
Abstract
An efficient and reproducible protocol is described for the regeneration of Astragalus melilotoides protoplasts isolated from hypocotyl-derived embryogenic calli. Maximum protoplast yield (11.74±0.6×105/g FW) and viability (87.07±2.8%) were achieved using a mixture of 2% (w/v) Cellulase Onozuka R10, 0.5% (w/v) Cellulase Onozuka RS, 0.5% (w/v) Macerozyme R10, 0.5% (w/v) Hemicellulase, and 1% (w/v) Pectinase, all dissolved in a cell protoplast wash (CPW) salt solution with 13% (w/v) sorbitol. First divisions occurred 3–7 days following culture initiation. The highest division frequency (9.86±0.68%) and plating efficiency (1.68±0.05%) were obtained in solid-liquid medium (KM8P) supplemented with 1.0 mg/l 2,4-dichlorophenoxyacetic acid, 0.5 mg/l 6-benzylaminopurine (BA), 0.2 mg/l kinetin, 0.2 M glucose, 0.3 M mannitol and 500 mg/l casein hydrolysate. Upon transfer to MS medium with 0.5 mg/l α-naphthaleneacetic acid and 1-2 mg/l BA, the protoplast-derived calli produced plantlets via somatic embryogenesis (56.3±4.1%) and organogenesis (21.6±0.6%). Somatic embryos or adventitious shoots developed into well-rooted plantlets on MS medium without any plant growth regulators or supplemented with 3.0 mg/l indole-3-butyric acid, respectively. About 81% of the regenerants survived in soil, and all were normal with respect to morphology and growth characters.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Astragalus melilotoides Pall., an important perennial forage legume, is widely cultivated in many of the arid regions of northern China for improving pasture and in crop rotation because of its drought-, cold- and disease resistance, soil- and water-holding capacity, and sand-fixation characteristics (Guo et al. 1987). Unfortunately, it is not as palatable as some of the other legumes such as alfalfa and sainfoin. Moreover, its long growth period and rather late maturity give rise to poor seeds with respect to both quality and yield. These disadvantages have hindered the further exploitation of this species.
Genetic manipulations such as somatic hybridization, direct gene transfer and mutant selection may provide useful approaches for the improvement of this species. An efficient system for plant regeneration from protoplasts is a crucial prerequisite for protoplast-based biotechnologies. To the best of our knowledge, only a few successful protoplast-to-plant regeneration systems have been established in the genus Astragalus at present (Luo and Jia 1998), although there have been numerous reports on plant regeneration from protoplasts in several other leguminous species (Xu et al. 1991; Puite 1992). In vitro regeneration of A. melilotoides from internode-segment-derived calli has been reported by Chen et al. (2001). We describe here a reliable system for plant regeneration of A. melilotoides from isolated embryogenic callus protoplasts via somatic embryogenesis and organogenesis.
Materials and methods
Plant material and callus induction
Seeds of Astragalus melilotoides were obtained from the Ningxia Shapotou Desert Control Center of the Chinese Academy of Sciences. The seeds were surface-sterilized in 70% ethanol for 30 s and then in 0.1% mercuric chloride solution for 10 min, washed with sterile distilled water at least six times and germinated aseptically in 100-ml glass Erlenmeyer flasks containing 30-ml aliquots of agar-solidified MS (Murashige and Skoog 1962) medium without plant growth regulators. For inducing callus formation, we cut the hypocotyls from 2-week-old seedlings (2–3 cm in length) into 5-mm-long segments and then inoculated these segments on agar-solidified MS medium supplemented with 2.0 mg/l 2,4-D, 0.5 mg/l BA and 500 mg/l CH. All culture media contained 3% (w/v) sucrose and 0.8% (w/v) agar, and the pH was adjusted to 5.6 before autoclaving at 121°C for 20 min. Following a 3-week culture on callus induction medium, friable and yellow-green embryogenic calli formed, which were then detached from the explants and subcultured every 3 weeks on MS medium supplemented with 1.0 mg/l 2,4-D, 0.25 mg/l BA, 500 mg/l CH, 3% (w/v) sucrose, and 0.8% (w/v) agar at pH 5.6. Seed germination, callus induction, and subculture were carried out at 25±1°C under a 16/8-h (light/dark) photoperiod with light supplied at an intensity of 30 μmol m−2 per second.
Protoplast isolation
Approximately 1 g of 10-day-old friable, yellow-green embryogenic callus was incubated in 10 ml of enzyme solution at 25±1°C in the dark. The five different enzyme digestion solutions tested contained different concentrations of Cellulase Onozuka R10 (Yakult Pharmaceutical, Tokyo, Japan), Macerozyme Onozuka R10 (Kinki Yakult, Mishinomiya, Japan), Cellulase Onozuka RS (Yakult Honsha, Tokyo, Japan), Pectinase (Serva), and Hemicellulase H 2125 (Sigma, St. Louis, Mo.), all in a cell protoplast wash (CPW) salt solution (Frearson et al. 1973) with 13% (w/v) sorbitol, pH 5.6 (Table 1). The enzyme solution was filter-sterilized through a 0.45-μm Millipore filter. After incubation for 10 h, the enzyme-protoplast mixture was gently shaken and filtered through a 45-μm stainless steel sieve under aseptic conditions to remove large debris and undigested tissue. The protoplasts subsequently released were collected by centrifugation at 70 g for 5 min. The pellets were resuspended in 3 ml of autoclaved protoplast wash solution consisting of 0.16 M CaCl2 and 0.05% (w/v) MES at pH 5.6, and the protoplast suspension was layered on the top of 5 ml autoclaved sucrose solution containing 16% (w/v) sucrose and 0.05% MES, pH 5.6. Following centrifugation at 70 g for 15 min, the purified protoplasts were concentrated on the interface between the two solutions, while debris was pelleted to the bottom of the sucrose solution. The protoplast band was collected using a pipette, suspended in the wash solution described above and then washed twice with the same solution and once with protoplast culture medium (see below) by centrifugation at 70 g for 5 min. Protoplast yield (protoplasts per milliliter of culture medium) was determined according to the improved method of Hou and Jia (2002). Protoplast viability was assessed by the FDA staining procedure of Widholm (1972). Different lengths of incubation time were tested to find the optimum period for protoplast isolation.
Protoplast culture
The purified protoplasts were cultured at a density of 4–5×105 protoplasts/ml in liquid KM8P medium (Kao and Michayluk 1975) containing 0.2 M glucose, 0.3 M mannitol, 500 mg/l CH, 1.0 mg/l 2,4-D, 0.5 mg/l BA, with or without 0.2 mg/l KIN. All media were filter-sterilized (0.45-μm pore size) and the pH adjusted to 5.6. The protoplasts were dispersed in a 6-cm petri dish containing 2.0 ml of KM8P medium. Liquid culture and solid-liquid double layer cultures were used. The solid-liquid double layer culture system consisted of 2 ml of protoplast suspension (in liquid KM8P medium) being layered onto 1.5 ml of the same medium solidified with 0.4% (w/v) agarose in a 6-cm petri dish. All cultures were incubated at 25±1°C in the dark. After 1 week of culture, the osmotic pressure of the liquid medium was gradually reduced by adding 0.2 ml of the initial protoplast culture medium to each dish once per week. The composition of the supplemental medium was the same as that of the initial culture medium, except that the concentration of glucose and mannitol were lowered to 50% and 30%, respectively. Protoplast division frequency (number of dividing protoplasts/total survival protoplasts ×100) and plating efficiency (the number of visible microcalli/total protoplasts plated ×100) were evaluated after the protoplasts were cultured for 2 weeks and 5 weeks, respectively. Cell divisions and the growth of protoplast-derived microcolonies were examined under an inverted microscope during the first 5 weeks of culture. After 5 weeks of liquid culture, the cell colonies were transferred onto a 0.4% agarose-solidified KM8P medium for further growth. The small visible calli, approximately 2–3 mm in diameter, were picked up with forceps and transferred onto a solidified MS medium supplemented with 1.0 mg/l 2,4-D, 0.5 mg/l BA, 500 mg/l CH, 3% (w/v) sucrose, and 0.8% (w/v) agar, pH 5.6. The calli produced were subcultured at 3-week intervals.
Plantlet regeneration
For inducing plantlet regeneration, the protoplast-derived calli were transferred onto MS medium containing 500 mg/l CH, 3% (w/v) sucrose, 0.8% (w/v) agar, and different combinations of NAA and BA, pH 5.6 (Table 4). Each treatment was repeated three times, and ten pieces of calli were used each time. After 5 weeks of culture, data on the frequency of somatic embryo formation and adventitious shoot regeneration were recorded. Cultures with embryoids were transferred onto a hormone-free MS medium for further development, and those with adventitious shoots were transferred on MS medium supplemented with 3.0 mg/l IBA for rooting. All cultures were maintained at 25±1°C under a 16/8-h (light/dark) photoperiod with cool-white fluorescence light supplied at an intensity of 30 μmol m−2 per second. The regenerated plantlets with well-developed roots were subsequently transplanted into soil.
Results and discussion
Establishment of embryogenic calli
The original calli that formed from the hypocotyl explants were yellow-brown. Three weeks after inoculation soft friable yellow-green embryogenic calli appeared on the surface. When detached from the explants and subcultured on MS medium with a lower concentration of 2,4-D (1.0 mg/l) and BA (0.25 mg/l) for two to three passages (3 weeks each passage), they rapidly proliferated and were ready for protoplast isolation. The establishment of such embryogenic calli was an excellent source of protoplasts capable of regeneration in A. melilotoides. Friable embryogenic calli and embryogenic suspensions have also been found to be a good source of material for protoplast isolation, culture and subsequent regeneration of plants in many species, such as wheat (Triticum aestivum L.) (Gu and Liang 1997; Li et al. 1999), rice (Oryza sativa L.) (Tsugawa and Suzuki 2000; Tang et al. 2001), and cassava (Manihot esculenta Crantz) (Sofiari et al. 1998).
Protoplast isolation
Protoplasts could be easily released using soft, friable, and young embryogenic calli. The freshly isolated protoplasts were bright and spherical with a densely organized cytoplasm (Fig. 1A) and showed no fluorescence when stained with calcoflower white (data not shown). In order to obtain large quantities of viable protoplasts, we tested different combinations of enzymes to find the optimum isolating conditions. It can be seen from Table 1 that culture in group B, which contained 2% (w/v) Cellulase R10, 0.5% (w/v) Cellulase RS, 0.5% (w/v) Macerozyme, 0.5% (w/v) Hemicellulase, and 1% (w/v) Pectinase, resulted in the highest yield (11.74±0.6×105/g FW) and viability (87.07±2.8%) of A. melilotoides protoplasts. When the concentration of Cellulase R10 was decreased to 1% or increased to 3%, the yield (groups A, E) and viability (group E) of the protoplasts significantly decreased. The removal of Cellulase RS (group C) or Pectinase (group D) from the enzyme mixture also resulted in lower quantities and/or poorer viability of the protoplasts.

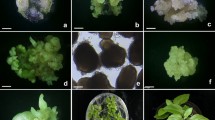
Regeneration of plantlets from callus-derived protoplasts of Astragalus melilotoides. A Freshly isolated protoplasts from callus culture (bar: 30 μm), B first division of a protoplast after 3 days of culture (bar: 40 μm), C, D second division of protoplast-derived cells after 8 days of culture (bar: 40 μm), E, F protoplast-derived cell colony after 3 weeks of culture (bar: 50 μm), G protoplast-derived microcalli on agarose-solidified KM8P medium after 3 weeks of culture (bar: 5 mm), H plantlets regenerated from protoplast-derived calli via either somatic embryogenesis or organogenesis (bar: 10 mm), I plantlets from protoplast-derived calli via somatic embryogenesis in early stage (bar: 1 mm), J a complete plantlet with well-developed roots and shoots (bar: 15 mm)
As shown in Table 2, protoplast yield and viability increased when the duration of enzyme digestion was increased from 8 h to 12 h—especially to 10 h. Protoplast yield and the percentage of viable protoplasts at 10 h reached 10.89±0.3×105/g FW and 88.01±3.3%, respectively. Thus, the source of the calli, concentration of the enzymes, and the length of the incubation period influenced protoplast yield and viability. We subsequently concluded that the optimal protocol for A. melilotoides protoplast yield and viability consisted of the incubation of 1 g of fresh embryogenic callus for 10 h in 10 ml of enzyme solution containing 2% (w/v) Cellulase R10, 0.5% (w/v) Cellulase RS, 0.5% (w/v) Macerozyme R10, 0.5% (w/v) Hemicellulase, and 1% (w/v) Pectinase, in CPW salts with 13% (w/v) sorbitol.
Protoplast culture
No visible changes in the shape of the cultured protoplasts were evident during the first day. After 2 days of culture, some protoplasts increased in volume and became elongated, indicating the formation of a new cell wall. The first divisions of the protoplasts occurred after 3-7 days in both liquid and solid-liquid double layer media (Fig. 1B), and the second divisions were observed after 8 days (Fig. 1C, D). There were no differences between the two culture methods with respect to any obvious effect on the time course of protoplast division. As shown in Table 3, the frequency of division after 2 weeks in solid-liquid culture was about twofold that observed in liquid culture under the same conditions. The highest division frequency (9.86±0.68%) was obtained in solid-liquid medium containing 1.0 mg/l 2,4-D, 0.5 mg/l BA and 0.2 mg/l KIN. In contrast, when the protoplasts were cultured on a medium without KIN, the frequency of protoplast division declined (Table 3). After 3 weeks of culture, protoplast-derived colonies were formed (Fig. 1E, F). A gradual reduction in the osmotic concentration of the culture medium promoted continuous division of protoplast-derived cell colonies. After 5 weeks, the cell colonies were transferred onto agarose-solidified KM8P medium and cultured further for an additional 3 weeks, after which microcalli about 2–3 mm in diameter developed on all media (Fig. 1G). These microcalli were transferred to MS medium supplemented with 1.0 mg/l 2,4-D, 0.5 mg/l BA, 500 mg/l CH, 3% (w/v) sucrose and 0.8% (w/v) agar where they grew faster and formed friable yellowish calli within 3–6 weeks. Plating efficiency on the solid-liquid medium with KIN reached 1.68±0.05% (Table 3). Thus, culture in the solid-liquid culture system with KIN resulted in both improved division frequency and plating efficiency. In addition, the combination of 2,4-D and a low level of KIN was beneficial for protoplast division and microcalli formation of A. melilotoides protoplasts. The result is similar to those observed in several other species—goosegrass (Yemets et al. 2003) and Taxus yunnanensis (Luo et al. 1999).
Plantlet regeneration
Plant regeneration via somatic embryogenesis and organogenesis was observed following the transfer of microcalli to MS medium supplemented with NAA and BA (Table 4, Fig. 1H). The highest induction frequency of somatic embryos (56.3±4.1%) was obtained within 3 weeks of calli being cultured on MS medium with 0.5 mg/l NAA and 1.0 mg/l BA. After an additional 2 weeks of culture, 84% of the somatic embryos developed into plantlets 0.5–2.0 cm in length (Fig. 1H, I). An average of about 5.4 somatic embryos was formed from 1 g callus on the same medium (Table 4). Following transfer onto hormone-free MS medium, the plantlets grew quickly (Fig. 1J), and a lower level of NAA and relatively higher level of BA enhanced somatic embryogenesis in A. melilotoides. This is in agreement with earlier reports in several other species (Sharma and Kumar 1994; Luo and Jia 1998). In addition, about 21.6% of calli produced adventitious buds and subsequently 1.0- to 1.5-cm-long shoots on the most favorable organogenesis medium, which contained 0.5 mg/l NAA and 2.0 mg/l BA (Table 4). After transfer to MS medium with 3.0 mg/l IBA, the adventitious shoots differentiated roots, and several plantlets were produced 2 weeks later. All well-developed plantlets were transplanted to soil, and about 81% of them survived under greenhouse conditions and showed no visible abnormalities with respect to morphology and growth characteristics.
In conclusion, an efficient and reproducible system for plant regeneration from protoplasts isolated from embryogenic calli of A. melilotoides has been established. The plantlets were regenerated from protoplast-derived calli simultaneously through somatic embryogenesis and organogenesis. This protocol may be useful for the improvement of A. melilotoides by means of somatic hybridization and protoplast-based genetic transformation.
Abbreviations
- BA::
-
6-Benzylaminopurine
- CH::
-
Casein hydrolysate
- CPW::
-
Cell protoplast wash
- 2,4-D::
-
2,4-Dichlorophenoxyacetic acid
- FDA::
-
Fluorescein diacetate
- IBA::
-
Indole-3-butyric acid
- KIN::
-
Kinetin
- MES::
-
2-(N-morpholino) Ethanesulphonic acid
- NAA::
-
α-Naphthaleneacetic acid
References
Chen G, Jia JF, Jin H, Hao JG (2001) Establishment of a high efficient regeneration system from internode segments of seedling in Astragalus melilotoides Pall. (in Chinese). Acta Bot Boreal Occident Sin 21:136–141
Frearson EM, Power JB, Cocking EC (1973) Isolation, culture and regeneration of Petunia leaf protoplasts. Dev Biol 33:130–137
Gu X, Liang GH (1997) Plantlet regeneration from protoplast-derived haploid embryogenic calli of wheat. Plant Cell Tissue Organ Cult 50:139–145
Guo BZ, Zhang HZ, Pan JT, Yang YC, Wu ZL, He TN, Zhou LH, Huang RF (1987) Flora of Qinghai economic plants, section I. Qinghai People’s Press, Xining, pp 312–314
Hou SW, Jia JF (2002) An easy way to count plant protoplast (in Chinese). Plant Physiol Commun 38:57
Kao KN, Michayluk MR (1975) Nutritional requirement for growth of Vicia hajastana cells and protoplasts at very low population density in liquid media. Planta 126:105–110
Li H, Machii H, Hagio T, Takezaki A, Hirabayashi T (1999) Plant regeneration from protoplasts of Triticum aestivum L. cv. Nakasoushu. Plant Cell Tissue Organ Cult 58:119–125
Luo JP, Jia JF (1998) Plant regeneration from callus protoplasts of the forage legume Astragalus adsurgens pall. Plant Cell Rep 17:313–317
Luo JP, Mu Q, Gu YH (1999) Protoplast culture and paclitaxel production by Taxus yunnanensis. Plant Cell Tissue Organ Cult 59:25–29
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Puite KJ (1992) Progress in plant protoplast research. Physiol Plant 85:403–410
Sharma A, Kumar A (1994) Somatic embryogenesis and plant regeneration from leaf-derived cell suspension of a mature tree Thevetia peruviana L. Plant Cell Rep 14:171–174
Sofiari E, Racmakers CJJM, Bergervoet JEM, Jacobsen E, Visser RGF (1998) Plant regeneration from protoplasts isolated from friable embryogenic callus of cassava. Plant Cell Rep 18:159–165
Tang K, Sun X, An D, Power JB, Cocking EC, Davey MR (2001) A simple and rapid procedure to establish embryogenic cell suspensions as a source of protoplast for efficient plant regeneration from two Chinese commercial rice cultivars. Plant Cell Tissue Organ Cult 66:149–153
Tsugawa H, Suzuki M (2000) A low-temperature method for maintaining plant regeneration activity in embryogenic callus of rice (Oryza sativa L.). Plant Cell Rep 19:371–375
Widholm JM (1972) The use of fluorescein diacetate and phenosafranine for determining viability of cultured plant cells. Stain Technol 47:189–194
Xu ZH (1991) Protoplast culture in pulses. In: Sun YR, An XP (eds) Plant protoplast culture. Science Press, Beijing, pp 72–85
Yemets AI, Klimkina LA, Tarassenko LV, Blume YB (2003) Efficient callus formation and plant regeneration of goosegrass [Eleusine indica (L.) Gaertn.]. Plant Cell Rep 21:503–510
Acknowledgements
We acknowledge the financial support of the National Science Foundation of China (contribution no. 30070366) and the Science Foundation of Gansu Province, China (contribution no. ZS011-A25–009-Z). We also thank Mr. Qing-Xiang Gao for his technical assistance with the photographs and helpful advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Altman
Rights and permissions
About this article
Cite this article
Hou, S.W., Jia, J.F. Plant regeneration from protoplasts isolated from embryogenic calli of the forage legume Astragalus melilotoides Pall.. Plant Cell Rep 22, 741–746 (2004). https://doi.org/10.1007/s00299-004-0760-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-004-0760-8