
Abstract
The first two mitochondrial genomes of marine diatoms were previously reported for the centric Thalassiosira pseudonana and the raphid pennate Phaeodactylum tricornutum. As part of a genomic project, we sequenced the complete mitochondrial genome of the freshwater araphid pennate diatom Synedra acus. This 46,657 bp mtDNA encodes 2 rRNAs, 24 tRNAs, and 33 proteins. The mtDNA of S. acus contains three group II introns, two inserted into the cox1 gene and containing ORFs, and one inserted into the rnl gene and lacking an ORF. The compact gene organization contrasts with the presence of a 4.9-kb-long intergenic region, which contains repeat sequences. Comparison of the three sequenced mtDNAs showed that these three genomes carry similar gene pools, but the positions of some genes are rearranged. Phylogenetic analysis performed with a fragment of the cox1 gene of diatoms and other heterokonts produced a tree that is similar to that derived from 18S RNA genes. The introns of mtDNA in the diatoms seem to be polyphyletic. This study demonstrates that pyrosequencing is an efficient method for complete sequencing of mitochondrial genomes from diatoms, and may soon give valuable information about the molecular phylogeny of this outstanding group of unicellular organisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are present in all eukaryotic cells. They appeared as the result of a single symbiotic event (Gray et al. 1999; Lang et al. 1999; Andersson et al. 2003) and have a common proteobacterial ancestor (Andersson et al. 1998; Emelyanov 2001, 2003). Recent studies have reported the sequences of mitochondrial genes and of full mitochondrial genomes from eukaryotes (Bullerwell and Gray 2004). The small size and low polymorphism of genes in mitochondrial genomes make them a good subject for molecular phylogenetic studies (Funk and Omland 2003), and these features should also be present in diatoms (Alverson 2008). Studies of diatom phylogeny were previously performed using fragments of particular mitochondrial genes, e.g., cox1 (Ehara et al. 2000; Imanian et al. 2007).
The diversity of diatoms and their important role in the biosphere call attention to their evolutionary history. At present, there are two species of diatoms, a centric marine species Thalassiosira pseudonana Hasle & Heimdal (Armbrust et al. 2004) and a raphid pennate marine species Phaeodactylum tricornutum Bohlin (Bowler et al. 2008) whose genomes are completely or almost completely sequenced. The availability of complete genomic information for these evolutionary distant diatom species will provide exciting new opportunities to apply all the powerful methods of molecular biology and genetic engineering to understand the function of these genomes. Of particular interest is the question of how the information stored in these genomes is transformed into the morphogenetic plan for their sophisticated cell walls, which are composed of diverse and genetically defined hierarchies of siliceous micro- and nanostructures.
The development of new superfast DNA sequencing procedures, such as pyrosequencing among others, makes fast and relatively inexpensive sequencing of genomes feasible. We recently began pyrosequencing the genome of the freshwater araphid pennate diatom Synedra acus var. radians (Kütz.) Skabitsch. It appeared that a single round of pyrosequencing of total S. acus genomic DNA produced a nearly complete mitochondrial genome. Completion of the sequencing required sequencing of a relatively small region of sequence repeats using the classical Sanger procedure. Here we report the complete sequence of the mitochondrial DNA of S. acus. This sequence was compared with those of the two other known diatom mitochondrial genomes, and with mitochondrial genomes of some other organisms. The new data were used for phylogenetic reconstructions. The complete mtDNA of S. acus was found to contain some interesting features that provide new insights into the molecular evolution of these species.
Materials and methods
Algal source and DNA isolation
A culture of S. acus was isolated from the phytoplankton of Listvennichny Bay of Lake Baikal, and diatom cells were cultivated in DM medium (Thompson et al. 1988) in a 100-L photobioreactor (Vereshchagin et al. 2008) up to a density of 5 × 104 cells/mL. Diatom cells were collected on a filter with 1.2 μm pores and washed with sterile medium to remove bacteria. Total DNA was isolated from 2 g of fresh biomass according to the procedure of Rochaix et al. (1988) as modified by Jacobs et al. (the second method, Jacobs et al. 1992). To remove RNA, 450 μL of TE buffer (10 mM Tris–HCl, pH 7.5 and 1 mM EDTA) was added to isolated DNA followed by 15 μL of 20% SDS and 3 μL of RNase (20 mg/mL). The mixture was incubated for 1 h at 37°C. A 3-μL aliquot of Proteinase K (20 mg/mL) was added, and the mixture was incubated for 1 h at 37°C. NaCl (100 μL of 5 M) was added, followed by the addition of 160 μL of 2.5% CTAB, 0.18 M NaCl. The mixture was incubated for 15 min at 65°C. Total DNA was purified first by chloroform extraction, then by phenol–chloroform extraction, and finally precipitated by ethanol and dissolved in TE for further use.
Sequencing and assembly
Mitochondrial genome sequencing was performed using total genomic DNA without prior isolation of mitochondrial DNA. The genome was sequenced on a Roche Genome Sequencer GS FLX using a standard protocol for a shotgun genome library. The GS FLX run resulted in the generation of about 90 MB of sequence data with an average read length of 230 bp. The GS FLX reads were assembled into contigs using “GS de novo assembler”. A single 43,384-bp contig was identified as the mtDNA based on extensive sequence similarity to known algal mitochondrial genomes. The depth of sequencing was approximately 20-fold. The remaining 3.3 kb region containing repeat sequences was amplified by PCR with primers MS-4F (TAT ATC TTA CTG GAT GCG GGA T) and MS-5R (TGA ACC TGT TTT AGT AGG TAA ACT) located in flanking unique regions, and sequenced by the primer-walking approach using ABI 3730 (Applied Biosystems, Foster City, CA) and the ABI Prism Big Dye terminator sequencing kit (Applied Biosystems). These two contigs were joined into a circular map. The complete mtDNA sequence of S. acus is available in GenBank (GU002153).
Genome analysis
Gene content was determined by BLAST similarity searches (Altschul et al. 1997) against the non-redundant database of National Center for Biotechnology Information. ORFs were localized using Clone Manager 6 (version 6.00), whereas tRNA-coding sequences were identified with tRNAscan-SE 1.23 (Lowe and Eddy 1997). Small and large ribosomal RNA subunit genes were identified by comparing S. acus mtDNA with rRNA genes from the mtDNAs of T. pseudonana and other organisms. Among ORFs, protein-coding genes were annotated by similarity with genes from T. pseudonana and other organisms.
Features of the mtDNA of T. pseudonana were retrieved based on annotation provided in GenBank NC_007405. The nucleotide sequence of P. tricornutum mtDNA was downloaded from ftp://ftp.jgi-psf.org/pub/JGI_data/Phaeodactylum_tricornutum/v2.0/Phatr2_assembly_organelle.fasta.gz. Potential protein-coding sequences and rRNA genes from this species were identified by BLAST searches against S. acus and T. pseudonana mtDNAs, whereas tRNA genes were identified using tRNAscan-SE 1.23 (Lowe and Eddy 1997).
Repeated sequences were identified by Complex TR (Hauth and Joseph 2002), Clone Manager 6 (version 6.00) and BLAST search (Altschul et al. 1997). Genome maps were generated using Circos 0.51 (Krzywinski et al. 2009).
Molecular phylogenetic analysis of cox1 sequences
We used mitochondrial cytochrome oxidase subunit I (cox1) sequences from the following heterokonts: oomycetes Phytophthora phaseoli (AAM98742), Phytophthora mirabilis (AAM98745), and Phytophthora infestans (NP_037600); yellow–green algae Vaucheria sessilis (BAA24976), Botrydium granulatum (BAA24968), and Mischococcus sphaerocephalus (BAA24972); brown algae Desmarestia viridis (YP 448662), Dictyota dichotoma (YP 448700), Fucus vesiculosus (YP 448623), Pylaiella littoralis (NP 150405), and Laminaria digitata (NP 659274); radial centric diatoms Rhizosolenia setigera (BAA86611), and Melosira ambiqua (BAA96357); diatoms from Thalassiosirales, bi- and multipolar centrics Ditylum brightwellii (BAA86608), Thalassiosira pseudonana (YP_316586), Thalassiosira nordenskioeldii (AB020229), and Skeletonema costatum (BAA86612); araphid pennates Synedra acus (GU002153, cox1), Grammonema striatula (BAA86609) and Thalassionema nitzschioides (BAA86613); raphid pennates Eunotia sp. (ABO77899), Cylindrotheca closterium (BAA86607), Nitzschia frustulum (BAA86610), Pinnularia sp. (ABO77877), Phaeodactylum tricornutum, Sellaphora capitata (ABO77886) and Sellaphora blackfordensis (ABO77887). In summary, the dataset contains an alignment of the 609-bp-long partial cox1 sequences of 27 species of diverse heterokonts, including 16 diatoms. Because many diatom cox1 genes have only partial sequence entries in the GenBank database, the alignment is much shorter than the full-length cox1 sequence. Cox1 nucleotide sequences were translated and aligned using the CLUSTALW 1.83 program. The RevTrans package (Wernersson and Pedersen 2003) was used to obtain the back-translated nucleotide sequence alignment. The nucleotide substitution model was chosen using jModeTest (Posada 2008) as GTR + I + Γ with four gamma rate categories. The maximum likelihood (ML) tree was constructed using the TREEFINDER package (Jobb et al. 2004) with 1,000 bootstrap replicates. MrBayes 3.1.2 (http://mrbayes.csit.fsu.edu) was applied for Bayesian phylogenetic analysis (BA). The BA was performed for two runs by four chains with 106 generations each. Chains were sampled every 100th generation. The average standard deviation of split frequencies at the end of the run was 0.014. The “burn-in” was set to 1,000 to construct a consensus tree. To check whether it was sufficient to assume the “burn-in” parameter was equal to 0.1, we considered the log-likelihood plot during the simulation. These plots resembled white noise, suggesting that our analysis gave an unbiased result. The TreeGraph2 (http://treegraph.bioinfweb.info) software was used to draw phylogenetic trees and to combine bootstrap supports.
Molecular phylogenetic analysis of RT domain sequences from group II intron ORFs
The initial alignment of the RT sequences was kindly provided by Dr Ryoma Kamikawa. The alignment contains RT sequences from a variety of eukaryotic group II introns. We added four RT domain sequences from the cox1 introns of S. acus and P. tricornutum to this set. The final dataset contained 143-aa long aligned sequences of 4 bacterial RT domain sequences used as an outgroup, 12 sequences of lower plants, 15 sequences of fungal introns, and 15 chromalveolate sequences including 6 diatom-specific sequences. We performed phylogenetic analysis according to the procedure of Kamikawa et al. (2009). Briefly, the dataset was analyzed using the PhyML 3.0 package (Guindon and Gascuel 2003) with the LG amino acid substitution model (Le and Gascuel 2008) with among-site rate variation approximated by a discrete gamma distribution with four rate categories (LG + Γ model). The initial tree was constructed using BioNJ. Subsequent tree topologies were searched by subtree pruning and regrafting (SPR). Bootstrap analysis was performed for 100 replicates.
Results
General characteristics of Synedra acus mtDNA
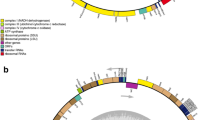
The mitochondrial DNA of S. acus maps as a circular molecule of 46,657 bp. It codes for the small subunit (rns) and the large subunit (rnl) of rRNAs within an operon, 24 transfer RNAs (tRNAs), and 33 predicted proteins. The genome map (Fig. 1a) shows dense packing of 59 genes. We did not test the possibility that the mtDNA molecule is really circular rather than concatemeric.

S. acus mtDNA. a Gene map of the S. acus mtDNA. Genes and ORFs are shown as solid blocks. The gene blocks shown outside and inside the circular map are transcribed in the clockwise and counter-clockwise directions, respectively. The color of the box designates the type of gene: the protein-coding genes are grey, the rRNA genes are light grey, the tRNA genes are black. The pseudogenes are marked by  . For intron-containing genes, the “-eN” suffix denotes the exon number. The exon–exon junctions are shown as black lines connecting the exon ends. The large repeat region is shown as the grey-hatched block. b Schematic diagram of the large noncoding region in the S. acus mtDNA. The noncoding region spans positions 42,005–301. Arrows/triangles indicate the relative order of repeat units and their orientation. The nucleotide sequence identity of the repeat units (a, b, c) is indicated by greyscale: black––100% identity, dark grey––more than 90% identity, light grey––less than 90% identity. Repeat unit c is part of unit b. Two repeat sequences that could form stem-and-loop structures (d) are shown as a hairpin symbols
. For intron-containing genes, the “-eN” suffix denotes the exon number. The exon–exon junctions are shown as black lines connecting the exon ends. The large repeat region is shown as the grey-hatched block. b Schematic diagram of the large noncoding region in the S. acus mtDNA. The noncoding region spans positions 42,005–301. Arrows/triangles indicate the relative order of repeat units and their orientation. The nucleotide sequence identity of the repeat units (a, b, c) is indicated by greyscale: black––100% identity, dark grey––more than 90% identity, light grey––less than 90% identity. Repeat unit c is part of unit b. Two repeat sequences that could form stem-and-loop structures (d) are shown as a hairpin symbols
Gene complements of the mtDNAs are similar in the three diatoms (Table 1); however, the order of the genes differs. There are two gene blocks in diatom mtDNAs that vary in the number of gene rearrangement events. The order of genes within one of the two major transcription units of the S. acus mtDNA (from trnR-UCU to nad11) is well conserved. The second unit (from trnQ-UUG to trnM-CAU) has its gene structure rearranged. While in S. acus and P. tricornutum this region of mtDNA constitutes the co-transcribed gene blocks, in T. pseudonana it is broken into two counter-oriented transcription units.
Comparison of tRNAs
S. acus mtDNA encodes 24 tRNA genes, including two separate genes for initiator (trnM(cau)f) and elongator (trnM(cau)e) methionine tRNAs. All tRNA sequences produce standard cloverleaf secondary structures. The set of S. acus tRNAs is sufficient to recognize codons for all of the natural amino acids except threonine. Like all heterokont mtDNAs (Gray et al. 2004), the mtDNA of S. acus lacks a trnT gene (Table 1).
Unlike T. pseudonana mtDNA, the S. acus and P. tricornutum mitochondrial genomes lack the tRNA gene trnW(uca) (Table 1). This tRNA is responsible for translation of the TGA codon as tryptophan in many mitochondrial genes of T. pseudonana and some other algae (Gray et al. 2004). The absence of the TGA codon from the mtDNA of S. acus precludes its use for encoding tryptophan and the execution of the termination function.
Protein-encoding genes
The identified S. acus mitochondrial protein-encoding genes include 13 ribosomal protein-coding genes (rps4, 8, 10–14, 19, and rpl2, 5, 6, 14, 16); genes for 3 subunits of the cytochrome oxidase (cox1–3); cob, which encodes apocytochrome B; 10 genes encoding subunits of the NADH dehydrogenase (nad1–7, 4L, 9 and 11); 3 genes encoding subunits of ATPase, atp6, 8, and 9; and a gene encoding a component of the twin-arginine translocation pathway, tatC. In addition, the S. acus mtDNA encodes two ORFs within cox1 introns and three unique ORFs (orf100, orf168, and orf182) whose functions were not revealed by similarity searches. Unlike the mtDNAs of the two other diatoms, the mtDNA of S. acus lacks three genes encoding ribosomal proteins: the rps2 gene is totally missing, while rps3 and rps7 are retained as truncated pseudogenes carrying frameshifts and deletions.
The complement of protein genes of S. acus is similar to those of T. pseudonana and P. tricornutum (Table 1). The rps2, rps3, and rps7 genes probably have been transferred to the nuclear genome and their mitochondrial versions are undergoing elimination. This fact is consistent with the finding that ribosomal genes show the highest level of ongoing loss in angiosperms (Adams and Palmer 2003).
Repeats
The two largest non-coding stretches of the genome are the 4,955 bp repeat-containing region between the start of the initiating trnM(cau)f and the end of nad2 and the 658 bp region without obvious structural features that is located between the start of cox2 and the end of trnM(cau)f. Analysis of the former region revealed the presence of several direct and inverted repeats (Fig. 1b). The largest repeat cluster (a) contains two imperfect 1,450/1,316 bp direct repeats separated by a 256 bp unique sequence. Both repeats contain sequences with high similarity at the ends. A tandem duplication of a 100-bp sequence in the first copy of the repeat accounts for most of the difference in the lengths of the repeat units. In addition to these long repeats, the trnM-nad2 intergenic region contains a few relatively short (<50 bp) direct repeats (b–b, and c–c); b–b is an inversion of c–c. The repeated sequences (d) contain short palindromes that are able to form stem-and-loop structures.
According to Levinson and Gutman (1987), formation, expansion, and contraction of massive tandem repeats involve multiple events of slipped mispairing during replication. However, the repeat region of S. acus mtDNA contains only a few tandem repeats and instead consists of two copies of a 1.3 kb sequence. This structure may have appeared due to a single duplication event following a mutation-driven sequence divergence. Densely packed genes and a single, long, repeat-containing intergenic region are also found in mtDNAs of the diatoms T. pseudonana and P. tricornutum. In T. pseudonana mtDNA, the 3.8-kb repeat region is flanked by a pair of 183-bp inverted repeats and consists almost entirely of multiple tandem copies of repeat units ranging from 37 to 75 bp and occurring 49 times. We identified approximately 36 kb-long repeats in P. tricornutum mtDNA using the same method as that applied to the S. acus sequence. This region has a complex structure and contains dozens of direct and inverted repeats arranged in tandem.
Phylogenetic analysis of cox1 genes
Figure 2 shows a phylogenetic tree based on BA of available nucleotide sequences from a fragment of the cox1 gene from 16 diatoms and a few other heterokonts. Oomycetes, yellow-green algae, brown algae, and diatoms form clearly resolved clades. ML reconstruction gives a similar phylogenetic tree, except for the topology of the brown algae clade (data not shown), although the whole-clade supports are vigorous. The robust monophyletic diatom clade consists of two sub-clades—Clade 1 and Clade 2. The latter falls into two distinct branches, one of them including Thalassiosirales, bi- and multi-polar centric diatoms (Clade 2a), and the other including all the sequences of pennates (Clade 2b). It is noteworthy that the araphid pennate species present in this tree (S. acus, G. striatula, and T. nizschioides) form a strongly supported branch.

A phylogenetic tree based on cox1 partial nucleotide sequences. The tree was constructed by BA. We used the GTR + I + Γ nucleotide substitution model with four gamma rate categories. The Bayesian clade credibility values that are based on posterior probabilities of taxon bipartitions are shown above the branches. ML tree (not shown) bootstrap values are shown on the BA tree (numbers below branches). The nomenclature of the diatom clades is given according to Medlin (2009)
Phylogeny reconstructions based on nuclear 18S RNA (Kooistra et al. 2003; Medlin and Kaczmarska 2004; Medlin 2009) and chloroplast genes (Andersen 2004) show that only raphid pennates are monophyletic; centrics and araphids are paraphyletic. The pennates are designated as Clade 2b, which includes both monophyletic raphid and polyphyletic araphid groups. The phylogenetic tree in Fig. 2 is in agreement with the recent diatom phylogeny concept (Medlin 2009). Our results also support previously reported phylogenies based on the sequences of cox1 genes of a smaller number of diatom species (Ehara et al. 2000; Imanian et al. 2007). The reason for the monophyly of the araphid lineage in Fig. 2 is that the cox1 dataset contains samples representing only one branch of this polyphyletic group. While the resolution of the fine phylogeny of diatoms does not reach the high levels of significance obtained for phylogenies based on nuclear genes, it should be noted that the cox1 dataset is considerably smaller than the 18S dataset.
Introns
There are three group II introns in the mtDNA of S. acus (Fig. 1). Two introns are inserted into the cox1 gene and have lengths of 2,564 and 2,416 bp, respectively. The third 778 bp intron is located within the rnl gene that encodes the large subunit of rRNA. The cox1 introns harbor ORFs of the so-called RT type (Lambowitz and Zimmerly 2004), which contain three distinct protein domains: reverse transcriptase, maturase, and endonuclease (Michel and Ferat 1995). The rnl intron does not contain an ORF.
A protein BLAST search against the non-redundant NCBI database shows that orf755 and orf590 of S. acus are most similar to RT proteins encoded by cox1 introns of brown alga Pylaiella littoralis (orf755) and of the haptophyte, Pavlova lutheri (orf590). They are also similar to intron-encoded proteins of some algae and fungi.
To reveal the origin of S. acus and other diatom group II introns, we reconstructed a phylogenetic tree based on the amino acid sequences of the RT domains located in the intron ORFs (Fig. 3). It is clear that RT sequences from diatom introns do not form a monophyletic clade; they are scattered over the clade formed by diverse eukaryotes. The S. acus orf590 sequence clusters with the P. lutheri cox1 intron 1, and the S. acus orf755 clusters with the P. littoralis cox1 intron 2.

Phylogenetic analysis of group II introns. The dataset containing aligned sequences of the RT domains from a variety of the group II introns was used to reconstruct a phylogenetic tree with the LG + Γ amino acid substitution model. The diatom-specific sequences are in bold. Bootstrap values below 50% are not indicated
Discussion
Within Chromista (the group including haptophytes, cryptophytes and heterokonts), the presence of an expanded repeat-rich intergenic region in mtDNA has been reported for both sequenced mtDNAs of cryptophytes (Hauth et al. 2005; Kim et al. 2008), for labyrinthulids (Burger et al. 2000), for two sequenced mtDNAs of diatoms (Armbrust et al. 2004; Bowler et al. 2008) and now for the sequence of S. acus. This suggests that the repeat regions appeared independently. It is believed that long intergenic regions may be involved in the regulation of replication and/or transcription. However, the exact function of long repeats in mtDNA remains unknown.
Phylogenetic analysis of the intronic RT domain sequences (Fig. 3) did not reveal a monophyletic origin of intron sequences of diatoms. We infer that introns were inserted into the cox1 genes of diatoms independently. The insertion of group II introns into mtDNA of diatoms, as well as into that of some other groups of eukaryotes (Kamikawa et al. 2009) can be considered a process of horizontal gene transfer between distant taxa. Such transfer might have been the basis for group II intron propagation that should have been more extensive in diversified and heterogeneous ecosystems.
The major advantages of mitochondrial genomes as tools for phylogenetic reconstructions are their small size and low polymorphism, as well as the orthology of mitochondrial genes (Alverson 2008). Until recently, it was impossible to use full mitochondrial genomes for this purpose. With the advent of fast sequencing methods, many new mitochondrial genomes will become available in the near future. The outstanding diversity of diatoms, which are believed to form up to 105–106 species, including cryptic ones (Mann 1994; Round 1996), together with the availability of a long paleontological record will make it possible to develop models of evolution of unprecedented precision once a number of full mitochondrial genomes are available. For this purpose, rearrangements of genes within the genome may become the most useful feature. It is also important that the patterns of gene rearrangement can be compared with the events of molecular evolution for many particular protein-coding genes, particularly if such mutations lead to adaptations to changing environment. The small number of mitochondrial genes and the relative simplicity of the mitochondrial genome may help to solve this keystone problem.
The study of the evolution of genomes instead of the evolution of genes will soon become a new instrument for understanding of the mechanisms of the biological speciation. This study demonstrates the feasibility of fast pyrosequencing of mtDNAs in terms of both time and expense.
Abbreviations
- ORF:
-
Open reading frame
- RT:
-
Reverse transcriptase
- ML:
-
Maximum likelihood
- BA:
-
Bayesian analysis
- RT:
-
Reverse transcriptase
References
Adams KL, Palmer JD (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol 29:380–395
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Alverson AJ (2008) Molecular systematics and the diatom species. Protist 159:339–353
Andersen RA (2004) Biology and systematics of heterokont and haptophyte algae. Am J Bot 91:1508–1522
Andersson SGE, Zomorodipour A, Andersson JO, Sicheritz-Pontén T, Alsmark UCM, Podowski RM, Näslund AK, Eriksson AS, Winkler HH, Kurland CG (1998) The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396:133–140
Andersson SG, Karlberg O, Canback B, Kurland CG (2003) On the origin of mitochondria: a genomics perspective. Philos Trans R Soc Lond B Biol Sci 358:165–177
Armbrust EV, Berges JA, Bowler C, Green BR, Martinez D, Putnam NH, Zhou S, Allen AE, Apt KE, Bechner M, Brzezinski MA, Chaal BK, Chiovitti A, Davis AK, Demarest MS, Detter JC, Glavina T, Goodstein D, Hadi MZ, Hellsten U, Hildebrand M, Jenkins BD, Jurka J, Kapitonov VV, Kröger N, Lau WW, Lane TW, Larimer FW, Lippmeier JC, Lucas S, Medina M, Montsant A, Obornik M, Parker MS, Palenik B, Pazour GJ, Richardson PM, Rynearson TA, Saito MA, Schwartz DC, Thamatrakoln K, Valentin K, Vardi A, Wilkerson FP, Rokhsar DS (2004) The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306:79–86
Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP, Rayko E, Salamov A, Vandepoele K, Beszteri B, Gruber A, Heijde M, Katinka M, Mock T, Valentin K, Verret F, Berges JA, Brownlee C, Cadoret JP, Chiovitti A, Choi CJ, Coesel S, De Martino A, Detter JC, Durkin C, Falciatore A, Fournet J, Haruta M, Huysman MJ, Jenkins BD, Jiroutova K, Jorgensen RE, Joubert Y, Kaplan A, Kröger N, Kroth PG, La Roche J, Lindquist E, Lommer M, Martin-Jézéquel V, Lopez PJ, Lucas S, Mangogna M, McGinnis K, Medlin LK, Montsant A, Oudot-Le Secq MP, Napoli C, Obornik M, Parker MS, Petit JL, Porcel BM, Poulsen N, Robison M, Rychlewski L, Rynearson TA, Schmutz J, Shapiro H, Siaut M, Stanley M, Sussman MR, Taylor AR, Vardi A, von Dassow P, Vyverman W, Willis A, Wyrwicz LS, Rokhsar DS, Weissenbach J, Armbrust EV, Green BR, Van de Peer Y, Grigoriev IV (2008) The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature 456:239–244
Bullerwell CE, Gray MW (2004) Evolution of the mitochondrial genome: protist connections to animals, fungi and plants. Curr Opin Microbiol 7:528–534
Burger G, O’Kelly C, Gray MW, Lang DF (2000) Thraustochytrium aureum mitocholdrial DNA, partial sequence. OGMP: GenBank access. no. AF288091
Ehara M, Inagaki Y, Watanabe KI, Ohama T (2000) Phylogenetic analysis of diatom coxI genes and implications of a fluctuating GC content on mitochondrial genetic code evolution. Curr Genet 37:29–33
Emelyanov VV (2001) Evolutionary relationship of rickettsiae and mitochondria. FEBS Lett 501:11–18
Emelyanov VV (2003) Common evolutionary origin of mitochondrial and rickettsial respiratory chains. Arch Biochem Biophys 420:130–141
Funk DJ, Omland KE (2003) Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu Rev Ecol Evol Syst 34:397–423
Gray MW, Burger G, Lang BF (1999) Mitochondrial evolution. Science 283:1476–1481
Gray MW, Lang BF, Burger G (2004) Mitochondria of protists. Annu Rev Genet 38:477–524
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hauth AM, Joseph DA (2002) Beyond tandem repeats: complex pattern structures and distant regions of similarity. Bioinformatics 1(Suppl):S31–S37
Hauth AM, Maier UG, Lang BF, Burger G (2005) The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucleic Acids Res 33:4433–4442
Imanian B, Carpenter KJ, Keeling PJ (2007) Mitochondrial genome of a tertial endosymbiont retains genes for electron transport proteins. J Eukaryot Microbiol 54:146–153
Jacobs JD, Ludwig JR, Hildebrand M, Kukel A, Feng T-Y, Ord RW, Volcani BE (1992) Characterization of two circular plasmids from the marine diatom Cylindrotheca fusiformis: plasmids hybridize to chloroplast and nuclear DNA. Mol Gen Genet 233:302–310
Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4:18
Kamikawa R, Masuda I, Demura M, Oyama K, Yoshimatsu S, Kawachi M, Sako Y (2009) Mitochondrial group II introns in the raphidophycean flagellate Chattonella spp. suggest a diatom-to-Chattonella lateral group II intron transfer. Protist 160:364–375
Kim E, Lane CE, Curtis BA, Kozera C, Bowman S, Archibald JM (2008) Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genomics 9:215
Kooistra WH, De Stefano M, Mann DG, Medlin LK (2003) The phylogeny of the diatoms. Prog Mol Subcell Biol 33:59–97
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Lambowitz AM, Zimmerly S (2004) Mobile group II introns. Annu Rev Genet 38:1–35
Lang BF, Gray MW, Burger G (1999) Mitochondrial genome evolution and the origin of eukaryotes. Annu Rev Genet 33:351–397
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
Levinson G, Gutman GA (1987) Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 4:203–221
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Mann DG (1994) The origins of shape and form in diatoms: the interplay between morphogenetic studies and systematics. In: Ingram DS, Hudson AJ (eds) Shape and form in plants and fungi. Academic Press, London, pp 17–38
Medlin LK (2009) Diatom (Bacillariophyta). In: Hedges SB, Kuman S (eds) The timetree of life. Oxford University Press, pp 127–130
Medlin LK, Kaczmarska I (2004) Evolution of the diatoms: morphological and cytological support for the major clades and a taxonomic revision. Phycologia 43:245–270
Michel F, Ferat J-L (1995) Structure and activities of group II introns. Annu Rev Biochem 64:435–461
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Rochaix J-D, Mayfield S, Goldschmidt-Clermont M, Erickson J (1988) Molecular biology of Chlamydomonas. In: Shaw CH (ed) Plant molecular biology: a practical approach. IRL Press, Oxford, pp 253–275
Round FE (1996) What characters define diatom genera, species and infra-specific taxa? Diatom Res 11:203–218
Thompson AS, Rhodes JC, Pettman I (eds) (1988) Culture collections of algae and protozoa. Titus Wilson & Son, Kendal
Vereshchagin AL, OYu Glyzina, Basharina TN, Safonova TA, Latyshev NA, Liubochko SA, Korneva ES, Petrova DP, Annenkov VV, Danilovtseva EN, Chebykin EP, Volokitina NA, Grachev MA (2008) Culturing of a fresh-water diatom alga Synedra acus in a 100 L photobioreactor and analysis of the biomass composition. Biotekhnologia 4:55–63
Wernersson R, Pedersen AG (2003) RevTrans—constructing alignments of coding DNA from aligned amino acid sequences. Nucleic Acids Res 31:3537–3539
Acknowledgments
We are grateful to Dr Chris Bowler for granting access to the P. tricornutum mtDNA sequence and to Dr Ryoma Kamikawa for the kindly presented alignment of RT domain sequences of group II introns. This work was financially supported by the Program of the Presidium of the Russian Academy of Sciences “Molecular and Cell Biology” (project # 22.3) and by the Federal Agency for Science and Innovations of Russia (contract 02.552.11.7045).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by D. Stern.
Rights and permissions
About this article
Cite this article
Ravin, N.V., Galachyants, Y.P., Mardanov, A.V. et al. Complete sequence of the mitochondrial genome of a diatom alga Synedra acus and comparative analysis of diatom mitochondrial genomes. Curr Genet 56, 215–223 (2010). https://doi.org/10.1007/s00294-010-0293-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-010-0293-3