
Abstract
In this report, we present a rare case of a 52-year-old man with a unique form of hypertrophic pachymeningitis involving the anterior part of the falx and who was positive for rheumatoid factor. The clinical symptom was only headache, without any cranial nerve palsies or ataxia. Diagnosis was made by gallium scintigraphy and magnet resonance imaging but was not confirmed by dural biopsy. Treatment with corticosteroid alone was extremely effective for him, while in most cases hypertrophic pachymeningitis recurs or progresses despite the treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cranial hypertrophic pachymeningitis (HP) is a rare disease characterizing an inflammatory hypertrophy of the dura mater that typically can cause progressive cranial nerve palsies, headache, and cerebellar dysfunction. Recently, with the spread of computed tomography (CT) and enhanced magnetic resonance imaging (MRI), the number of detected HP cases is increasing. However, we only found less than 200 cases on a MEDLINE search using the heading “hypertrophic pachymeningitis.” Several specific etiologies of this disorder have been reported, including tuberculosis, malignant lymphoma, syphilis, rheumatoid arthritis (RA), Wegener’s granulomatosis, sarcoidosis, and aspergillosis. However, many cases have been diagnosed as idiopathic. In this report, we present a patient with a unique form of idiopathic HP and discuss the clinical, radiographic, and etiologic findings.
Case report
A 52-year-old-man suffered upper respiratory symptoms and all-sided headache in December, 2000. He was treated with nonsteroidal anti-inflammatory drugs and oral antibiotic agents at the outpatient clinic in our hospital, but the severe headache did not improve. On January 23, 2001, he was admitted to our hospital due to worsening headache accompanied by elevated C-reactive protein (CRP). A smoker for 30 years, he had neither a past medical history nor family history. He was afebrile and showed normal results on physical examinations. In neurologic examinations, he had no meningeal signs, papilledema, visual field defects, cranial nerve palsies, pyramidal signs, sensory abnormalities, ataxia, or involuntary movements. Deep tendon reflexes were also normal.
The patient’s laboratory and immunologic findings were as follows: white blood cell count 11,500/mm3 (neutrophil 77.5%, eosinophil 0.8%, basophil 0.2%, lymphocyte 16.0%, monocyte 5.5%), red blood cell count 376×104/mm3, hemoglobin 11.3 g/dl, hematocrit 35.8%, platelet count 69.2×104/mm3, erythrocyte sedimentation rate 117 mm/h, CRP 10.9 mg/dl, CH50 57.8 U/ml (normal 29–48 U/ml), C3 166.2 mg/dl (normal 86–160 mg/dl), C4 60.3 mg/dl (normal 17–45 mg/dl), immunoglobulin (Ig)G 1378 mg/dl, IgA 246 mg/dl, IgM 135 mg/dl, and ferritin 264.3 ng/ml. Antinuclear antibody and disease-specific autoantibodies were negative. Antimyeloperoxidase antineutrophil cytoplasmic antibody (MPO-ANCA) and antiproteinase-3 antineutrophil cytoplasmic antibody (PR3-ANCA) were within normal ranges. Rheumatoid factor was positive (39.7 IU/ml, normal <20.0 IU/ml). Treponema and human T cell leukemia virus antibody tests were negative. Angiotensin-converting enzyme (ACE) was within the normal range. Coagulation and urinalysis test results were normal. A lumbar puncture revealed an opening pressure of 170 mmH2O, and CSF showed a glucose level of 80 mg/dl, protein level of 20 mg/dl, and white blood cell count of 18/mm3 (predominantly lymphocyte). Cytologic examination and culture of CSF showed no abnormalities. The purified protein derivative (PPD) skin test was positive (10×10mm), and no findings on chest X-ray suggested tuberculosis.
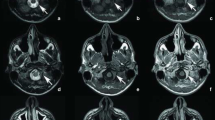
After admission, a slightly tender superficial temporal artery was noticed, and temporal arteritis was greatly suspected. However, temporal artery biopsy showed negative results for arteritis. Brain CT (Fig. 1A) revealed thickened dura mater on the parietal lobes. Gallium scintigraphy (Fig. 1B) detected accumulation among the falx. Magnetic resonance imaging (Fig. 2) of the brain showed marked thickening of the dura mater, which was most prominent over the anterior part of the falx. T1- and T2-weighted images showed thickened meningitis with low intensity, and there was high intensity by gadolinium enhancement on T1-weighted images.

Pretherapeutic brain images of our patient. A Computed tomography shows thickened dura matter on the parietal lobes. B Gallium scintigraphy (1 February 2002) shows strong accumulation among the dura of anterior part of the falx

Brain axial magnet resonance images show marked thickening of the dura mater, especially at the anterior part of the falx. The lesion is hypointense on T1-weighted (A) and T2-weighted images (B) and diffusely enhanced by gadolinium on coronal T1-weighted image (C)
Except for the location of the lesion, MRI showed typical findings of hypertrophic pachymeningitis. Biopsy of the dura matter for microscopic diagnosis could not be performed due to lack of patient consent, because a craniotomy was necessary to reach the lesion. Prednisolone (30 mg/day) was administered, and the severe headache immediately disappeared. C-reactive protein, CSF, and gallium scintigraphy findings improved to normal (Fig. 3A). Brain MRI also showed reduced size of the dural lesion (Fig. 3B). Prednisolone was gradually tapered without any signs of recurrence. Two years after the onset of disease, the patient was asymptomatic and had been without steroid therapy for 8 months.

Brain imaging 3 weeks after initiation of corticosteroid treatment. A Gallium scintigraphy shows no significant accumulation among the dura of the anterior part of the falx. B Magnetic resonance images show reduced size of the dural lesion. Left axial T1-weighted image, right coronal T1-weighted image enhanced by gadolinium
Discussion
Berger et al. [1] suggest that cases referred to as “idiopathic intracranial pachymeningitis” represent a continuum of fibrotic syndromes affecting the nerve system. Intracranial lesions of multifocal fibrosclerosis usually result from extension of the tentorium cerebelli and the posterior part of the falx [2, 3, 4, 5]. In our patient, the lesion was limited to the dura of the anterior part of the falx, and this is why neither cranial nerve palsies nor ataxia was observed. In other, previously described cases of HP, multiple cranial nerve palsies and ataxia in addition to headache are the most commonly observed clinical manifestations [2, 3, 4, 5, 6].
The diagnosis of idiopathic cranial pachymeningitis is performed on clinical and radiologic grounds. Unenhanced CT shows thickened, hyperdense dura typically along the tentorium, tentorial ridge, falx, and prepontine brainstem. After administration of iodinated contrast materials, the hypertrophic dura is enhanced markedly. T1- and T2-weighted MRI typically show hypointensity of the thickened meninges that are enhanced intensely after gadolinium-pentetic-acid administration, which makes the above findings more evident [6, 7, 8, 9]. These findings on MRI are thought to represent the fibrous tissue and inflammatory regions. Taking advantage of gallium scintigraphy, we could notice and suspect this disease.
Through a review of the literature, we found only a few cases of HP evaluated using gallium scintigraphy [10, 11]; in our case, it exactly detected the lesion, which was clearly improved by steroid therapy. Gallium scintigraphy might be useful for investigating the location and development of HP.
Histopathologic studies of dural biopsy are necessary to distinguish HP from other granulomatous, tumorous, and infectious diseases. However, extensive investigations, even dural biopsy, do not always provide a specific diagnosis [8]. Thus, histopathologic studies sometimes show nonspecific granulomatous pachymeningitis with plasma cells, giant cells, lymphocytes, and areas of necrosis but not with caseation [2, 6]. Since dural biopsy could not be performed, we cannot discuss histopathologic findings in this case.
The treatment of HP is not well established, due to the rarity of the disease and uncertainty regarding its etiology. Corticosteroid therapy is often effective initially, but idiopathic HP in most cases recurs or progresses when the corticosteroid is tapered or discontinued. Radiation therapy and immunosuppressive agents such as azathioprine and cyclophosphamide have been reported to have variable efficacy [2, 4, 6, 7]. Our patient, fortunately, responded well to corticosteroid monotherapy and became independent of corticosteroid. Surgical treatment with removal of the hypertrophic dura provides temporary relief of symptoms [7], and surgery is required in patients who have hydrocephalus or suffer mass effects.
Several specific etiologies of HP have been reported, including tuberculosis, malignant lymphoma, syphilis, rheumatoid arthritis (RA), autoimmune vasculitis, Wegener’s granulomatosis, sarcoidosis, and aspergillosis, but none of them corresponds to the findings of our patient. We could not find the obvious cause of the disease in this case, which might represent so-called idiopathic HP. Idiopathic HP is considered to be an autoimmune disorder because of its association with other autoimmune disorders and the effect of corticosteroid on it. Eighteen cases of HP with seropositive rheumatoid factor have been described previously, and most of them have clinical features of RA, Sjögren’s syndrome, or other systemic autoimmune diseases [8, 10, 11, 12, 13, 14, 15].
Our patient had no multiple joint pain or deformity and did not satisfy the criteria for RA. Moreover, the possibility of infection, tumor, or systemic autoimmune diseases could be excluded based on clinical course and various examinations including laboratory tests, CSF, CT, and MRI. However, it is difficult to rule out Wegener’s granulomatosis, chronic infections, atypical lymphoma, and neurosarcoidosis. Generally, Wegener’s granulomatosis causes signs of nasopharyngeal or sinus inflammation, and peripheral neuropathies are common. Unlike idiopathic HP, which progresses slowly and is associated with low mortality, Wegener’s granulomatosis usually progresses rapidly and is almost always fatal unless treated aggressively with corticosteroid and cyclophosphamide [6].
Hypertrophic cranial pachymeningitis may be the presenting symptom of an adjacent paranasal sinus or ear infection, but there were no clinical or radiologic criteria supporting a chronic infection of the ear, nose, or throat in our patient. Although cytologic examination of CSF revealed only mature lymphocytes, dural biopsy is necessary to exclude atypical lymphoma, which may always be partially alleviated with corticosteroid therapy, unlike our patient. On the other hand, the diagnosis of neurosarcoidosis is difficult when neurologic involvement is the first or only manifestation of the disease.
Since we have no reliable diagnostic test for neurosarcoidosis [7], on which corticosteroids sometimes have a marked effect, we cannot absolutely exclude this disorder in our patient. The elevated rheumatoid factor might suggest the involvement of immunologic etiology. However, the rheumatoid factor titer was not reduced by steroid therapy, unlike previously described cases, which we compared to investigate the possible relationship of HP to other autoimmune disorders and cytokine action and to establish the treatment for HP.
Conclusion
We report here a case of HP with seropositive rheumatoid factor. To our knowledge, this is the first such case with a uniquely manifested location involving only the anterior part of the falx and no symptoms except severe headache. Administration of corticosteroid markedly improved the headache as well as CRP and radiographic findings.
References
Berger JR, Snodgrass S, Glaser J, Post MJD, Norengerg M, Benedetto P (1989) Multifocal fibrosclerosis with hypertrophic intracranial pachymeningitis. Neurology 39:1345–1349
Masson C, Hénin D, Hauw JJ, Rey A, Raverdy P, Masson M (1993) Cranial pachymeningitis of unknown origin: a study of seven cases. Neurology 43:1329–1334
Hassin GB, Zeitlin H (1940) Syphilitic cerebral hypertrophic pachymeningitis. Arch Neurol 43:362–371
Michel D, Girard PF, Tommasi M, Masson R, Trillet M, Piccinali JP (1969) Intracranial granulomatous pachymeningitis with pseudotumoral symptomatology. Apropos of four cases. [French.] J Med Lyon 50:545–577
Feringa ER, Weathebee L (1975) Hypertrophic granulomatous cranial pachymeningitis causing progressive blindness in a chronic dialysis patient. J Neurol Neurosurg Psychiatry 38:1170–1176
Mamelak AN, Kelly WM, Davis RL, Rosenblum ML (1993) Idiopathic hypertrophic cranial pachymeningitis: report of three cases. J Neurosurg 79:270–276
Botella C, Orozco M, Navarro J, Riesgo P (1994) Idiopathic chronic hypertrophic craniocervical pachymeningitis: case report. Neurosurgery 35:1144–1149
Yur WTC, Drew JM, Rizzo M, Ryals TJ, Sato Y, Bell WE (1990) Evaluation of pachymeningitis by contrast enhanced MR imaging in a patient with rheumatoid disease. AJNR Am J Neuroradiol 11:1247–1248
Martin N, Masson C, Henin D (1989) Hypertrophic cranial pachymeningitis: assessment with CT and MR imaging. AJNR Am J Neuroradiol 10:477–484
Sugiyama Y, Shimizu M, Hoshi A, Aoki T, Matsuura Y, Tochikubo S et al (1999) An old man presenting with fluctuating bilateral multiple cranial nerve palsies and positive test for perinuclear antineutrophil cytoplasmic antibody [Japanese]. Brain and Nerve (Tokyo) 51:825–832
Higgins WL, Marano GD (1986) Gallium imaging of rheumatoid pachymeningitis. Clin Nucl Med 11:350–351
Otsuka M, Kuwata Y, Fujiwara T, Yamada S, Ueki A (1997) A case of rheumatoid hypertrophic pachymeningitis. Clin Neurol (Tokyo) 37:834–840
Li JY, Lai PH, Lam HC et al (1999) Hypertrophic cranial pachymeningitis and lymphocytic hypophysitis in Sjögren’s syndrome. Neurology 52:420–423
Manabe Y, Narai H, Warita H, Hayashi T, Sakai K, Abe K (2001) Rheumatoid factor positive hypertrophic cranial pachymeningitis in association with hypopituitarism and multiple cranial nerve palsies. Internal Medicine 40:964–967
Bathon JM, Moreiand LW, Di Bartolomeo AG (1989) Inflammatory central nervous system involvement in rheumatoid arthritis. Semin Arthritis Rheum 18:258–266
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kanemoto, M., Ota, Y., Karahashi, T. et al. A case of idiopathic hypertrophic cranial pachymeningitis manifested only by positive rheumatoid factor and abnormal findings of the anterior falx. Rheumatol Int 25, 230–233 (2005). https://doi.org/10.1007/s00296-004-0482-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-004-0482-9