. Phylogenetic and MEME analyses of conserved motifs indicated the presence of maize corresponding RDR orthologs in each subgroup from rice and Arabidopsis RDR gene families. These results suggested that the RDR gene families of maize, rice and Arabidopsis diverged from the same common ancestor and therefore performed a similar function in all three taxa.
EST expression profiles of Zmdcl, Zmago and Zmrdr genes in silico
Gene expression in silico data from EST databases play an increasingly vital role in providing gene expression research information. This facilitates the identification of gene function and future functional genomic studies in plant growth and development. In this study, we investigated the expression profiles of Zmdcl, Zmago and Zmrdr genes through several approaches by EST database. These results revealed that Zmdcl, Zmago and Zmrdr genes exhibited distinct expression patterns in different tissues or organs. One explanation is that some of these investigated genes may have temporal and spatial expression pattern, which varies with tissue types, developmental stages or genotypes of maize. For example, some genes exhibit a tissue-specific expression pattern in embryo relative to other tissues, which suggests that they may function in embryogenesis. Moreover, some genes investigated in this study exist in gene rearrangement or partial motifs absence in gene structure. These changes might result in distinct expression pattern, and the genes were also deemed to belong to pseudogenes.
The expression pattern of Zmdcl, Zmago and Zmrdr gene families under stress treatment
In this study, the expression analyses of semi-quantitative RT-PCR showed that maize Zmdcl, Zmago and Zmrdr genes exhibited different expression levels under two different abiotic stress treatments. For 16 candidate genes, the expression levels increased after applying 15% PEG or 0.15 M NaCl treatment than for controls, suggesting that these genes might play important roles in plant RNAi regulation, especially those showing strong response to the two abiotic stress conditions in this study. In contrast, those genes (Zmdcl1, Zmdcl4, Zmago1a, Zmago1c, Zmago5c, Zmago10a, Zmrdr3 and Zmrdr5) that showed lower expression in stress treatments had a high possibility to contribute to maize RNAi regulation by only expressing under specific conditions or in specific tissues other than seedling leaves, and these remain to be further confirmed experimentally. Furthermore, those genes (Zmago1e, Zmago2, Zmago5b and Zmago18a) exhibiting distinct expression patterns under different stress conditions might play vital roles in evolving specialized regulatory mechanisms in response to different abiotic stresses (Xie et al. 2004).
References
Astier-Manifacier S, Cornuet P (1971) RNA-dependent RNA polymerase in Chinese cabbage. Biochim Biophys Acta 232:484–493
Bailey TL, Elkan C (1995) The value of prior knowledge in discovering motifs with MEME. Proc Int Conf Intell Syst Mol Biol 3:21–29
Baumberger N, Baulcombe DC (2005) Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits mRNAs and siRNAs. Proc Natl Acad Sci USA 102:11928–11933
Bernstein E, Caudy AA, Hammond SM, Harmon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409(6818):363–366
Carmell MA, Xuan Z, Zhang M, Hannon GJ (2002) The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev 16:2733–2742
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Science 301:336–338
Chan SWL, Zilberman D, Xie Z, Johansen LK, Carrington JC, Jacobsen SE (2004) RNA silencing genes control de novo DNA methylation. Science 303:1336
Chapman EJ, Carrington JC (2007) Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet 8:884–896
Cogoni C, Macino G (1999) Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature 399:166–169
Dalmay T, Hamilton A, Rudd S, Angell S, Baulcombe DC (2000) An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 101:543–553
Deleris A, Gallego-Bartolome J, Bao J, Kasschau KD, Carrington JC, Voinnet O (2006) Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. Science 313:68–71
Djupedal I, Ekwall K (2009) Epigenetics: heterochromatin meets RNAi. Cell Res 19:282–295
Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, Chandler VL (2000) Mediator of paramutation1 is required for establishment and maintenance of paramutation at multiple maize loci. Plant Cell 12:2101–2118
Eddy SR (2008) A probabilistic model of local sequence alignment that simplifies statistical significance estimation. PloS Comput Bio l4(5):e1000069
Fagard M, Boutet S, Morel J-B, Bellini C, Vaucheret H (2000) AG01, QDE-2, and RDE-1 are related proteins required for post-transcriptional gene silencing in plants, quelling in fungi, and RNA interference in animals. Proc Natl Acad Sci USA 97:11650–11654
Fahlgren N, Montgomery TA, Howell MD, Allen E, Dvorak SK, Alexander AL, Carrington JC (2006) Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr Biol 16:939–944
Finnegan EJ, Matzke MA (2003) The small RNA world. J Cell Sci 116:4689–4693
Girard A, Sachidanandam R, Hannon GJ, Carmell MA (2006) A germline specific class of small RNAs binds mammalian Piwi proteins. Nature 442(7099):199–202
Großhans H, Filipowicz W (2008) The expanding world of small RNAs. Nature 451:414–416
Gu Z, Cavalcanti A, Chen FC, Bouman P, Li WH (2002) Extent of gene duplication in the genomes of drosophila, nematode, and yeast. Mol Biol Evol 19:256–262
Hammond SM, Boettcher S, Caudy AA, Kobayashi R, Hannon GJ (2001) Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 293:1146–1150
Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, Jacobsen SE (2006) Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 38(6):721–725
Höck J, Meister G (2008) The Argonaute protein family. Genome Biol 210(9):1–8
Hunter C, Sun H, Poethig RS (2003) The Arabidopsis heterochronic gene ZIPPY is an ARGONAUTE family member. Curr Biol 13:1734–1739
Hutvagner G, Simard MJ (2008) Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol 9:22–32
Iyer LM, Koonin EV, Aravind L (2003) Evolutionary connection between the catalytic subunits of DNA-dependent RNA polymerases and eukaryotic RNA-dependent RNA polymerases and the origin of RNA polymerases. BMC Struct Biol 3(1):1
Jia Y, Lisch DR, Ohtsu K, Scanlon MJ, Nettleton D, Schnable PS (2009) Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet 5(11):e1000737
Jovel J, Walker M, Sanfacon H (2007) Recovery of Nicotiana benthamiana plants from a necrotic response induced by a nepovirus is associated with RNA silencing but not with reduced virus titer. J Virol 81:12285–12297
Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S (2008) Genome-wide identification, organization and phylogenetic analysis of dicer-like, argonaute and RNA-dependent RNA polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genomics 451(9):1471–2164
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 5:150–163
Lai EC (2003) microRNAs: Runts of the genome assert themselves. Curr Biol 13:925–936
Lisch D, Carey CC, Dorweiler JE, Chandler VL (2002) A mutation that prevents paramutation in maize also reverses Mutator transposon methylation and silencing. Proc Natl Acad Sci USA 99:6130–6135
Liu B, Li P, Li X, Liu C, Cao S, Chu C, Cao X (2005) Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol 139:296–305
Luo Z, Chen Z (2007) Improperly terminated, unpolyadenylated mRNA of sense transgenes is targeted by RDR6-mediated RNA silencing in Arabidopsis. Plant Cell 19(3):943–958
Lynn K, Fernandez A, Aida M, Sedbrook J, Tasaka M, Masson P, Barton MK (1999) The PINHEAD/ZWILLE gene acts pleiotropically in Arabidopsis development and has overlapping functions with the ARGONAUTE1 gene. Development (UK) 126:469–481
MacRae IJ, Doudna JA (2007) Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol 17:138–145
Margis R, Fusaro AF, Smith NA, Curtin SJ, Watson JM, Finnegan EJ, Waterhouse PM (2006) The evolution and diversification of dicers in plants. FEBS Lett 580(10):2442–2450
Matzke M, Kanno T, Huettel B, Daxinger L, Matzke AJ (2007) Targets of RNA-directed DNA methylation. Curr Opin Plant Biol 10:512–519
McGinnis KM, Springer C, Lin Y, Carey CC, Chandler V (2006) Transcriptionally silenced transgenes in maize are activated by three mutations defective in paramutation. Genetics 173:1637–1647
Millar AA, Waterhouse PM (2005) Plant and animal microRNAs: similarities and differences. Funct Integr Genomics 5:129–135
Moazed D (2009) Small RNAs in transcriptional gene silencing and genome defence. Nature 457:413–420
Mourrain P, Beclin C, Elmayan T, Feuerbach F, Godon C, Morel J-B, Jouette D, Lacombe AM, Nikic S, Picault N (2000) Arabidopsis SGS2 and SGS3 genes are required for posttranscriptional gene silencing and natural virus resistance. Cell 101(5):533–542
Moussian B, Schoof H, Haecker A, Jurgens G, Laux T (1998) Role of the ZWILLE gene in the regulation of central shoot meristem cell fate during Arabidopsis embryogenesis. EMBO J 17:1799–1809
Nicholson AW (2003) The ribonuclease III family: forms and functions in RNA maturation, decay, and gene silencing. In: Hannon GJ (ed) RNAi: a guide to gene silencing, vol 8. Cold Spring Harbor Laboratory Press, New York, pp 149–174
Peters L, Meister G (2007) Argonaute proteins: mediators of RNA silencing. Mol Cell 26(8):611–623
Rivas FV, Tolia NH, Song JJ, Aragon JP, Liu J, Hannon GJ, Joshua-Tor L (2005) Purified Argonaute2 and an siRNA form recombinant human RISC. Nat Struct Mol Biol 12(4):340–349
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schauer SE, Jacobsen SE, Meinke DW, Ray A (2002) DICER-LIKE1: Blind men and elephants in Arabidopsis development. Trends Plant Sci 7:487–491
Schmitz RJ, Hong L, Fitzpatrick KE, Amasino RM (2007) DICER-LIKE 1 and DICER-LIKE 3 redundantly act to promote flowering via repression of FLOWERING LOCUS C in Arabidopsis thaliana. Genetics 176:1359–1362
Schnable PS, Ware D, Fulton RS et al (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326:1112–1115
Sijen T, Fleenor J, Simmer F, Thijssen KL, Parrish S, Timmons L, Plasterk RH, Fire A (2001) On the role of RNA amplification in dsRNA-triggered gene silencing. Cell 107(4):465–476
Smardon A, Spoerke JM, Stacey SC, Klein ME, Mackin N, Maine EM (2000) EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr Biol 10:169–178
Thompson J, Higgins D, Gibson T (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Vaucheret H, Vazquez F, Crete P, Bartel DP (2004) The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev 18:1187–1197
Wassenegger M, Heimes S, Riedel L, Sanger HL (1994) RNA-directed de novo methylation of genomic sequences in plants. Cell 76(3):567–576
Xie Z, Fan B, Chen C, Chen Z (2001) An important role of an inducible RNA-dependent RNA polymerase in plant antiviral defense. Proc Natl Acad Sci USA 98:6516–6521
Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, Jacobsen SE, Carrington JC (2004) Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2(5):E104
Yang S, Zhang X, Yue J, Tian D, Chen J (2008) Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol Genet Genomics 280:187–198
Yigit E, Batista PJ, Bei Y, Pang KM, Chen CC, Tolia NH, Joshua-Tor L, Mitani S, Simard MJ, Mello CC (2006) Analysis of the C. elegans Argonaute family reveals that distinct argonautes act sequentially during RNAi. Cell 127(4):747–757
Yu D, Fan B, MacFarlane SA, Chen Z (2003) Analysis of the involvement of an inducible Arabidopsis RNA-dependent RNA polymerase in antiviral defense. Mol Plant Microb Interact 16:206–216
Zaratiegui M, Irvine DV, Martienssen RA (2007) Noncoding RNAs and gene silencing. Cell 128:763–776
Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W (2004) Single processing center models for human dicer and bacterial RNase III. Cell 118:57–68
Zilberman D, Cao X, Jacobsen SE (2003) Argonaute 4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299:716–719
Zilberman D, Cao X, Johansen LK, Xie Z, Carrington JC, Jacobsen SE (2004) Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr Biol 14:1214–1220
Acknowledgments
This study was supported by grants from the National High-Tech Research and Development Program (863 Program) (No. 2008AA10Z408) and the National Natural Science Foundation of China (No. 11075001 and No. 30771309). We wish to thank the two anonymous reviewers for their helpful comments on this manuscript. We also acknowledge Professor Beijiu Cheng for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by B. Li.
Y. Qian and Y. Cheng contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Qian, Y., Cheng, Y., Cheng, X. et al. Identification and characterization of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families in maize. Plant Cell Rep 30, 1347–1363 (2011). https://doi.org/10.1007/s00299-011-1046-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-011-1046-6