
Abstract
Novel side chain dendronized polyurethane was synthesized by the sequential conjugation of amine-terminated fourth generation polyester dendrons with carboxyl groups at the side chain of polyurethane and deprotection of acetonide groups at the surface of dendrons. The effect of each step on the properties of the polymers was measured by GPC, DSC, SEM, AFM, and water contact angle measurement. The results showed that immobilization of polyester dendrons improve the compatibility of hard and soft segments in polyurethane and decrease the tendency of crystallinity due to the bulkiness of the substituents. After deprotection of the acetonide groups, both bulk and surface hydrophilicity of the polymer were evidently enhanced. Cell toxicity analysis showed that the side chain dendronized polyurethane synthesized in this study has exhibited excellent biocompatibility to human embryonic kidney 293T cells. The cells can effectively adhere to and proliferate on the surface of the polymer film. Overall, the present study demonstrated an efficient modification method for polyurethanes to improve their cell compatibility with high potential in biomedical applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyurethanes (PUs) are popularly investigated in biomedical applications because of their long-term biostability, excellent mechanical properties, and moderately good biocompatibility [1–4]. In recent years, many attentions have been focused on employing PUs as new tissue engineering materials with all their versatility in terms of physical properties and biocompatibility [1, 5]. However, cell compatibility of PUs is relatively poor, which has become one of the major drawbacks that hinder their further biomedical applications [6–8]. Various methods have been developed to improve cell compatibility of PUs including plasma treatment [9], introduction of gelatin [10], or grafting biological agents (such as RGD) to their surfaces [11]. But these methods are limited to surface modification without affecting their bulk properties which may be not quite suitable for the preparation of tissue engineering materials.
Polyester dendrimers/dendrons, prepared by a stepwise synthetic procedure have attracted many attentions in drug delivery systems and other biomedical areas due to their unique properties including highly regular branching pattern, well-defined architecture, mono molecular weight distribution, and excellent biocompatibility [12–14]. It would be interesting if these polyester dendrons could be conjugated with PU chains. Introduction of polyester dendrons not only makes the PU chains surrounded by a strictly controlled number of hydroxyl groups that can be used for the attachment of drugs, solubilizing groups, targeting moieties, or other molecules to tune the biological properties of the polymer, but also grafts spacers between the PU chains that may be contribute to modify the physical properties of the polymer.
In this study, such method of polyester dendrons immobilization was adopted and the characteristics of the modified polymers were explored. The aim of the present study was to evaluate if this method could be utilized to improve the cell compatibility for PUs.
Experimental
Materials
1,6-Hexane diisocyanate (HDI) was purchased from Bayer with purity greater than 99.5%. Polycarbonate diols (PCDL) with a number average molecular weight of 2,000 g mol−1 was kindly provided by Asahi Kasei Corporation and dried in vacuum at 50 °C for 24 h prior to use. 2,2-bis(hydroxymethyl) propionic acid (bis-MPA) and Dowex H+ resin were purchased from Acros and used as received. The amine-terminated fourth generation polyester dendrons (G4-NH2) were synthesized according to the previous method by a divergent route based on anhydride chemistry [15, 16]. Dimethylformamide (DMF) and tetrahydrofuran (THF) were refluxed over calcium hydride for 3 days and distilled prior to use. Other reagents were commercially available and used as received.
Synthesis of the PU base polymer
In an oven dried four-neck flask equipped with a stir bar, reflux condenser, and nitrogen on command, 0.84 g (5 mmol) of HDI and three drops of dibutyltin dilaurate (DBTDL) were dissolved in 50 mL of DMF and maintained at 80 °C with continuous stirring. 5 g (2.5 mmol) of PCDL dissolved in 50 mL of DMF was added dropwise within 4 h to form a prepolymer solution. After which, 0.335 g (2.5 mmol) of chain extender (bis-MPA) dissolved in 10 mL of DMF was added to the prepolymer solution and was stirred at 80 °C for 2 h to form the polyurethane solution. Subsequently, the solution was poured into a large amount of water and the precipitate was collected after dried in vacuum at 50 °C for 24 h. The obtained polymer was then re-dissolved in dichloromethane and the solution was centrifuged at 13,000 rpm for 1 h to remove the heavy metal catalyst. Finally the polymer solution was precipitated into a large amount of chilled methanol, filtered and dried in vacuum at room temperature.
Synthesis of PU-dendrons conjugate (PU-G4)
PU (2 g, n[COOH] = 0.74 mmol, determined by titration) and CDI (1.05 g, 6.5 mmol) were dissolved in 100 mL of THF and stirred at room temperature for 12 h to activate the carboxyl groups at the side chain of the polymer. After which, 5.2 g (2.43 mmol) of G4-NH2 dissolved in 20 mL of THF was added slowly within 5 h. The mixture was then allowed to stir at room temperature for additional 24 h. Once the reaction was complete, the catalyst was filtered off and the solution was centrifuged at 13,000 rpm for 10 min, and finally precipitated into excess methanol. This process was repeated again to remove any residue of unreacted CDI. After dried in vacuum at room temperature for 24 h, PU-G4 was collected as a white solid.
Deprotection of the acetonide groups at the surface of the dendrons
Two grams of PU-G4 was dissolved in a mixture of 75 mL of THF and 25 mL of MeOH. Three teaspoon of Dowex H+ resin was added and the mixture was stirred at room temperature for 24 h. The Dowex H+ resin was filtered off and washed with small amount of THF. Then the THF solution was condensed to about 30 mL and precipitated into methanol to give 1.76 g of PU-G4-OH as a white solid.
Characterization
1H-NMR spectra were recorded at room temperature on a Bruker DRX-500 using CDCl3 as the solvent. Gel permeation chromatography (GPC) was carried out on a Waters HPLC system equipped with a Model 2690D separation module, a Model 2410 refractive index detector, and Shodex columns (K802.5, K803, and 805). THF was used as eluent at the flow rate of 1.0 mL/min. Calibration was fulfilled with narrow-molar-mass distributed polystyrene standards. Differential scanning calorimetric (DSC) analysis was carried on a DSC2910 thermal analysis system (TA Instruments Inc., USA) at a heating rate of 10 °C/min under nitrogen atmosphere. Surface morphologies of the cast films were examined by a scanning electron microscope (SEM, JSM-6360LV, JOEL). Atom force microscope (AFM) was used to analyze the microphase-separation between the soft and hard segments.
Functionality of side chain carboxyl groups with G4 dendrons in PU-G4
The average functionality of side chain carboxyl groups with G4 dendrons in PU-G4 was determined by titration. Generally, 0.2 g of PU-G4 and a few drops of phenolphthalein were dissolved in 20 mL of 1:1 dichloromethane:methanol. Then, 0.05 M NaOH/EtOH was added slowly until the polymer solution become reddish. The volume of NaOH/EtOH consumed was obtained and the amount of carboxyl groups in PU-G4 was calculated. The functionality of carboxyl groups (FCG) in PU-G4 was calculated based on:
wherein CG1 and CG2 are the content of carboxyl groups in PU-G4 and PU, respectively.
Equilibrium water content and contact angel measurement
Polymer films (3 for per sample) of 2 cm diameter disks were cut from cast films, dried at 50 °C in a vacuum oven, and weighted. The samples were then immersed in distilled water/and phosphate buffered saline (PBS, pH = 7.4) at 37 °C for 1, 2, 4, 12, 24, and 48 h, respectively. At each time point, the samples were removed and blotted lightly with filter paper to remove excess water. The weight of the hydrated samples was then determined and the water adsorption was calculated.
Contact angle measurements were carried out using the methods described earlier [17]. For each sample, twelve data points were taken for mean and standard deviation calculations.
In vitro cell responses evaluation
Cell responses to PU, PU-G4, and PU-G4-OH were evaluated by seeding human embryonic kidney (HEK) 293T cells (0.9 × 105 cells/mL) on the polymer films in the medium (Dulbecco’s modified Eagle’s medium, DMEM, Gibco) supplemented with 10% Newborn Calf Serum (NCS, Gibco). Polymer films of 0.7 cm diameter disks were cut from cast films and putted into 96-well tissue culture plates. Next, 0.5 mL of medium was added to the wells and mixed with the cells. The cells were then cultured in a humidified incubator equilibrated with 5% CO2–95% air. Cell attachment was evaluated after 4 days. The morphology of cells attached onto the polymer surface was observed using a phase contrast microscope. The cells were also stained with Hoechst No. 33342 and observed using a fluorescent microscope. Cell proliferation was determined by MTT assay.
Results and discussions
Synthesis
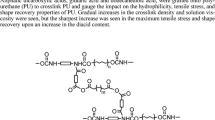
In this study, the PCDL soft segments were reacted with HDI with the molar ratio of NCO/OH = 2 to prepare the isocyanate terminated prepolymer, which further reacted with bis-MPA to form the base polyurethane with carboxyl groups at the side chain. Then the amine-terminated fourth generation polyester dendrons were reacted with the carboxyl groups to immobilize the dendrons to the PU chains. Purification could be easily carried out by precipitating the polymer solution in methanol. There will be no residue of unreacted dendrons in the polymer matrix since they can be easily dissolved in methanol and will not precipitate from the solution, which was also confirmed by the monomodal GPC trace of PU-G4 (not shown). The synthetic route was illustrated in Scheme 1.

Synthesis of PU-dendron conjugate
Structure analysis
The structure of PU, PU-G4, and PU-G4-OH were analyzed by 1H-NMR (Fig. 1). After the immobilization reaction, new peaks (n1–4, m4, i, j) attributed to the dendrons appeared, indicating the successful immobilization of polyester dendrons. The number of available carboxyl groups in PU base polymer determined by titration was found to be quite close to that in feed bis-MPA, meaning no side reaction of isocyanate with carboxyl groups happened. This might be explained by the much higher reactivity of isocyanate with hydroxyl groups than with carboxyl groups. The average functionality of side chain carboxyl groups of PU with G4 dendrons was illustrated in Table 1. From the GPC results, there were about 20 precursor molecules connected by the bis-MPA chain extend. In other words, about nineteen carboxyl groups exist in each PU chain. After the immobilization reaction, over 80% carboxyl groups were functionalized by the polyester dendrons.

1H-NMR spectra of PU, PU-G4, and PU-G4-OH
The next step consisted in deprotecting the acetonide protective groups at the surface of the dendrons. This was easily realized by reacting it with Dowex H+ resin at room temperature. This reaction deprotected all the acetonide groups at the surface of the dendrons without any side reactions, as supported by the 1H-NMR spectrum (Fig. 1). The peak for the acetonide groups at 1.34–1.41 ppm (i, j) completely disappeared, and that of –O–CH2 (m4) shift from 3.65 to 3.74 ppm, while the ratio between other peaks attributed to PU chains and dendrons remained constant after the deprotection, suggesting no other reactions occurred. After deprotection, a wide absorption peak at about 2.12 ppm that can be attributed to terminal hydroxyl groups appeared, indicating the formation of the expected structure. Although the loss of surface methyl groups leads to a slight decrease in the polymer molar mass (Table 1), it cannot be analyzed quantitatively as the interactions between the polymer chains and the solvent are different after the modification of the terminal groups.
Thermal analysis
Thermal analysis of the PUs obtained was performed to provide insights into the morphological structure of the material. Figure 2 showed the heating DSC thermograms for the PUs. Both DSC traces were from the second heat in order to minimize effects of thermal history. No evident Tg was detected above −75 °C for each sample. The melt point and ΔH m for PU, PU-G4, and PU-G4-OH was 44.9 °C, 40.86 J/g, 42.61 °C, 21.71 J/g, and 39 °C, 28.02 J/g, respectively. The decrease in melt point and ΔH m for PU-G4 and PU-G4-OH than PU was most probably resulted from the introduction of polyester dendrons, which grafted spacers between the PU chains. A little exothermal peak corresponding to the crystallinity of polycarbonate chains can be observed in PU and PU-G4, but could not be observed in PU-G4-OH. The total disappearance of crystallinity of polycarbonate chains in PU-G4-OH could be explained by the bulkiness of the substituents and the formation of large amount of hydroxyl groups which can easily form hydrogen bonds and restrict the mobility of the polymer chains, as was also supported by the increase in ΔH m for PU-G4-OH than PU-G4. In this work, these side chains may also provide pseudo-crosslinking sites via hydrogen bonding between the PU molecules.

DSC curves of PU, PU-G4, and PU-G4-OH
Surface morphology
SEM micrographs in Fig. 3 illustrated the surface morphology changes of PU films before and after each step of treatment. The films were fabricated by casting 0.02 g/mL polymer/CH2Cl2 solution onto the glass slides and evaporating the solvent directly at room temperature. As shown in Fig. 3, the surface of PU base polymer was rough with irregular pores and the film was semitransparent. After the first step of polyester dendrons immobilization, the polymer surface became relatively smooth and the film was transparent, which could be interpreted by the improved compatibility of the hard and soft segments after connecting the soft polyester dendrons with the PU chains between two hard segments. After the second step of deprotecting the acetonide groups at the surface of the dendrons, a semi-porous structure with ordered pore size for PU-G4-OH was observed and the film was also transparent. It is well-known that the “breath figure” method can generate ordered structure porous polymer films by evaporating volatile organic solvent under humid condition. [18, 19] Clearly, changes in mechanical properties also contributed to the structures observed. The surface morphology certainly affected the related properties of the polymer including wettability, contact angle, and cell responses.

SEM images of the polymer surfaces made at ×2,000. a PU is a semitransparent material with a rough surface. b The surface of PU-G4 displays a relatively flat surface and the material is transparent. c The surface of PU-G4-OH shows a semi-porous structure with ordered pore size and the material is transparent
Microphase separation
Atom force microscopy (AFM) images can provide useful information for understanding morphological features between hard and soft segments. Figure 4 showed the tapping-mode AFM result of the surfaces of cast films of PU, PU-G4, and PU-G4-OH. The hard domains in appears to be the bright regions, whereas the dark regions denote to the soft segments. For all the samples, ordered microphase separation can be observed. The irregular soft spherical domains were randomly distributed in the polymer matrix with different dimensions. Among these three samples, PU-G4 showed the smallest mean surface roughness. However, the domain structure on the surface of PU that we believe possessed the greatest microphase separation could be visualized by AFM, where the z-roughness was greater than both PU-G4 and PU-G4-OH, which also correlated with the SEM result. The high degree of microphase separation for PU is due to limited miscibility of the PCDL soft segments with the hard segments. As the polyester dendrons were connected to PU chain between two hard segments, which greatly improved the compatibility of the hard and soft segments, the corresponding images for PU-G4 showed a surface morphology that was virtually distinguishable from that of base PU. As mentioned above, irregular soft domains were randomly distributed in the order hard domains for PU because of the strong force between soft segments and hard segments, whereas the distinction between light and dark regions appeared unclear for PU-G4, indicating improved compatibility of hard and soft segments. After deprotection of the terminal acetonide groups, PU-G4-OH consisting of large amount of hydroxyl groups seems to provide more strong hydrogen bonding, and subsequently led to exhibit clear boundary between hard segments and soft segments again.

AFM images of PU (a), PU-G4 (b), and PU-G4-OH (c)
There is a close relationship between surface roughness and surface hydrophilicity. Surface roughness enhances surface hydrophobicity or hydrophilicity depending on the initial equilibrium contact angle of the substrate with water [20].
Hydrophilicity
The water uptake for the samples in PBS (A) and deionized water (B) at 37 °C as a function of time was shown in Fig. 5. Water uptake was measured to determine the bulk hydrophilicity of the PUs because this parameter was expected to have an important influence on cell responses. The chemical composition (content of hydroxyl groups) was the main factor controlling the amount of absorbed water. For PU-G4 almost no water could be absorbed into the polymers matrix within 48 h. After deprotecting the acetonide groups, a more hydrophilic character can be expected due to the presence of higher amount of hydrophilic hydroxyl groups, thus greatly increasing the water uptake.

Water absorption of different PU polymers in PBS (a), deionized water (b), and contact angle results (c)
The water contact angle measurement of PU, PU-G4, and PU-G4-OH was shown in Fig. 5c. The immobilization reaction led to a slight decrease in water contact angle from 79° for PU to 71° for PU-G4. The decrease in contact angle indicated that the polymer surface became more hydrophilic after conjugation with polyester dendrons. After deprotection of the acetonide groups, the contact angle greatly decreased to 49°. The decrease of contact angle was an index of the chemical changes occurring on the film surface, which renders the surface more hydrophilic with respect to the virgin film. It was expected that the higher hydrophilicity for PU-G4-OH must be induced by the formation of large amount of hydroxyl groups and rough surface with a large surface area.
Cell compatibility
Preliminary studies were performed to evaluate in vitro cell compatibility of the PU, PU-G4, and PU-G4-OH using HEK293T cells for different time intervals (1, 2, and 4 days). Figure 6 showed the morphology (A, B) and MTT absorption (C) for HEK 293T cells adhered to and proliferated on the surface of these polymer films 4 days post-seeding. There are only quite few cells could adhere to and proliferate on the surface of PU and PU-G4 and the difference was not significant (p > 0.05). However, PU-G4-OH showed much faster HEK 293T cells adhesion and proliferation. The number of attached HEK 293T cells on the polymer surface 4 days post-seeding increases evidently on PU-G4-OH than PU and PU-G4, which is consistent with the MTT absorption data in Fig. 6c. Cell morphology also confirmed improved cell compatibility for PU-G4-OH than PU and PU-G4: in the case of PU and PU-G4, most of the cells present a spherical shape and were not uniformly distributed on the film surfaces, presenting small cell clusters, whereas on PU-G4-OH film, they appeared to be flat, polygonal in shape, and were better spreadout than those adhering to PU and PU-G4 samples. This result suggested that the hydrophilic surface of PU-G4-OH was more favorable for the cell spreading and growth than the hydrophobic surface of PU and PU-G4. There are three major factors in influencing cell responses on biomaterials: chemical, surface morphology, and mechanical factors [17]. Deprotection of the acetonide groups greatly improved the hydrophilicity of the polymer, which allowed the culture medium penetrated more easily into the polymer films and was helpful for cell growth. The adhesion and proliferation of cells on these materials indicates that the use of PU-G4-OH is much more preferable for the activation of cellular processes.

HEK 293T cell attachment and proliferation on PU (A1, B1), PU-G4 (A2, B2), and PU-G4-OH (A3, B3). a Phase contrast images, b nuclei of attached HEK 293T cells stained by using Hoechst No. 33342 and visualized by a fluorescence microscope 4 days post-seeding; c MTT absorption for 1, 2, 4 days. The scale bar in B3 stands for 100 μm and it is applicable for all micrographs in a, b. In c, no significant difference (p > 0.05) is between PU and PU-G4, p < 0.01 between PU or PU-G4 and PU-G4-OH at day 1, day 2, and day 4
Conclusions
This paper describes the synthesis and characterization of a novel side chain dendronized polyurethane with prescribed structures and properties. The results showed that the immobilization of polyester dendrons greatly improved the compatibility of hard and soft segments in polyurethane and decrease the tendency of crystallinity due to the bulkiness of the substituents. Deprotection of the acetonide groups enhanced both bulk and surface hydrophilicity of the polymer film, which greatly improved the cell biocompatibility in comparison with base PU and PU-G4. Overall the results demonstrated an efficient modification method for PUs to improve their cell compatibility with high potentials for diverse biomedical applications.
References
Santerre JP, Woodhouse K, Laroche G, Labow RS (2005) Understanding the biodegradation of polyurethanes: from classical implants to tissue engineering materials. Biomaterials 26:7457
Anne S, Ajay DP, Lynn MF, Laura APW (2008) Biostability and biological performance of a PDMS-based polyurethane for controlled drug release. Biomaterials 29:2987
Matthew RW, Richard B, Cay K (2006) PCL-CPU composite vascular scaffold production for vascular tissue engineering: attachment, proliferation and bioactivity of human vascular endothelial cells. Biomaterials 27:3608
Gerard L, Joan C, Ronda MG, Virginia C (2007) Poly(ether urethane) networks from renewable resources as candidate biomaterials: synthesis and characterization. Biomacromolecules 8:686
Sibylle G, Laszlo K, Katarzyna G, Sylwester G, Mauro A (2003) The use of biodegradable polyurethane scaffolds for cartilage tissue engineering: potential and limitations. Biomaterials 24:5163
Li YH, Huang YD (2007) The study of collagen immobilization on polyurethane by oxygen plasma treatment to enhance cell adhesion and growth. Surf Coat Technol 201:5124
Ryeon H, Baek HS, Lee MH, Woo YI, Han DW, Han MH, Baik HK, Choi WS, Park KD, Chung KH, Park JC (2008) Surface modification for enhancing behaviors of vascular endothelial cells onto polyurethane films by microwave-induced argon plasma. Surf Coat Technol 202:5768
Bo-Lennart J, Anders L, Anette O, Åke Ö (2002) Characterization of air plasma-treated polymer surfaces by ESCA and contact angle measurements for optimization of surface stability and cell growth. J Appl Polym Sci 86:2618
Van Wachem PB, Hendriks M, Blaauw EH, Dijk F, Verhoeven MLPM, Cahalan PT, Van Luyn MJA (2002) (Electron) microscopic observations on tissue integration of collagen-immobilized polyurethane. Biomaterials 23:1401
Zhu YB, Gao CY, He T, Shen JC (2004) Endothelium regeneration on luminal surface of polyurethane vascular scaffold modified with diamine and covalently grafted with gelatin. Biomaterials 25:423
Lin HB, Sun W, Deane FM, Carlos G, Kenneth S, Peter IL, Stuart LC (1994) Synthesis, surface, and cell-adhesion properties of polyurethanes containing covalently grafted RGD-peptides. J Biomed Mater Res 28:329
Duncan R, Izzo L (2005) Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev 57:2215
Gillies ER, Dy E, Frchet JMJ, Szoka FC (2005) Biological evaluation of polyester dendrimer: poly(ethylene oxide) “bow-tie” hybrids with tunable molecular weight and architecture. Mol Pharm 2:129
Frauenrath H (2005) Dendronized polymers—building a new bridge from molecules to nanoscopic objects. Prog Polym Sci 30:325
Tian L, Hammond PT (2006) Comb-dendritic block copolymers as tree-shaped macromolecular amphiphiles for nanoparticle self-assembly. Chem Mater 18:3976
Malkoch M, Malmstrom E, Hult A (2002) Rapid and efficient synthesis of aliphatic ester dendrons and dendrimers. Macromolecules 35:8307
Wang SF, Kempen DH, Simha NK, Lewis JL, Windebank AJ, Yaszemski MJ, Lu LC (2008) Photo-cross-linked hybrid polymer networks consisting of poly(propylene fumarate) and poly(caprolactone fumarate): controlled physical properties and regulated bone and nerve cell responses. Biomacromolecules 9:1229
François B, Pitois O, François J (1995) Polymer films with a self-organized honeycomb morphology. Adv Mater 7:1041
Widawski G, Rawiso B, François B (1994) Self-organized honeycomb morphology of star-polymer polystyrene films. Nature 369:387
Sun T, Wang G, Feng L, Liu B, Ma Y, Jiang L, Zhu D (2004) Reversible switching between superhydrophilicity and superhydrophobicity. Angew Chem Int Ed 43:357
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rao, J., Gong, F., Cheng, S. et al. Novel side chain dendronized polyurethane: synthesis, characteristics, and cell responses. Polym. Bull. 62, 867–879 (2009). https://doi.org/10.1007/s00289-009-0058-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-009-0058-7