
Abstract
Four novel wholly para-oriented aromatic poly(ether-amide-hydrazide)s containing various pendant groups on their aromatic rings were synthesized from p-aminosalicylic acid hydrazide (PASH) with an equimolar amount of either 4,4′-(1,4-phenylenedioxy)dibenzoyl chloride (1a), 4,4′-(2,5-tolylenedioxy)dibenzoyl chloride (1b), 4,4′-(2-tert-butyl-1,4-phenylenedioxy)dibenzoyl chloride (1c), or 4,4′-(2,5-biphenylenedioxy)dibenzoyl chloride (1d) via a low temperature solution polycondensation reaction. A polyamide-hydrazide without the ether and pendant groups, poly[4-(terephthaloylamino)salicylic acid hydrazide, PTASH, is also investigated for comparison. It was synthesized from PASH and terephthaloyl chloride by the same synthetic route. The polymer intrinsic viscosities ranged from 4.5 to 2.47 dlg−1 in N,N-dimethyl acetamide (DMAc) at 30 °C and decreased with the introduction of the ether and pendant groups into the polymer. All the polymers were soluble in DMAc, N,N-dimethyl formamide (DMF), and N-methyl-2-pyrrolidone (NMP) and their solutions could be cast into flexible films with good mechanical strengths. Further, they exhibited a great affinity to water sorption. Their solubility and hydrophilicity increased with introduction of the ether and pendant groups into the polymer. The prepared polymers could be thermally cyclodehydrated under nitrogen atmosphere into the corresponding poly(ether-amide-1,3,4-oxadiazole)s approximately in the region of 300–450 °C. The introduction of the flexibilizing ether linkages and the pendant groups into the polymer improves the solubility of the resulting poly(ether-amide-1,3,4-oxadiazole)s compared to poly(amide-1,3,4-oxadiazole) free from these groups.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Wholly aromatic polyamide-hydrazides have attracted great attention as a class of high performance materials with high thermal and thermo-oxidative stability [1–5]. They are extensively used in reverse osmosis applications [6–14]. Their appropriately drawn fibers showed very high mechanical strength and moduli [15–17]. Further, their modified transition metal chelates have shown good electrical conductivity [18–21]. Aromatic polyamide-hydrazides are well known as precursors to polyamide-1,3,4-oxadiazoles which are one of the most important classes of chemically and thermally stable heterocylic polymers [22, 23]. Polyamide-1,3,4-oxadiazoles are considered to be an interesting alternative for the development of high-temperature- and flame-resistant fibers [24, 25] and thermally stable membranes for gas separation [26]. In addition, they have been widely investigated in the field of polymer light-emitting diodes as well as other fields of polymer electronics [27, 28]. Unfortunately, these polymers are difficult to process because of low solubility in organic solvents and high melting or softening temperatures. Many efforts have been made to improve these characteristics to make such polymers more processable by the introduction of flexible linkages in the backbone [29, 30] or bulky pendant groups on the aromatic rings [31]. Thus, it was of interest to investigate the possibility of preparing and evaluating a series of novel wholly para-oriented aromatic poly(ether-amide-hydrazide)s containing bulky pendant groups on their aromatic rings and their corresponding poly(ether-amide-1,3,4-oxadiazole)s. p-Aminosalicylic acid hydrazide (PASH), 4,4′-(1,4-phenylenedioxy)dibenzoyl chloride (1a), 4,4′-(2,5-tolylenedioxy)dibenzoyl chloride (1b), 4,4′-(2-tert-butyl-1,4-phenylenedioxy)dibenzoyl chloride (1c), and 4,4′-(2,5-biphenylenedioxy)dibenzoyl chloride (1d) were selected as monomers to be used in this study. Besides the afore-mentioned properties of wholly aromatic polyamide-hydrazides, one can expect the greater hydrophilicity of the prepared polymers, owing to the presence of ether and hydroxyl groups in addition to the amide and hydrazide linkages in their repeating units, which is of prime importance for their processing as synthetic fibers and membranes. Further, we hoped that the incorporation of both ether and pendant groups would result in oxadiazole polymers with enhanced solubility and acceptable other characteristics. For a comparative study, a related polymer without the ether and the pendant groups (poly[4-(terephthaloylamino)salicylic acid hydrazide]), PTASH, was also investigated. A detailed description of its preparation and properties has already been reported [32].
Experimental
Monomer synthesis
4,4′-(1,4-Phenylenedioxy)dibenzoyl chloride (1a) (m.p. 207–209 °C), 4,4′-(2,5-tolylenedioxy)dibenzoyl chloride (1b) (m.p. 165–167 °C), 4,4′-(2-tert-butyl-1,4-phenylenedioxy)dibenzoyl chloride (1c) (m.p. 126–128 °C), and 4,4′-(2,5-biphenylenedioxy)dibenzoyl chloride (1d) (m.p. 118–121 °C) were synthesized according to the method originally developed by Hsiao et al. [33]. Terephthaloyl chloride was obtained from Acros, Germany. p-Aminosalicylic acid hydrazide (PASH) was prepared according to the method described by Drain et al. [34].
Polymer synthesis
All polymers were prepared by essentially the same experimental procedure, which will be given here, as an example, for the preparation of polymer 2d. p-Aminosalicylic acid hydrazide (PASH), 0.84 g (5 mmol), was dissolved in 25 ml of dry NMP and cooled at −10 °C for 15 min. 2.32 g (5 mmol) solid diacyl chloride (1d) was added slowly under constant stirring over a period of 1 h. Stirring was continued for another 2 h at the aforementioned temperature. Then, the temperature of the polymerization reaction was allowed to rise gradually to room temperature and maintained for 24 h with stirring. Afterward, a clear, slightly yellow, highly viscous solution was obtained. Finally, the polymer solution was slowly poured into 100 ml of rapidly stirred methanol upon which a fibrous white precipitate of polymer 2d immediately formed. The polymer was isolated by filtration, washed successively with methanol and acetone, and dried in a vacuum oven at 75 °C to constant weight.
Film preparation and cyclodehydration of the hydrazide polymers
Films were prepared by casting on glass plates from 5 wt% polymer solutions in DMAc. Solvent evaporation was performed at a constant temperature of 120 °C for 4 h. The films were then immersed in deionized water overnight to remove any residual solvent. Finally, the films were dried in vacuum oven at 75 °C to constant weight. The thickness of the films was controlled to be 50 μm.
The cyclodehydration to poly(ether-amide-1,3,4-oxadiazole)s was carried out via the heating of the fabricated films of the corresponding hydrazide polymers at 450 °C for 1 h under nitrogen atmosphere.
Polymer characterization
Elemental analyses of the prepared polymers were done in a Perkin-Elmer (Model 2410 Series II) C, H, N Analyzer (USA) at the Microanalytical Unit, Cairo University (Egypt).
Infrared spectra of the polymer films were measured on a Perkin-Elmer Infrared Spectrophotometer (FTIR 1650). All spectra were recorded within the wave number range of 4000–400 cm−1 at 25 °C.
Intrinsic viscosities were measured for polymer solutions in DMAc with a 0.5 gdl−1 concentration at 30 °C using a suspended-level Ubbelohde viscometer with negligible kinetic energy correction. Flow times were measured at five different concentrations of the polymer sample. All the plots obtained were linear. Intrinsic viscosity was determined by usual extrapolation of ηsp/c to zero concentration and expressed in dlg−1.
Solubility of the polymers in various amide solvents namely: DMF, DMAc, and NMP was determined at 25 °C. It was performed by gradual addition of the polymer to the solvent and stirred well till saturation. The maximum solubility of the polymers was calculated as percent weight of polymer per hundred ml of solvent (% wt/v).
The percent moisture regained by the polymer sample was determined by allowing a dried polymer sample to absorb moisture for 24 h in a humidity chamber maintained at 85% relative humidity at 23 °C. Then, a weighed amount of the moisture-absorbed sample was dried in an air oven at 110 °C to constant weight. The percent moisture regained by the polymer sample was calculated on the basis of weight loss.
A Rigaku Denki Co. Ltd., RADB system diffractometer with a Ni monochromater was used to record the X-ray diffractograms of these polymers films. X-ray source was CuKα (40 kV/15 mA). The sample was maintained stationary, while scattering angles from 3 to 900 were scanned in the reflection mode at a scanning rate of 2° min−1.
Tensile strength, elastic modulus, and elongation at break (%) of the polymer films were measured on Shimadzu Autograph in air at room temperature.
Differential scanning calorimetry (DSC) measurements were performed on a Shimadzu differential scanning calorimeter with a resolution of 0.04 μW (Model DSC-50). The scan ranging between 30 and 600 °C was investigated, using a constant scanning rate of 10 °C min−1. The samples analyzed were 3 ± 0.2 mg in all the cases. All the DSC measurements were carried out in nitrogen atmosphere with a flow rate of 30 ml min−1.
Thermogravimetric analysis (TG) curves of all the polymers were recorded on a Shimadzu TGA-50H Thermogravimetric Analyzer with a resolution of 0.04 μW, under nitrogen and air atmospheres. The scanning rate was 10 °C min−1 over a scanning range from 30 to 800 °C. The samples’ weights ranged from 3 to 5 mg, and the gas flow rate was 30 ml min−1.
Results and discussion
Polymer synthesis
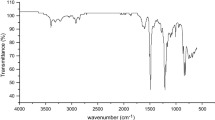
Four novel wholly aromatic poly(ether-amide-hydrazide)s bearing bulky pendant groups on their aromatic rings have been synthesized by reacting PASH with stoichiometric amount of either 1a, 1b, 1c, or 1d via a low temperature (−10 °C) solution (in anhydrous NMP as a solvent) polycondensation as shown in Scheme 1. It is established that, in such an acidic condition, the rate by which hydrazide group of aminobenzhydrazide reacted with acid chloride is about seven times greater than that of the amino group with the same acid chloride [35, 36]. These properties indicate that when this polymerization is performed by gradual addition of solid acid chloride in a solution of aminobenzhydrazide monomer, the hydrazide groups of the latter react first, and the so-called wholly ordered polymer with alternating amide and hydrazide linkages is formed. Further, the elemental analysis values of the hydrazide polymers 2a–2d and PTASH agreed well with the theoretical ones calculated for the expected repeating units (Scheme 1) as represented in Table 1. All the IR spectra of the hydrazide polymers showed common absorption stretching vibration bands at the following wave numbers: (1) 3700–3000 cm−1 (intensive and broad) assigned the overlapped –NH, –OH (phenolic) and the possible interchain hydrogen bonding; (2) 2300–2250 cm−1 (weak) is attributed to possible enol-type configuration of hydrazide and amide groups; (3) 1670–1650 cm−1 (strong) corresponded to the amide carbonyl groups; (4) 1600 cm−1 indicated the aromatic carbon–carbon double bonds; (5) 1540–1520 cm−1 is due to =NH of amide II; (6) 1500 cm−1 indicated carbon–carbon single bond in ring; (7) 1420 cm−1 is attributed to carbon–oxygen (phenolic); (8) 1330–1320, 1290–1280, 1260 cm−1 corresponded to carbon–hydrogen combined with =NH of amide III; and (9) 1100 cm−1 assigned the ether linkages stretching. Figure 1 shows the IR spectrum of polymer 2a as an example. The intrinsic viscosity values for the prepared poly(ether-amide-hydrazide)s which stayed in the range of 3.14 and 2.47 dlg−1 are listed in Table 1. These values reflect the high molecular weight of the polymers. The polymers 2a and 2b exhibited higher intrinsic viscosities relative to those of the 2c and 2d. This experimental finding may be attributed to the higher chain symmetry and packing efficiency of the non-substituted and the methyl-substituted polymers which permit greater interchain hydrogen bonding formation. On the other hand, polymer 2a showed lower viscosity than that of PTASH as a result of incorporation of the flexibilizing ether linkages in the former. All of the poly(ether-amide-hydrazide)s formed flexible, transparent, and colorless films from DMAc. The prepared poly(ether-amide-hydrazide)s were thermally cyclodehydrated into their corresponding poly(ether-amide-1,3,4-oxadiazole)s (Scheme 1). The films of the hydrazide polymers changed from being colorless to being darkened after cyclodehydration. This may be attributed to the conjugation and/or formation of charge-transfer complex between the oxadiazole ring and the aromatic unit. Poly(ether-amide-1,3,4-oxadiazole)s showed lower viscosities than those of the corresponding hydrazide polymers as shown in Table 1. This may be attributed to the decreased intermolecular interactions and/or stress buildup caused by chain shrinkage during the cyclization. These results are supported by those reported by Hensema et al. [37]. Thermal cyclodehydration of the hydrazide group to the 1,3,4-oxadiazole ring was monitored with FTIR. As a representative study, a thin film sample of polymer 2a was heated at 450 °C under nitrogen atmosphere for 1 h. Changes of the IR spectrum of this sample is illustrated in Fig. 1. After thermal treatment, the hydrazide polymer 2a was almost completely cyclodehydrated to the corresponding poly(1,3,4-oxadiazole) 3a (Scheme 1), as confirmed by the reduction of the intensity of the absorption peak at 3700–3000 cm−1, indicating deprotonation of the –NH of the hydrazide groups and partial loss of hydrogen bonding. Further, the reduction of the intensity of the carbonyl vibration band (at 1670–1650 cm−1) suggested partial loss of carbonyl double bond character. Moreover, IR spectrum showed the appearance of new absorption bands at 1620 cm−1, corresponding to C = N stretching vibration and at 1020 and 960 cm−1 which are assigned to =C–O–C= group [1–3]. The cyclodehydration reaction was further proved experimentally by identifying the nature of the residual products formed in the later stages of thermal treatment. For this purpose, elemental analyses of the oxadiazole polymers 3a-3d and PTASH′, which had been thermally cyclodehydrated in nitrogen at 450 °C for 1 h, were carried out. The results are presented in Table 1. It may be observed that the elemental analysis data are in excellent agreement with those calculated for the expected repeating units of polymers 3a, 3b, 3c, and 3d (Scheme 1) and PTASH′. TG and DSC measurements, as will be discussed later, were also used to investigate the cyclodehydration reaction. 

Typical synthetic scheme for poly(ether-amide-hydrazide)s and the corresponding poly(ether-amide-1,3,4-oxadiazole)s

FRIR spectra of ply (ether-amide-hydrazide) 2a and its corresponding poly(ether-amide-1,3,4-oxadiazole) 3a
Polymer characterization
Solubility
The results of the solubility of the poly(ether-amide-hydrazide)s are listed in Table 2. All these polymers were highly soluble in polar solvents such as DMAc, DMF, and NMP at room temperature. The polymers 2a and 2b showed lower solubilities relative to those of the polymers 2c and 2d. The attachment of bulky tert-butyl- and phenyl- groups on the aromatic rings introduces lateral disorder by forcing the chain apart and interferes with the intermolecular cohesion of the existing polar linkages and leads to increased chain packing distance and decreased interchain interactions such as hydrogen bonding; thus the polymers 2c and 2d were more soluble than the other polymers. Further, the introduction of the flexibilizing ether linkages into polymer 2a increases its solubility with respect to PTASH. On the other hand, the oxadiazole polymers showed decreased solubilities with respect to the corresponding hydrazide prepolymers (Table 2). This is due to the increased chain stiffness. The introduction of flexible ether linkages and the bulky pendant groups increased the solubility of the oxadiazole polymers 3a–3d as compared with that of the oxadiazole polymer free from these groups (PTASH′).
Percent moisture regain
All the polymers showed strong hydrophilicity as indicated from their high moisture regain values which lie between 22 and 24.64% (at 23 °C, relative humidity 85%) (Table 2). The hydrophilicity of these polymers can be related to their polar structures that possess ether, amide, hydroxyl, and hydrazide groups in each repeating unit. These polar groups form hydrogen bonds with water readily promoting its adsorption by these polymers. The hydrophilicity is increased with increasing the size of the group X on the aromatic ring of the evaluated polymers. This observation may be explained by the polymers 2a and 2b being more strongly hydrogen bonded than 2c and 2d polymers as a result of their greater chain symmetry and shorter interchain distance. However, the separation between the macromolecular segments of polymers 2c and 2d would be further expected, particularly in the presence of water, and consequently the inter- or intra-hydrogen bonds would be partially restricted. Thus, they are free to interact with water. The enhancement of the moisture regain of the polymer 2a relative to that of the PTASH is attributed to the incorporation of the flexibilizing ether linkages into the chains of the former. Owing to the decreased chain polarity, the oxadiazole polymers exhibited a lower percent moisture regain relative to that of the corresponding hydrazide polymers (Table 2). The high hydrophilicity of the hydrazide polymers guides us to select them as reverse osmosis candidate materials. Their fabrication into semipermeable membranes for reverse osmosis performance will be reported in the forthcoming article.
Crystallinity
Figure 2 shows the wide-angle X-ray diffraction patterns of the evaluated polymers. In this figure, polymers 2a and 2b showed a higher crystalline pattern relative to that of polymers 2c and 2d. Thus, methyl substitution seems to result in a little influence. The introduction of bulky pendent tert-butyl or phenyl group destroys the backbone symmetry and regularity and interferes with the intermolecular hydrogen bonding and the tight packing of the polymer chains, thus resulting in a decrease in crystallinity. Moreover, polyoxadiazole 3a (as a representative example) showed increased crystallinity with respect to its corresponding hydrazide precursor 2a (Fig. 3). This result may be attributed to the similarity of 1,3,4-oxadiazole to p-phenylene structure which is known to be a planner rigid unit.

Wide-angle X-ray diffractograms of poly (ether-amide hydrazides)

Wide-angle X-ray diffractograms of poly(ether-amide-hydrazide) 2a and its corresponding poly (ether-amide-1,3,4-oxadizole) 3a
Mechanical properties
Table 3 summarizes the mechanical properties of the prepared polymer films. The tensile strengths, elongation at break, and initial moduli of the poly(ether-amide-hydrazide)s were in the range of 200.78–154.43 MPa, 55.31–113.25%, and 3.89–3.11 GPa, respectively. In general, elastic modulus as well as tensile strength of the polymers are decreased as the size of the substituent X on the aromatic rings increases. On the other hand, the ductility is increased by increasing the size of the substituent X as displayed by the values of elongation at break. The thermally converted poly(ether-amide-1,3,4-oxadiazole) films showed better results for both tensile strengths and initial moduli relative to those of the corresponding hydrazide precursors. This may be attributed to the increased stiffness of the oxadiazole polymers.
Thermal stability
Thermal stability and degradation behavior of a series of novel wholly para-oriented aromatic polyamide-hydrazides containing the flexibilizing ether linkages in their main chains and bulky pendant groups on their aromatic rings have been investigated by differential scanning calorimetry (DSC) technique. A polyamide-hydrazide without the flexibilizing linkages and the pendant groups is also studied for comparison. The typical DSC thermograms of the evaluated polymers are represented in Fig. 4. All these thermograms were recorded at 10 °C min−1 heating rate, and under prepurified nitrogen with a constant flow rate of 30 ml min−1. DSC curves indicate a common thermal behavior of these polymers which all exhibit three endotherms. The first endotherm is small and is attributed to the evaporation of the absorbed surface water. The second endotherm is large and broad due to the thermally induced cyclodehydration reaction of the polymers hydrazide groups into 1,3,4-oxadiazole rings by the loss of water molecules [1, 5] as represented in Scheme 1. The third endotherm is due to the decomposition of the poly(ether-amide-1,3,4-oxadiazole)s which were formed in situ. It can be noted from Fig. 4 that the areas under the curves are not the same. This may be attributed to the differences in the amount of the absorbed surface water, and water released during the cyclodehydration reaction. On the other hand, there is a slight shift of the decomposition temperature and of the cyclodehydration peak to a lower temperature as the flexibilizing ether linkages introduced into the backbone chains as well as the bulky pendant groups on their aromatic rings as compared with PTASH. Finally, it could be noted that it was not possible to observe any melting transitions of the evaluated polymers during the DSC measurements.

DSC thermograms of poly(ether-amide-hydrazide)s. All the thermograms were recorded in nitrogen at a heating rate of 10 °C min−1 and a gas flow rate of 30 ml min−1
TG measurements were also performed on the evaluated polymers to examine the influence of their structural differences on their degradation behavior under the purely thermal and thermo-oxidative conditions. All these measurements were carried out at 10 °C min−1 heating rate, under the constant streams of prepurified nitrogen and air (the flow rates of which were in all cases 30 ml min−1), and the results obtained are shown in Figs. 5 and 6, respectively. These results are in fairly good agreement with those of DSC. It can be seen from these results that in both degradation atmospheres all polymers showed a characteristic similar thermal behavior which consisted of three distinctive steps in which appreciable weight losses were observed. During the first weight-loss step, which occurred in both investigated atmospheres between 90 and 120 °C, all of the samples exhibited relatively small losses of only about 1–2% of their original weights, as shown in Figs. 5 and 6. These weight losses were clearly attributable to evaporation of absorbed moisture from the surface of the polymer samples. The second step in which all of the investigated samples showed considerable losses, occurred in different temperature ranges for various polymers in nitrogen and in air atmospheres as listed in Table 4. This step reflected the occurrence of the thermally induced cyclodehydration reaction of the polymers into the corresponding poly(ether-amide-1,3,4-oxadiazole)s, 3a–3d, by losing water (Scheme 1). The amount of water evolved during the cyclodehydration reaction was 3–6 wt% (based on the weight of the perfectly dried polymer samples), which seems to be in good agreement with the theoretical values calculated for the expected polymers repeating units (Table 4). The third weight-loss step is steep and assigned to the decomposition of the poly(ether-amide-1,3,4-oxadiazole)s, which were formed in situ. It is worth mentioning that the beginnings, maxima, and the ends of these steps agreed well with the corresponding DSC values. Moreover, the resulting poly(ether-amide-1,3,4-oxadiazole)s start to decompose in the temperature range above 490–520 °C in nitrogen and above 480–520 °C in air, without weight loss at lower temperatures. They lost 29.5–36% of their original weights at 800 °C in nitrogen. On the other hand, smaller residues were obtained at 800 °C in air (Table 4). These results indicate that the decomposition rate of the poly(ether-amide-1,3,4-oxadiazole)s in air is faster than that obtained in nitrogen, and the former atmosphere appeared more destructive as confirmed by the weight loss of the samples at 800 °C as well as at particular temperatures (Table 4). The differences between the weight losses of various polymers (Table 4) may be attributed to the differences of the amount of absorbed moisture and water eliminated during the cyclodehydration reaction. Thus, the incorporation of the evaluated ether linkages and the pendant groups did not seem to significantly influence the thermal stability of the oxadiazole polymers. The observed high thermal stability may be attributed to the chemical structure of the polymer which possesses an aromatic, 1,3,4-oxadiazole rings, amide, ether and phenolic –OH groups in its repeating unit. These groups are known to be highly resistant to elevated temperatures in addition to the strong hydrogen bonding established between the amide and the phenolic –OH groups of the neighboring chains. This indicates that polyamide-hydrazides can be used as precursor polymers for preparation of thermally stable 1,3,4-oxadiazole containing polyamides.

Typical TG thermograms patterns of poly(ether-amide-hydrazide)s. All the thermograms were recorded in nitrogen at a heating rate of 10 °C min−1 and a gas flow rate of 30 ml min−1

Typical TG thermograms patterns of poly(ether-amide-hydrazide)s. All the thermograms were recorded in air at a heating rate of 10 °C min−1
Conclusions
High molecular weight easily processed new wholly para-oriented aromatic poly (ether-amide-hydrazide)s containing various pendant groups have successfully been synthesized as highly viscous film forming solutions. All the prepared polymers are highly thermally stable and could be thermally cyclodehydrated into the corresponding poly(ether-amide-1,3,4-oxadiazole)s at elevated temperature ranges. The introduction of the flexibilizing ether and bulky pendant groups leads to polymers of a higher solubility in DMF, DMA, and NMP and of higher affinity to water sorption and relatively reduced mechanical and intrinsic viscosity with respect to the polymer free from these linkages. Although the incorporation of the flexibilizing ether linkages into the polymer main chains and various pendant groups on their aromatic rings did not seem to significantly influence the thermal stability of the polyoxadiazoles, they remarkably enhanced their solubility in comparison to that of polymer free from these linkages. Further, oxadiazole polymers showed decreased solubility with respect to the corresponding hydrazide precursors.
References
Culbertson BM, Murphy R (1967) Aromatic poly(amide-hydrazides) and poly(amide-1,3,4-oxadiazoles). J Polym Sci Polym Lett 5:807–812
Dvornic PR (1984) Wholly aromatic polyamide-hydrazides. III. Thermal stability and degradation behaviour of poly[4-(terephthaloylamino)benzoic acid hydrazide]. Bull Soc Chim Beograd 49:589–606
Mohamed NA (1994) Novel wholly aromatic polyamide-hydrazides. V. Structure-molecular-weight-thermal-stability relationships. Polym Degrad Stab 44:33–42
Mohamed NA, Al-Dossary AOH (2003) Structure-property relationships for novel wholly aromatic polyamide-hydrazides containing various proportions of para-phenylene and meta-phenylene units. II. Thermal stability and degradation behaviour. Polym Degrad Stab 79:61–75
Al-Ghamdi RF, Fahmi MM, Mohamed NA (2006) Thermal stability and degradation behaviour of novel wholly aromatic azopolyamide-hydrazides. Polym Degrad Stab 91:1530–1544
Sourirajan S, Matsura T (1979) Environmental Protection Agency Symposium Proceedings Textile and Technology No. 299132 (US Publ No EPA-60012-79-104); EPA: Chicago, II, 1979, p 73
Dvornic PR (1986) Wholly aromatic polyamide-hydrazides. IV. Structure-property relationships for polymers containing p-phenylene and m-phenylene units. J Polym Sci Part A 24:1133–1160
McKinney R Jr, Rhodes JH (1971) Aromatic polyamide membranes for reverse osmosis separations. Macromolecules 4:633–637
Satre MD, Ghatge ND, Ramani MPS (1990) Aromatic polyamide-hydrazides for water desalination. I. Syntheses and RO membrane performance. J Appl Polym Sci 41:697–712
Dvornic PR (1991) Wholly aromatic polyamide-hydrazides. V. Preparation and properties of semipermeable membranes from poly[4-(terephthaloylamino)benzoic acid hydrazide]. J Appl Polym Sci 42:957–972
Satre MD, Ghatge ND (1993) Aromatic polyamide-hydrazides for water desalination. II. RO membrane performance of terpolymers. Desalination 91:121–133
Nakamae K, Mohamed NA (1993) Novel wholly aromatic polyamide-hydrazide. II. Preparation and properties of membranes for reverse osmosis separation. J Appl Polym Sci Appl Polym Symp 52:307–317
Mohamed NA (1997) Novel wholly aromatic polyamide-hydrazides. 6. Dependence of membrane reverse osmosis performance on processing parameters and polymer structural variations. Polymer 38:4705–4713
Mohamed NA, Al-Dossary AOH (2003) Structure-property relationships for novel wholly aromatic polyamide-hydrazides containing various proportions of para-phenylene and meta-phenylene units. III. Preparation and properties of semi-permeable membranes for water desalination by reverse osmosis separation performance. Eur Polym J 39:1653–1667
Preston J, Black WB, Hofferbert WL Jr (1973) High modulus wholly aromatic fibers. I. Wholly ordered polyamide-hydrazides and poly-1,3,4-oxadiazole-amides. J Macromol Sci-Chem 7:45–65
Preston J, Black WB, Hofferbert WL Jr (1973) High modulus wholly aromatic fibers. II. Partially ordered polyamide-hydrazides. J Macromol Sci-Chem 7:67–98
Black WB, Preston J, Morgan HS, Raumann G, Lilyguist MR (1973) Some physical and mechanical properties of some high-modulus fibers prepared from all-para aromatic polyamide-hydrazides. J Macromol Sci-Chem 7:137–171
Mohamed NA, Nakamae K (1993) Novel wholly aromatic polyamide-hydrazide. 3. Preparation and characterization of semiconductors from poly[4-(terephthaloylamino) salicylic acid hydrazide]-metal chelates. Polymer 34:3940–3947
Mohamed NA (1998) Novel wholly aromatic polyamide-hydrazides. VII. Metallization of polymers through transition metal complexation. Eur Polym J 34:387–398
Mohamed NA, Al-Dossary AOH (2003) Structure-property relationships for novel wholly aromatic polyamide-hydrazides containing various proportions of para-phenylene and meta-phenylene units. IV. Preparation and characterization of metalized plastic films through transition metal complexation. Polym Testing 22:785–800
Mohamed NA (2007) Structure-property relationships for novel wholly aromatic polyamide-hydrazides containing various proportions of para-phenylene and meta-phenylene units. V. Evaluation of the electrical surface conductivity of several metalized plastic films. Polym Testing 26:471–481
Cassidy PE (ed) (1980) Thermally stable polymers. Marcel Dekker, New York, p 179
Nanjan MJ (1988) Polyhydrazides and polyoxadiazoles. In: Mark HF, Bikales NM, Overberger CG, Menges G, Kroschwitz JI (Eds) Encyclopedia of polymer science and engineering, vol 12, 2nd ed., Wiley, New York, pp 322–339
Frazer AH, Wallenberger FT (1964) Poly(1,3,4-oxadiazole) fibers: new fibers with superior high temperature resistance. J Polym Sci Part A 2:1171–1179
Yang HH (ed) (1989) Aromatic high-strength fibers. Wiley, New York, pp 315–348
Gebben BMH, Mulder V, Smolders CA (1989) Gas separation properties of a thermally stable and chemically resistant polytriazole membrane. J Membr Sci 46:29–41
Song S-Y, Jang MS, Shim M-K, Hwang D-H, Zyung T (1999) Highly efficient light-emitting polymers composed of both hole and electron affinity units in the conjugated main chain. Macromolecules 32:1482–1487
Lee Y-Z, Chen S-A (1999) Oxadiazole-containing phenylene vinylene ether linkage copolymer as blue-green luminescent and electron transport material in polymer light-emitting diodes. Synth Met 105:185–190
Hsiao S-H, Yu C-H (1998) Novel aromatic polyhydrazides and poly(amide-hydrazide)s based on multiring flexible dicarboxylic acids. J Polym Sci Part A 36:1847–1854
Mohamed NA, Fahmi MM (2009) Synthesis and characterization of novel wholly para-oriented aromatic polyamide-hydrazides containing sulfone-ether linlages. J Appl Polym Sci 113:767–776
Ubale VP, Patil AS, Maldar NN (2007) Polyhydrazides based on 2,5-bis(4-carboxymethylenephenyl)-3,4-diphenyl thiophene. Eur Polym J 43:1038–1045
Mohamed NA, Nakamae K (1993) Novel wholly aromatic polyamide-hydrazide. I. Syntheses and properties of films based on a polymer of p-aminosalicylic acid hydrazide via polycondensation with terephthaloyl chloride. J Appl Polym Sci Appl Polym Symp 52:297–306
Hsiao S-H, Dai L-R, He M-H (1999) Synthesis and properties of novel aromatic polyhydrazides and poly(amide-hydrazide)s based on the bis(ether benzoic acid)s from hydroquinone and substituted hydroquinones. J Polym Sci Part A 37:1169–1181
Drain DJ, Martin DD, Mitchell BW, Seymour DE, Spring FS (1949) 4-Aminosalicylic acid and its derivatives. J Chem Soc 1498–1503
Morrison RW, Preston J, Randall JC, Black WB (1973) Self-regulating polycondensation. II. A study of the order present in polyamide-hydrazides derived from terephthaloyl chloride and p-aminobenzhydrazide. J Macromol Chem 7:99–118
Randall JC, Morrison RW, Preston J (1973) Self-regulating polycondensation. III. NMR analysis of oligomers derived from terephthaloyl chloride and p-aminobenzhydrazide. J Macromol Sci Chem 7:119–133
Hensema ER, Boom JP, Mulder MHV, Smolders CA (1994) Two reaction routes for the preparation of aromatic polyoxadiazoles and polytriazoles: syntheses properties. J Polym Sci Part A 32:513–525
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fahmy, M.M., Al-Ghamdi, R.F. & Mohamed, N.A. Synthesis, characterization, and thermal stability of novel wholly para-oriented aromatic poly(ether-amide-hydrazide)s bearing pendant groups and their corresponding poly(ether-amide-1,3,4-oxadiazole)s. Polym. Bull. 66, 609–625 (2011). https://doi.org/10.1007/s00289-010-0296-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-010-0296-8