
Abstract
Dendritic cells (DCs) determine the outcome of the immune response based on signals they receive from the environment. Presentation of antigen under various contexts can lead to activation and differentiation of T cells for immunity or dampening of immune responses by establishing tolerance, primarily through the priming of regulatory T cells. Infections, inflammation and normal cellular interactions shape DC responses through direct contact or via cytokine signaling. Although it is widely accepted that DCs sense microbial components through pattern recognition receptors (PRRs), increasing evidence advocates for the existence of a set of signals that can profoundly shape DC function via PRR-independent pathways. This diverse group of host- or commensal-derived metabolites represents a newly appreciated code from which DCs can interpret environmental cues. In this review, we discuss the existing information on the effect of some of the most studied metabolites on DC function, together with the implications this may have in immune-mediated diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Dendritic cells (DCs) constitute a bridge between innate and adaptive immunity. They are able to receive and interpret signals either from invading pathogens or dying cells, and to respond to them through the acquisition of a mature phenotype. Mature DCs gain the ability to migrate to the lymph nodes (LNs) where they initiate and shape adaptive responses via antigen presentation, the expression of costimulatory molecules and cytokine secretion. These properties enable them to interact with and activate naïve T cells, inducing protective immune responses against non-self-antigens. In addition, DCs participate in a series of processes that act at different levels to ensure the maintenance of immunological tolerance (reviewed in [1]).
One of the major tolerogenic mechanisms accomplished by DCs involves their interaction with regulatory T cells (Tregs), which are crucial in controlling excessive immune responses and preventing autoimmunity [2–4]. These two cell populations are in close contact and regulate one another. DCs participate in maintaining Treg homeostasis [5, 6]. Moreover, DCs can produce soluble factors, such as TGF-β, that are essential for the generation of induced Tregs [7, 8]. Therefore, it has been hypothesized that disruption of DC homeostasis may result in Treg imbalance and autoimmunity. This view is supported by studies evaluating the consequences of DC depletion, in which ablation of CD11c-expressing cells using various genetic mouse models led to increased Th1 and Th17 responses and concomitant lower Treg frequencies [9, 10]. DC deficiency in humans is also related to lower Treg numbers [11, 12] and mice constitutively lacking DCs spontaneously develop severe autoimmunity, characterized by autoantibody production and tissue infiltration of autoreactive CD4+ T cells [10]. Newer studies using transgenic mice with improved DC targeting specificity show that defined DC subsets are required for the generation of peripheral Tregs and oral tolerance, confirming the idea that DCs are essential for establishing and maintaining immune homeostasis [13]. Still, the precise contribution of DCs to immune-mediated diseases remains unclear, though it is conceivable that changes in their phenotype, including their activation state, migratory capacity and production of immunomodulatory factors, could affect the state of tolerance. Indeed, studies from patients with different autoimmune diseases, including Rheumatoid Arthritis (RA), Multiple Sclerosis (MS), Systemic Lupus Erythematosus (SLE), Psoriasis, and Inflammatory Bowel Disease (IBD) have revealed several abnormalities in DC activation or function as factors mediating inflammatory sequelae (reviewed in [14]).
The microenvironment where DCs reside and become activated can dramatically modulate their function. Several small metabolites have been shown to exert profound changes on DCs [15]; some have even proved to be essential for DC homeostasis and function. This diverse group includes diet-derived and endogenously produced compounds, the latter being synthesized by neighboring cells or the host microflora. In the human gastrointestinal tract, 1013–1014 commensal microorganisms contribute to the breakdown, absorption and synthesis of nutrients and metabolites [16]. Diet can further influence the concentration of these products by direct and indirect mechanisms. For instance, the amount of daily fiber intake can promote colonization by specific bacterial families [17], suggesting that the composition of the microbiota and their products heavily relies on diet [18]. In addition, the intestinal flora produces a vast array of diet-independent metabolites. These products can alter the homeostasis and development of the mucosal immune system. Therefore, metabolites are emerging as a new type of signal shaping the immune system. Interestingly, increasing evidence suggests that the drastic changes in Western diet over the past few decades could account for the rise in autoimmune diseases observed in industrialized countries [19]. It would therefore be intriguing to speculate that changes in diet impairing the tolerogenic capacity of DCs may enhance predisposition to immune-mediated disorders.
Further supporting this view, it is now clear that autoimmunity is not only caused by genetic factors; studies of identical twins have shown that the environment has a major influence on development of autoimmune diseases [20]. Under-developed countries present lower a incidence of autoimmune diseases, arguing in favor of a link between lifestyle and immune status. The widely accepted hygiene hypothesis contends that increased exposure to certain pathogens, particularly helminths, reprograms the immune system rendering it less prone to autoimmunity [21]. Recent reports from experiments in mice demonstrate that helminths can alter the composition of the gut microbiota, promoting the expansion of particular bacterial families [22–26]. In addition, human studies also showed a correlation between helminth infection and altered microbiota composition [27, 28].
In this review, we will focus on some of the most studied metabolites, their effect on DCs, and their implication for immune-mediated diseases. Finally, we will also discuss their potential use in immune therapy.
Different regions, different folks: DC subsets and their localization
Because of their functional specialization and broad tissue localization, DCs comprise a highly heterogeneous cell population whose classification and study have been challenging and controversial. Another confounding factor is that some of the markers historically used for identifying DCs, such as CD11c [29] are also expressed by other cell types [30–35]. Recent advances have allowed for the identification of DC precursors and crucial transcription factors required for initiating the DC differentiation programs [36, 37]. This key information has made it possible to pinpoint which of the previously described subpopulations truly classify as DCs, and to determine defined lineages within the total DC population.
In human and mouse, DCs can be broadly subdivided into classical DCs (cDCs) and plasmacytoid DCs (pDCs) [38]. In this review we will focus on murine cDCs unless stated otherwise. cDCs can be found in most lymphoid and non-lymphoid tissues. Lymphoid tissue-resident cDCs remain in the lymphoid organ where they originated for their entire life, whereas non-lymphoid tissue cDCs usually have the ability to migrate to the draining LNs to present the antigens they acquired at their site of origin [39]. Within the cDC subtype, two distinct lineages have been identified, cDC1 and cDC2 [38]. cDC1 DCs in lymphoid tissues express CD8α and represent around 20% of splenic DCs and 70% of thymic DCs. In humans, this DC subset is characterized by the expression of BDCA-3 (CD141) [40–45]. Splenic CD8α+ DCs are more efficient at cross-presenting cell-bound and soluble antigens on MHC class I than other DC subsets [46, 47]. They also express receptors such as CD36 and Clec9A that enable them to carry out phagocytosis of dead cells [48, 49]. These unique characteristics make them essential to prime CD8+ T cell responses against tumors and intracellular pathogens [50–52], but may also permit the attenuation or exacerbation of autoimmune reactions (reviewed in [53]). In addition, cDC1 DCs are good inducers of Th1 responses due to their ability to secrete considerable amounts of IL-12 upon stimulation [52, 54]. In non-lymphoid tissues the CD103+CD11b− cDC1 subset is the counterpart to the CD8α+ cDC1 population and their ontogeny and functional characteristics are closely related [55].
The cDC2 subset, characterized by CD11b expression, is the most abundant in lymphoid organs excluding the thymus. The human counterpart of this subset is defined by the expression of BDCA-1 (CD1c) [56, 43]. In contrast to cDC1 DCs, this subset is poorly characterized, partly because of its heterogeneity. DCs belonging to this subtype are thought to be specialized in driving CD4+ T cell responses through antigen presentation via MHC class II [57, 58]. Non-lymphoid tissue CD11b+ DCs, historically considered as bona fide DCs, are now described as a heterogeneous subpopulation including DCs but also macrophages. This generalization has hindered our understanding of the contribution of the CD11b+ compartment to tissue immunity [56]. The latest identification of new markers for DCs and macrophages has enabled an efficient discrimination between these different subsets [59, 60]. Therefore, while the precise study of cDC2 DCs is still in progress, current evidence suggests they may play a role in the induction of Th2 and Th17 responses [56, 60].
Peripheral lymphoid organs also comprise a non-lymphoid tissue DC population that has arrived to the lymph node through the afferent lymphatics. Known as migratory cDCs, they are characterized by higher MHC class II and lower CD11c expression than resident DCs in the steady state. This subset is responsible for delivering tissue-derived antigens in order to reinforce tolerance or initiate adaptive responses [61, 62].
DCs are also localized in most non-lymphoid tissues, especially in those which are in close contact with the environment. In the intestine, cDCs are present in the lamina propria (LP), gut-associated lymphoid tissue (i.e. Peyer’s Patches) and mesenteric lymph nodes (mLNs). cDCs in these locations have a dual role, eliciting robust immune responses to pathogens and at the same time, inducing tolerance to food antigens and the commensal flora. In the LP, which constitutes the main mucosal immune effector site, most cDCs express CD103 and can be classified according to their expression of CD11b into CD11b− (cDC1) and CD11b+ (cDC2). There is also a CD103− CD11b+ population which expresses intermediate levels of CX3CR1. These subsets can also be found in the mLNs, where DCs migrate to prime naïve T cells in a CCR7-dependent manner (reviewed in [63]). Lung DC populations share similarities with LP DCs, though cDC1 are generally CD103+CD11b− and cDC2 are CD103−CD11b+ in this organ (reviewed in [64]). In the skin, cDCs are found in the dermis and can also be categorized by their expression of CD11b. CD11b− DCs are developmentally related to the splenic CD8α+ subpopulation, whereas CD11b+ DCs belong to the cDC2 subset and can be distinguished from CD11b+ monocytes by their lack of CD64 expression [65]. The epidermis is colonized by Langerhans Cells (LCs); this subset cannot be classified as bona fide DCs, since they do not arise from blood-circulating DC precursors but from hematopoietic progenitors that populate the skin before birth and are not dependent on FLT3L [66, 67]. These characteristics strongly resemble those of tissue resident macrophages. However, unlike this cell subset, LCs have the ability to migrate to LNs and present antigen, thus functionally resembling DCs [68]. Overall, this broad tropism suggests that DCs are exposed to different environments based on their tissues; consequently, various sets of cues may impart distinct functions to DCs in specific sites.
In contrast to other immune cells, which can be easily obtained ex vivo in large numbers, DCs are very rare (1–5% in non-lymphoid organs) [64]. This renders assays that require high cell numbers and purity, labor intensive and expensive. As a result, researchers commonly employ in vitro culture systems to yield high numbers of DCs for functional studies. The GM-CSF-based protocol for the generation of bone marrow-derived DCs is the most widely used [69, 70]. This culture is predominantly composed by CD11c+ cells with different levels of MHC II expression; traditionally it has been assumed that this was due to different degrees of maturation but careful investigation has revealed that it might not consist of a single population. A recent study showed that the GM-CSF method gives rise to a CD11c+ MHCII+ heterogeneous population of myeloid cells that comprises both macrophages and DCs [71]. Although clustering analysis indicates that the DC fraction is not closely related to the cDC1 or cDC2 subsets found in vivo, it shares some signatures with migratory DCs. Worth mentioning, this subpopulation represents only around 30% of the CD11c+ MHCII+ cells present in the culture. Conversely, bone marrow culture with FLT3L allows for the differentiation of different DC subsets (pDCs, cDC1 and cDC2) equivalent to the populations present in the spleen under steady-state conditions [72]. Therefore, this culture system closely resembles DC differentiation from bone marrow precursors in vivo. However, due to its high complexity, if the study of a single population is intended, FACS sorting may be required. Furthermore, a new method for the generation of large numbers of cDC1 DCs with high purity is now available [73]. This protocol requires the addition of GM-CSF and FLT3L and yields a 90% pure population of CD103+ DCs, thus eliminating the need for further purification steps for certain applications. Human cDC1 can be obtained following a similar procedure [45, 74]. DC subsets obtained from these cultures are functionally and phenotypically similar to those found in vivo, making them particularly useful for studying populations that exist in very low numbers in the body, such as CD103+ DCs. Furthermore, this may be potentially relevant for new immunotherapeutic approaches since these DCs display improved migratory capacity which results in enhanced antigen delivery to the LNs [73]. Advances in methods to isolate DCs ex vivo and genetic models to conditionally target genes in DC subsets will be instrumental in helping understand DC mechanisms.
Endogenous metabolites and metabolic processes in DCs
In the past few years, the idea of a link between immune cell function and metabolism has gained momentum, giving rise to the field of immunometabolism. Immune cells can respond to changes in the environment, such as oxygen and nutrient availability, by changing their core metabolic program. Interestingly, it seems that this metabolic reprogramming can be accompanied by or even required for changes in DC cellular function.
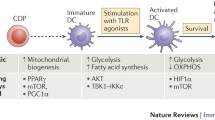
Under resting conditions, DCs mainly rely on oxidative phosphorylation (OXPHOS) for energy [75, 76]. This process occurs within mitochondria, whereby cells utilize NADH generated from the catabolism of nutrients (e.g. glucose, amino acids and fatty acids) to produce ATP through a series of reduction-oxidation reactions. Engagement of Toll-like receptor (TLR) on GM-CSF derived DCs leads to upregulation of aerobic glycolysis. This process – known as the “Warburg effect” – involves the conversion of glucose into lactate [76–78] and is dependent on the PI3K/Akt pathway. In cancer cells, the Warburg effect accompanies rapid proliferation, whereas DCs are generally non-proliferative and do not divide further upon TLR ligation. Notably, the rise in aerobic glycolysis observed in GM-CSF derived DCs is accompanied by a decrease in mitochondrial oxygen consumption and ATP levels [76, 78]. Expression of Hypoxia-inducible factor 1-alpha (HIF-1α) is also required to trigger these metabolic changes; TLR ligands can induce the expression of this transcription factor through an as yet unknown mechanism [79]. Analysis of Hif1a −/− DCs revealed that these cells do not upregulate glycolysis in response to TLR ligation and are unable to induce T cells responses, highlighting the importance of this signaling pathway for DC immunogenicity [79, 80, 75].
Initial reports on DC metabolism argued that DCs induce glycolysis in order to counteract the inhibition of mitochondrial respiration caused by nitric oxide (NO), a product of nitric oxide synthase (iNOS) [78]. Thus, DCs would upregulate their glycolytic rate to maintain the ATP levels needed for their function and survival. However, this observation is restricted to GM-CSF-derived cells, since bona fide DCs do not express iNOS in response to TLR stimulation [71, 75]. In fact, according to Helft et al., not even the DC subset within GM-CSF cultures expresses iNOS upon LPS activation, indicating that the NO produced in this system likely comes from the accompanying macrophage subpopulation [71]. Thus, it would be interesting to determine if in vivo, NO generated by other cells during the inflammatory response could indirectly induce changes in the metabolic program of adjacent DCs. More recent studies claim that TLR-driven activation triggers an early induction of glycolysis required for providing biosynthetic precursors for fatty acid synthesis (FAS) [77]. Glycolysis-derived pyruvate can enter the tricarboxylic acid (TCA) cycle in the mitochondria and yield citrate which can be transported back to the cytosol where it serves as a precursor for FAS. This process sustains the expansion of the ER and Golgi membranes, thus promoting the translation and transport of new proteins involved in DC activation [77]. Nevertheless, since these studies were performed using pharmacological inhibitors which often have unwanted off-target effects, it will be crucial to make use of genetic models to study the contribution of these metabolic pathways to DC function.
The high cell numbers and purity required to conduct most metabolic analyses render it difficult, if not impossible, to study the metabolic features of DCs in vivo. The only information available comes from ex vivo-isolated DCs. Pantel et al. showed that in vivo activation of splenic cDCs via polyinosinic:polycytidylic acid (poly I:C) administration results in type I interferon-dependent upregulation of glycolysis and decreased mitochondrial respiration. [75]. A new study argues that upon activation, both pDCs and cDCs upregulate OXPHOS via an induction of fatty acid oxidation in a type I interferon-dependent manner [81, 82]. Since these findings contradict previous reports [82] this process needs to be studied in further detail. It is possible that interaction of DCs with neighboring cells via the production of metabolites (e.g. NO) could influence their metabolic program, thus accounting for the differences observed.
Interestingly, products of metabolic processes can act as environmental cues on DCs, triggering changes in their core metabolic program and/or their functional characteristics. For instance, DCs can sense the presence of succinate (reviewed in [83]) through the receptor GPR91, which acts as an activating stimulus [84]. Moreover, reactive oxygen species (ROS) which are constitutively produced by the mitochondrial respiratory chain can exert signaling functions by inducing post-translational modifications on proteins [85]. By-products of microbial metabolism also induce changes in DC function; these signals can come either from invading pathogens or the commensal microflora [86, 87]. We are only beginning to understand the metabolic processes that follow DC activation and their importance for DC function. Nonetheless, the information available suggests that modulation of metabolic core pathways could become the next generation therapy for the treatment of immune-mediated diseases. In the following sections we will discuss some of the best studied metabolites influencing DC metabolism and function.
ATP and adenosine
Because of its central role in driving virtually every cellular process, ATP is often referred to as the energy currency of the cell. However, ATP can also be released to the extracellular environment by necrotic and apoptotic cells and in response to different types of stress, or even as part of physiological processes [88–90]. Once in the extracellular space, ATP can be degraded to ADP, AMP and adenosine by ectonucleotidases such as CD39 and CD73 (reviewed in [91]). Adenosine can be further metabolized by adenosine deaminase (ADA), yielding inosine, which comparatively has no activity [92]. Adenosine can also be taken back into cells through nucleoside transporters (reviewed in [93, 94]).
Released ATP, as well as its degradation products, can be sensed by purinergic receptors. These can be classified into two families: P1, composed of G protein-coupled adenosine receptors (ARs), and P2, which includes receptors that bind to ATP, ADP and other nucleotides. The P2 family can be further subdivided in P2Y, which comprises metabotropic G protein-coupled receptors and P2X, formed by oligomeric ion channels [95].
Adenosine receptor signaling
Early reports show that human immature DCs express the ARs A1R, A2AR and A3R, whereas LPS-stimulated DCs downregulate the expression of A1R and A3R, displaying only detectable mRNA levels of A2AR [96]. Activation of A2AR inhibits IL-12 and TNF-α production and enhances IL-10 secretion on mature DCs but has no influence on basal levels of secretion of these cytokines [97]. In this way adenosine treatment of mature DCs renders them less efficient at priming Th1 responses [97]. In line with these results, activation of ARs in human DCs also inhibited their capacity to induce CD8+ T cell responses [98]. Similar effects of adenosine receptor activation were observed in murine DCs, although they seem to be dependent on A2BR rather than A2AR [99, 100].
Adenosine also has chemotactic properties on DCs [96, 101]; a recent study showed that Tregs can degrade extracellular ATP to generate adenosine through their expression of CD39 and CD73 [101]. Adenosine then attracts DCs to remove apoptotic cells and promotes their interaction with Tregs to ensure tolerance. This process may be relevant for clearance of dying cells under physiological conditions.
DCs can also express CD39, which allows them to directly dampen immune responses (reviewed in [102]). CD39 expression is dependent on IL-27, a cytokine belonging to the IL-12 family secreted by DCs upon TLR ligation [103] that can act directly on T cells, preventing Th17 responses while promoting the development of IL-10-producing Tregs [104–107]. IL-27 acts in an autocrine or paracrine manner, inhibiting the maturation of DCs [108, 109]. CD39 upregulation leads to a decrease in extracellular ATP levels, which results in lower ATP-dependent activation of the NLRP3 inflammasome. This process may be important in the context of the experimental autoimmune encephalitis model (EAE), the murine model for MS, since treatment of mice with IL-27-conditioned DCs reverses the chronic status after the disease is already established [110].
Conversely, growing evidence supports the notion that DCs may also have mechanisms to deplete adenosine from their microenvironment, implying they could release themselves and other cells in their vicinity from the immunomodulatory effects of this nucleoside [111–113]. DCs express surface ADA, capable of eliminating the signaling actions of adenosine [113]. In line with these findings, blockade of ADA led to a more pronounced effect of adenosine and synthetic AR agonists on DC maturation [113]. Direct in vitro addition of ADA to human immature DCs resulted in upregulation of costimulatory molecules (CD80, CD83, CD86) and higher secretion of Th1-polarizing cytokines, further supporting the concept of a negative regulatory mechanism of adenosine signaling in DCs [114]. This mechanism is likely to be relevant during inflammation, where a significant rise in adenosine levels is expected; in this context, expression of ADA by DCs would enhance their maturation status, thus boosting their immunogenicity. However, it could have a detrimental effect in immune-mediated diseases. For instance, non-obese diabetic (NOD) mice DCs display elevated levels of ADA in comparison to other mouse strains [111]. Furthermore, DCs from NOD mice lacking ADA expression fail to trigger autoimmunity when adoptively transferred, confirming the importance of ADA expression on DCs in the T cell priming phase, as well as in the regulation of the magnitude of the T cell response [111].
In the intestine, ARs are essential for keeping inflammation in check. In a chemically-induced colitis model, genetic ablation or pharmacological inhibition of A2BR resulted in higher severity [115]. In the context of the same model, CD39-deficient mice showed more severe symptoms than wildtype mice, further underscoring the immunomodulatory role of adenosine in the gut [116].
Another setting where immunomodulation by adenosine seems to play a relevant role is cancer. The tumor microenvironment displays high concentrations of adenosine [117, 118], which promotes secretion of VEGF by DCs and inhibits their maturation, leading to increased angiogenesis, inefficient antigen presentation and lower activation of effector T cells [119]. Transfer of AR-stimulated DCs into tumor-bearing mice promoted tumor growth [119]. In addition, treatment with AR antagonists inhibited tumor growth and metastasis formation, due to improved T cell activation and recruitment to the tumor site [120]. This seems to be a consequence of adenosine signaling blockade in the DC compartment, more specifically in the CD8+CD11b− subset, which showed higher activation levels after treatment. The use of adenosine receptor antagonists, particularly for A2AR, has been put forward as a next-generation checkpoint blockade therapy [121].
P2XR and P2YR signaling
ATP and other nucleotides can be released into the extracellular space by apoptotic cells through different types of channels, e.g. pannexins, or they can also be released by cell damage or lysis [122–124]. These molecules have chemoattractant properties and have been described to act as a “find me” signal that recruits phagocytes in a P2XR/P2YR-dependent manner in order to ensure clearance of dying cells [88]. DCs express both types of receptors; interestingly, ATP has chemotactic activity on immature DCs but not on mature DCs [125, 126].
Regarding the effect of ATP on DC maturation, there seems to be inconsistencies on what has been reported so far, but that is likely due to different ATP concentrations being used. High ATP concentrations (mM range) induce secretion of TNF-α and IL-1β by LPS-stimulated DCs, but are also highly cytotoxic [127–129]. This is connected to the ability of ATP to activate the NLRP3 inflammasome in a P2X7R-dependent fashion [130–132]. These effects require high ATP concentrations since, compared to other members of the P2XR family, P2X7R has low affinity for ATP [133]. In DCs, this process may be important for inducing anti-tumor immunity: ATP released by dying tumor cells following chemotherapy can trigger inflammasome activation, resulting in proteolytic processing and secretion of IL-1β, which promotes the generation of IFNγ-producing tumor antigen-specific CD8+ T cells [134]. On the other hand, μM concentrations of ATP induce the expression of costimulatory molecules but at the same time inhibit the production of IL-1β, TNF-α, IL-6 and IL-12. The reduced IL-12 production by ATP-treated DCs results in their impaired ability to prime Th1 responses [135]. However, since these effects are very similar to the ones observed for adenosine, and as DCs can express CD39, it would be important to rule out the contribution of ATP degradation.
Commensal intestinal bacteria release large amounts of ATP into the extracellular space [136–138]. Germ-free mice have considerably lower ATP content in their feces, and antibiotic treatment of specific pathogen-free mice reduces fecal concentration of ATP [139]. ATP levels in the intestinal lumen strongly correlate with the frequency of LP Th17 cells, implying an association between bacterial ATP and Th17 cell differentiation. A subpopulation of antigen-presenting cells in the LP (CD70highCD11clowCD11b+CD103−CX3CR1+) expresses higher levels of P2XR/P2YR than other DC subsets and is able to drive Th17 differentiation of naïve CD4+ T cells through the production of IL-6, IL-23 and TGF-β [139]. In addition, their ability to induce the Th17 subset is significantly enhanced by ATP supplementation, indicating a role for ATP in the generation of naturally occurring Th17 cells in the LP. This could have important implications in the context of IBD, where variations in the composition of the microbiota might lead to altered extraluminal ATP concentrations and thus influence disease severity. Indeed, in the context of a T cell-mediated colitis model, ATP administration resulted in higher Th17 cell numbers and worsening of clinical symptoms [139]. Furthermore, intestinal biopsies from patients suffering from Crohn’s disease displayed higher expression of the P2X7R, which mainly co-localized with DCs and macrophages [140]. In summary, ATP levels can directly and indirectly influence inflammation.
Short-chain fatty acids
Although for many years it was unclear how gut-colonizing bacteria modulate cell function, current studies indicate a role for bacterial-derived metabolites as emissaries linking the gut flora and the host (reviewed in [141]). Short-chain fatty acids (SCFAs), defined as C1–6 organic fatty acids, represent the main metabolic product of anaerobic bacterial fermentation in the gut [142]. They are produced through the breakdown of dietary fiber, carbohydrates and proteins and serve as an energy source for both commensal bacteria and the colonic epithelium [143]. Acetate is the most abundant SCFA found in the colon, followed by propionate and butyrate in an approximate constant ratio of 60:20:20 [144, 145]. Their concentration is highest in the caecum and decreases towards the distal colon, reflecting the availability of substrate for fermentation throughout the gut [146]. The majority of SCFAs is taken up by colonic epithelial cells through passive diffusion or active transport by solute carriers [147], where they can regulate different cellular processes such as epithelial cell proliferation and differentiation [148]. While butyrate is mostly metabolized by colonocytes, acetate and propionate can enter the circulation, serving as a substrate for gluconeogenesis or de novo lipid synthesis [149]. SCFAs influence a wide range of physiological functions including electrolyte and water absorption, regulation of gut motility, as well as leptin and peptide YY secretion (reviewed in [150]). These actions occur by directly acting on target cells or indirectly through a gut-brain neuronal circuit [151].
Accumulating evidence suggests that SCFAs also have profound effects on the immune system where they act via two major pathways. The best characterized effect is the modulation of gene expression through inhibition of class I and IIa histone deacetylases (HDACs) [152]. More recently, SCFAs were found to be the natural ligands for the orphan G-protein coupled receptors GPR41 (free fatty acid receptor 3 – FFAR3) and GPR43 (free fatty acid receptor 2 – FFAR2) [153]. Butyrate also acts on the niacin receptor GPR109a (also known as hydroxycarboxylic acid receptor 2) [154]. FFARs for SCFAs are coupled to pertussis toxin-sensitive Gαi/o subunits. Activation of these receptors results in inhibition of adenylate cyclase and decreased cAMP [155]. FFAR2 also couples to Gαq and promotes calcium release, a prerequisite to induce cell migration [156].
It is currently accepted that within the intestinal microbiota, specific bacterial species can positively influence health status and improve disease resistance against inflammatory diseases such as colitis, arthritis or asthma [125]. For example, IBD, a chronic inflammatory disorder of the gastrointestinal tract, is associated with an imbalance in healthy microbiota especially from the genera Bifidobacterium and Bacteroides [157], and a parallel reduction in SCFAs [158]. Furthermore, germ-free mice recolonized with Bacteroides fragilis [159] present less severe signs of colonic inflammation in the DSS colitis model, a well-established murine model of human IBD. Although the beneficial effects of B. fragilis colonization have been ascribed to Polysaccharide A (PSA), as PSA alone is able to protect mice from experimental colitis by suppressing IL-17 production and promoting the development of IL-10-producing Tregs in the gut [160], feeding of germ-free mice with acetate can mimic the protective effect [161]. In 2009, Sina et al. demonstrated that mice lacking the FFAR2 receptor are more susceptible to acute DSS colitis [162]. In this model, SCFA-induced neutrophil recruitment through FFAR2 contributed to contain systemic bacterial dissemination and was therefore crucial to prevent mortality. Yet, Ffar2 −/− mice were protected from chronic DSS colitis [162]. Ffar2 −/− mice presented lesser signs of colonic inflammation including less tissue damage and neutrophil infiltration in both models. Strong mucosal neutrophil infiltration and over-activation is a hallmark of IBD (reviewed in [163]). Along the same line, several groups have shown that SCFAs promote neutrophil migration and ROS production in a FFAR2-dependent manner [155, 164–166, 161]. However, using a different transgenic mouse strain, Maslowski and colleagues found that absence of FFAR2 results in more severe inflammation as well as higher levels of neutrophilic infiltration and activation during the acute phase of DSS treatment [161]. The protective effect of FFAR2 was attributed to its expression on immune cells as revealed using bone marrow chimeras. In the same study, greater morbidity and impaired weight recovery was observed in Ffar2 −/− mice during chronic DSS colitis [161]. The reasons for these discrepancies between studies are not yet clear. Since FFAR2 deficiency in neutrophils does not seem to affect their ability to migrate to other chemoattractants [166], increased neutrophilic recruitment in the latter model might be due to the presence of other potent chemokines at the site of inflammation. Besides, one should take into account the strong inter-laboratory variability of the DSS colitis models, mainly due to the different colitogenic potential of the DSS batches, but also to the mouse strain used, as well as environmental factors such as the hygiene conditions of the mouse facility [167] which in turn would impact the microbiota. In this sense, while Bacteroidetes produce preferentially acetate and propionate, Firmicutes generate mainly butyrate. Acetate and propionate are highly potent at activating FFAR2 whereas butyrate has less activity. Conversely, FFAR3 has higher affinity for butyrate than propionate and marginally binds acetate. Thus, the net effect of deleting one receptor may be subjected to several variables, including the presence or absence of specific commensals as well as diet composition. A further limitation to define the role of FFAR2 in SCFA activity in vivo relates to the wide expression pattern of this receptor in immune cells. ImmGen gene expression data suggests that in addition to neutrophils, Ffar2 is also expressed in eosinophils, monocytes, splenic, mesenteric and small intestine DCs, as well as in resident macrophages from the lung [33].
In macrophages, SCFAs suppress inflammatory cytokine production through the attenuation of HDAC activity and concomitant modulation of gene expression [168]. Along the same line, a study using immature and LPS-matured human monocyte-derived DCs revealed that both butyrate and propionate strongly reduced the expression of several pro-inflammatory chemokines and cytokines at the transcriptional level [169]. Similar observations were made with murine bone marrow-derived DCs, where acetate, butyrate and propionate inhibited the LPS-induced expression of costimulatory molecules CD80, CD86 and CD40 and production of pro-inflammatory cytokines [170, 171]. DCs exposed to SCFAs also displayed a strong Treg-inducing capacity [172]. Analysis of the gene expression profile of murine in vivo FLT3L-expanded splenic DCs exposed to butyrate or the HDAC inhibitor Trichostatin A showed repression of LPS-responsive genes, particularly Il12a, Il6 and Relb, a member of the NF-κB family mediating DC maturation [172]. Thus, in contrast to neutrophil migration and activation, which are dependent on GPCR activation, the anti-inflammatory effects of SCFAs on macrophages and DCs are linked to the modulation of gene expression, an effect that might be related to HDAC inhibition. Accordingly, the effect of butyrate on Treg induction by splenic DCs was independent of GPR109a expression, yet the contribution of FFAR2 to these effects was not investigated [172]. Conversely, a later study reported that Gpr109a −/− colonic DCs and macrophages express less RALDH1 and IL-10 and as a consequence fail to induce Tregs [173]. Furthermore, Gpr109a −/− mice showed lower Treg numbers and frequencies in colonic LP, as well as enhanced susceptibility to acute DSS colitis and colonic inflammation [173]. Of note, while both colonic and splenic macrophages exhibit similar amounts of GPR109a, colonic DCs express more GPR109a than splenic DCs, possibly explaining the contradictory results. Further studies using conditional knockout mice will be required to better understand the effects of SCFAs on specific cell populations in vivo.
SCFAs have also been reported to act directly on T cells promoting either immunity or tolerance depending on the cytokine milieu. Using the TNBS T cell-dependent colitis model, more severe symptoms were observed in Ffar2 −/− mice, accompanied by exacerbated intestinal Th17 responses [161]. The same mice were also more susceptible in the models of K/BxN serum-induced inflammatory arthritis and OVA-induced allergic airway inflammation [161]. Furusawa et al. showed that a high fiber diet increases caecal levels of SCFAs, which results in enhanced differentiation of colonic IL-10-producing Tregs (cTregs) and ameliorates the development of adoptive transfer colitis [174]. This effect was mediated by commensal fermenting bacteria, since no expansion of Foxp3+ Tregs could be observed under germ-free conditions [174]. In the same vein, microbiota-deficient broad spectrum antibiotic-treated mice displayed reduced SCFA concentrations in the stool and less cTregs [172, 175]. Butyrate administration to mice which selectively lack peripherally-derived Tregs but display intact thymic Treg differentiation, due to deletion of the conserved non-coding DNA sequence (CNS) 1 in the Foxp3 gene, did not result in Treg expansion. These findings suggest that butyrate is only able to act on extrathymic Tregs. Mechanistically, the authors demonstrated that these effects were not due to improved proliferation or survival of Tregs, but enhanced histone acetylation in the promoter and conserved non-coding regions of the Foxp3 locus, thereby enhancing the accessibility of other transcription factors to enhancer elements [174]. These results are supported by previous studies showing the inhibitory effect of Class IIa HDAC during Treg induction [176–179]. In addition, butyrate promotes the acetylation of Foxp3 itself increasing its stability [174]. Nevertheless, HDAC inhibition might not be the only mechanism involved in cTreg induction by SCFAs. Ffar2 −/− mice failed to expand their cTreg compartment after propionate administration, advocating for an involvement of this receptor [175].
While these findings link SCFAs and fermenting commensals with the induction of cTregs, thus providing a new molecular mechanism for their anti-inflammatory effect, the effect of SCFAs on other T helper (Th) cell subsets is less well understood. Several groups reported no major effects of SCFAs on Th subsets [175, 174, 172]. In contrast, other studies indicate that propionate and butyrate potentiate IFNγ and IL-17 production during Th1 and Th17 differentiation, although these cells produce more IL-10 and have suppressive capacity [180]. These effects were not mediated by FFAR2 or FFAR3 as T cells do not express FFAR2 or FFAR3 at functionally relevant levels [180]. Instead, SCFAs act by inhibition of HDAC activity and subsequent enhancement of the mTOR-S6K pathway [180].
Given their ability to be transported to the systemic circulation, SCFAs can also exert functions in other organs besides the intestine. An interesting study linked fiber consumption to lung homeostasis, showing that low fiber intake renders mice more susceptible to allergic airway inflammation (AAI). This effect is mediated by SCFAs, since a high fiber diet promoted the growth of SCFA-producing bacteria, resulting in increased systemic SCFA levels, and administration of propionate in a model of AAI ameliorated the disease symptoms in a FFAR3-dependent manner [17]. Although no SCFAs were detected in the lungs, activation levels of DCs from lung-draining lymph nodes negatively correlated with fiber intake. Indeed, propionate can directly act on DC precursors in the bone marrow, promoting the differentiation of cells with lower Th2-inducing potential [17]. Another study reported that SCFAs are able to reach the brain, where they can influence microglial function [181]. Absence of microbiota leads to disruption of microglial homeostasis under steady state conditions and stunted microglial activation in response to inflammatory stimuli. SCFA administration was able to revert these effects, though no expression of FFAR2 was detected in microglia. Therefore, SCFAs might either modulate microglia through a FFAR2-independent mechanism and/or act in an indirect way, targeting an intermediary cell subset that will respond by producing factors that affect microglial function. Analysis of the microglial compartment in Ffar2 −/− mice advocates for a FFAR2-dependent mode of action, but the specific population which participates in this dialog remains elusive.
Collectively, this evidence points to a tight relationship between microbiota and tolerance. Yet, studies attempting to use SCFAs as a therapeutic approach in inflammatory situations have rendered contradictory results (reviewed in [182]). Thus, further investigation will be required to better understand their mechanism of action on specific cell types and to prove their anti-inflammatory potential as new therapeutic tools.
Medium-, long-chain and polyunsaturated fatty acids
In addition to the above mentioned GPCR recognizing SCFAs, interest has been put on other receptors recognizing medium-chain (C6-C12) and long-chain fatty acids (C10-C18) as well as polyunsaturated fatty acids (PUFAs), usually acquired from the diet. For example, GPR120 (FFAR4), which recognizes long-chain unsaturated fatty acids such as docosahexaenoic acid (DHA), possesses potent anti-inflammatory actions effects by preventing the secretion of pro-inflammatory cytokines in macrophages [183]. The effects mediated by GPR120 may explain, at least in part, the beneficial effects of omega-3 fatty acids contained in the Mediterranean diet, long recognized for its positive effects on health. Still, PUFAs can also exert their function through peroxisome proliferator-activated receptors (PPARs), a group of nuclear receptors that act in an anti-inflammatory manner, both by directly regulating gene expression as well as through interference with the prototypic inflammatory mediator NF-kB (reviewed in [184]). GPR40 and GPR84 represent two other examples of previously considered orphan receptors that are now jumping to fame because of their recently described effects on the immune system and the identification of their endogenous ligands [185, 186]. Studies targeting these receptors in specific cells types, and particularly in DCs, as well as investigating their role in immune-mediated diseases will contribute to our understanding of their function and their potential therapeutic applications.
Vitamin A
Vitamin A (all-trans retinol, VitA) is a fat-soluble vitamin that has many essential functions for the life of all vertebrates and its deficiency has several detrimental effects on human health. Since animals lack the required machinery for de novo VitA synthesis, they must obtain it from their diet, either from plants and microorganisms in the form of carotenoids, or by ingesting animal-derived products that contain retinyl esters. After absorption in the intestine, both sources are transported to the liver in the form of retinyl esters. The majority travels incorporated into chylomicrons and is taken up by hepatocytes, where they are hydrolyzed. Free retinol can then associate with retinol-binding protein (RBP), which allows the secretion of the retinol-RBP complex into plasma. Another fate of free non-esterified retinol is storage within lipid droplets in hepatic perisinusoidal stellate cells, which occurs especially under VitA-sufficient conditions (reviewed in [187]). Although the main site of VitA depot is the liver (>90%), its presence has been detected in other organs such as skeletal muscle and bone marrow. Plasmatic concentrations of retinol are tightly regulated and maintained around 2 μM regardless of fluctuations in daily VitA intake (reviewed in [187]).
Target cells are responsible for carrying out the conversion of all-trans retinol to all-trans retinoic acid (ATRA), which is the active form of VitA. This two-step process involves oxidation of all-trans retinol to all-trans retinal followed by oxidation of retinal to ATRA. The first step can be performed by cytosolic alcohol dehydrogenases (ADH) and membrane bound short-chain dehydrogenases/reductases (SDR). The second step can be executed by at least three different enzymes, known as retinal dehydrogenases 1, 2 and 3 (RALDH1–3) [187].
ATRA exerts most of its functions through the activation of Retinoic Acid Receptors (RARs), which are ligand-activated nuclear receptors [188, 189]. There are three different subtypes of RARs (α, β and γ), which display higher homology between different species than among themselves, implying they have specific roles in retinoic acid signaling. RARs function as heterodimers, associated with another nuclear receptor, Retinoid X Receptor, which also exists in three different forms (α, β and γ). Upon ligand binding, RAR-RXR heterodimers associate with specific DNA sequences known as RAR elements (RARE) and retinoid X response elements (RXRE) located in promoter regions of target genes. RAR-RXR heterodimers can then modulate gene transcription by recruiting negative or positive regulatory proteins. More than 500 genes have been suggested to be regulated by ATRA, either by direct or indirect mechanisms, which accounts for the pleiotropic effects of this metabolite [190].
ATRA participates in maintaining the homeostasis of cDC subsets. The fate commitment of pre-cDCs into different DC subpopulations is dependent on the concentration of this metabolite. In particular, the development of splenic CD11b+CD8α− and small intestine LP CD11b+CD103+ DCs is severely impaired in absence of VitA [191, 192], whereas other DC subsets and progenitors are able to develop normally. ATRA can also guide the differentiation of in vitro-generated CD103+ DCs [73] into the cDC1 and cDC2 subsets found in the intestinal LP, CD103+CD11b− and CD103+CD11b+ [193]. Addition of ATRA to human monocyte-derived DCs during their development imprints them with mucosal-like properties. ATRA-conditioned DCs express CD103 [194, 195] and constitutively produce the anti-inflammatory cytokine IL-10. They also display higher levels of CCR7 than their non-treated counterparts, suggesting that these DCs would exhibit enhanced migration to the draining LNs [194]. Some studies also reported the induction of RALDH2 after ATRA administration [195].
In the gut, a specific subset of intestinal DCs expresses RALDHs which allow them to convert retinol into ATRA [196]. This subpopulation of DCs is characterized by the expression of CD103 and is present in Peyer’s Patches, mLNs and LP. Regarding the signals required to induce ATRA-synthesizing ability in gut-associated DCs, the available information suggests it is mediated by a mechanism dependent on TLR1/2-MyD88 signaling. Expression of ATRA-synthesizing enzymes can be induced in DCs by TLR2 ligation [197, 198]. Furthermore, DCs isolated from Tlr2 −/− mice express lower levels of ATRA-producing enzymes and show an impaired capacity to imprint gut-homing molecules in T cells. The same was observed for Myd88 −/− DCs [198].
ATRA exerts several functions in the intestine; first of all, it is essential for maintaining homeostasis of the mucosal immune system, by inducing the gut homing receptors α4β7 integrin and CCR9 in T cells and B cells. Depletion of VitA in mice caused a significant reduction of α4β7 integrin+ CD4+ T cells in secondary lymphoid organs and complete absence of T cells in the LP, indicating that this metabolite is essential for T cell migration to gut tissues [196]. Moreover, ATRA is required by CD103+ DCs in the gut to induce the generation of intestinal Foxp3+ Tregs via a mechanism which depends on TGF-β [199, 7, 200, 201]. Induction of Tregs by intestinal CD103+ DCs may play an important role in maintaining tolerance to dietary antigens and commensal flora. ATRA-conditioned DCs can induce gut homing IL-10 producing Tregs which is dependent on their ability to produce ATRA [195]. Furthermore, ATRA seems to regulate the balance between Th17 and Treg differentiation. Despite their opposing roles, both of these subsets require TGF-β for their development. Notwithstanding, the additional presence of ATRA is able to inhibit Th17 differentiation in vitro. This mechanism could be particularly relevant in mucosal tissues, where tight regulation of immune responses by ATRA would be crucial for maintaining integrity of the intestinal barrier [202–204]. However, the ATRA concentration used in this work appears to be 1000-fold higher than physiological levels in plasma (μM versus nM) [205], suggesting that these results should be interpreted with caution. Later studies have shown that nM concentrations of ATRA do not impair Th17 differentiation [206–208]. In fact, addition of an ATRA receptor antagonist inhibited the differentiation of Th17 cells in vitro [206], while VitA-deficient mice showed a dramatic decrease of Th17 cells in the gut [207, 208], which could be explained by altered numbers and composition of the microbiota. Therefore, low levels of ATRA may be required for the development of intestinal Th17 cells.
Interestingly, the study of a mouse reporter strain that expresses luciferase upon RA signaling revealed high levels of RAR activity in CD4+ T cells upon activation. Furthermore, RA signaling proved to be essential to mediate rejection in a skin allograft model [209]. Ablation of RA signaling caused a shift from Th1/Th17 to a Th2 phenotype [209]. Furthermore, Th1 and Th17 mucosal and systemic responses were severely impaired in VitA-deficient mice, further underscoring the role of RA signaling in adaptive immunity [210]. These findings could explain the considerable amount of data showing a correlation between VitA deficiency and impaired responses to several pathogens. Several studies have revealed the importance of VitA in fighting infections. VitA deficiency in children from developed and developing countries has been linked to greater mortality caused by infectious diseases [211, 212]. Clinical trials showed that VitA supplementation in neonates and children reduced mortality by 12 and 25%, respectively [213, 214]. Mucosal IgA responses in the gut and respiratory tract are impaired in VitA-deficient individuals, possibly due to the essential role of ATRA in imprinting homing receptors to lymphocytes and in the induction of IgA responses [215]. Insufficient levels of VitA cause deficient immune responses to vaccines and respiratory and gastrointestinal pathogens. It has been reported that VitA deficiency in mice leads to decreased numbers of antigen-specific CD8+ T cells in the lower respiratory tract which express unusually high levels of CD103 [216]. This high CD103 expression has been suggested to interfere with T cell migration to the lower respiratory tract, thus accounting for the lower numbers observed. VitA-deficient mice also showed an altered IgA/IgG production ratio in response to intranasal inoculation of a Sendai virus vaccine [217]. Co-administration of VitA with the vaccine was able to improve the mucosal IgA response in VitA-deficient mice [218, 219]. Further work is needed to determine how the interplay between DCs and T cells can be tuned by VitA levels to regulate mucosal immunity.
Vitamin D
Vitamin D (VitD) is mostly produced in the skin, where a cholesterol derivative, 7-dehydrocholesterol, is converted upon exposure to sunlight via a UVB-dependent reaction. VitD can also be acquired from the diet in limited amounts. Once it enters systemic circulation, VitD must undergo 2 hydroxylation steps to be transformed into the metabolically active form, 1,25-dihydroxyvitamin D3 (1,25(OH)2D3). The first hydroxylation occurs mostly in the liver and it is catalyzed by 25-hydroxylases. The resulting product, 25-hydroxyvitamin D3, is the main circulating VitD metabolite and has a half-life of approximately 15 days. 25-hydroxyvitamin D3 is then further hydroxylated by 1-⍺ hydroxylases to yield the final active metabolite of VitD, 1,25(OH)2D3. This reaction mainly takes place in the kidneys and is tightly regulated by parathyroid hormone levels and serum calcium and phosphorous levels (reviewed in [220, 221]).
VitD has various described immunomodulatory effects. The first reports of the effect of 1,25(OH)2D3 on immune cells [222–224], as well as the expression of a high-affinity receptor for this metabolite in different immune subpopulations [225, 226] were made over two decades ago. In line with these studies, VitD administration showed a therapeutic effect in different mouse models of autoimmune disease, including encephalomyelitis [227, 228] and type 1 diabetes [229].
1,25(OH)2D3 exerts its effects on immune cells by binding to the Vitamin D Receptor (VDR), a ligand-activated nuclear receptor that acts as a transcription factor upon binding of 1,25(OH)2D3. When this metabolite binds to the receptor, it induces its heterodimerization with RXRs. The heterodimer binds to VitD response elements in the promoter of VitD target genes, inducing or inhibiting their transcription [230]. Analysis of Vdr −/− mice revealed lymph node hypertrophy and elevated numbers of mature DCs in the subcutaneous lymph nodes, highlighting the importance of the VitD-VDR signaling axis in maintaining DC homeostasis [231].
Studies of in vitro-generated human [232–234] and murine [231, 235] DCs conditioned with 1,25(OH)2D3 or VitD analogs revealed that these cells are resistant to maturation induced by inflammatory stimuli. The involvement of VDR signaling in the effect mediated by 1,25(OH)2D3 on DCs was confirmed in experiments with Vdr −/− DCs, where the immunomodulatory effects of this metabolite were lost upon deletion of the receptor [231]. Although there are some contradicting reports regarding their phenotype under steady state conditions, they unanimously show that 1,25(OH)2D3-conditioned DCs fail to acquire a fully mature phenotype in response to inflammatory stimuli such as LPS, TNF-α and CD40 ligation. Treatment with 1,25(OH)2D3 prevents the upregulation of MHC II and costimulatory molecule (CD40, CD80, CD86) expression [236, 232, 233, 231, 237]. 1,25(OH)2D3-conditioned DCs produce significantly lower levels of IL-12 [232, 233, 231, 237], but higher amounts of the anti-inflammatory cytokine IL-10 [232, 234]. This results in a decreased capacity to induce T cell responses in a mixed-lymphocyte reaction setting [236, 234, 235, 232, 233, 237]. In addition, 1,25(OH)2D3-conditioned DCs are able to induce IL-10-producing Tregs that display potent immunosuppressive activity [237]. In conclusion, these studies showed that 1,25(OH)2D3 treatment induces a tolerogenic phenotype in DCs and introduced the idea of using 1,25(OH)2D3-conditioned DCs for immunotherapy. Although tolerogenic DCs can also be obtained by administering other compounds such as glucocorticoids, 1,25(OH)2D3-treated DCs present the advantage of inducing Tregs with antigen specificity [237]. As a whole, this opens the door to using 1,25(OH)2D3-treated DCs loaded with antigen as immunotherapy, excluding the possibility of unwanted off-target effects. Indeed, in a model of allogeneic pancreatic islet transplantation, adoptive transfer of antigen-loaded DCs only prevented graft rejection if DCs were pre-treated with 1,25(OH)2D3 [238]. Furthermore, treatment with 1,25(OH)2D3 enabled antigen-loaded DCs to promote prolonged survival of skin grafts expressing the same antigens [231].
Regarding the mechanism through which 1,25(OH)2D3 exerts its effects on DCs, there are several reports implicating inhibition of NF-κB by this metabolite [239–242]. A recent study showed that VDR is able to interact directly with IKKβ, preventing assembly of the IKK complex and consequently inhibiting phosphorylation and degradation of IκBα. This results in impaired translocation of NF-κB to the nucleus and hence lower transcriptional activity [243]. In addition, VDR activation could induce metabolic reprogramming in DCs. Proteomic analysis revealed that 1,25(OH)2D3 treatment induces dramatic changes in DC glucose metabolism, including proteins involved in glycolysis, the TCA cycle and pentose phosphate pathway [244]. Lipid metabolism was also modulated by this metabolite, in particular fatty acid oxidation and elongation in the mitochondria, glycerophospholipid metabolism and phospholipid degradation [244]. Later studies confirmed these findings, showing that 1,25(OH)2D3 upregulates oxidative metabolism and aerobic glycolysis through the PI3K/Akt/mTOR pathway. Interestingly, 1,25(OH)2D3-treated DCs rely on glycolysis to induce and maintain their tolerogenic phenotype [245, 246].
Serum levels of the active form of VitD 1,25(OH)2D3 are in the picomolar range, which is around 100 times lower than the dose used in the studies described above. Conversely, the concentration of the monohydroxylated precursor is approximately 1000 fold higher [247], implying that the effect of VitD on DCs could be influenced by its local conversion by neighboring cells or even DCs themselves. This local production could lead to accumulation of this metabolite in the microenvironment, thus reaching effective concentrations to act on its target cells. Although the liver and kidneys are the major sites of VitD modification, the expression of VitD-metabolizing enzymes in immune cells [248, 249], including DCs [250] has been reported. In studies conducted by several groups, human in vitro-generated DCs and freshly isolated DCs from blood were capable of producing 1,25(OH)2D3 when an external source of 25-hydroxyvitamin D3 was added [250–252]. 1,25(OH)2D3 production was further increased when LPS was added to the culture. This observation was accompanied by the discovery of 1-⍺ hydroxylase expression in these cell subsets, which was inducible by LPS stimulation [250]. Nevertheless, the VitD-metabolizing ability of the different DC subsets in vivo remains unknown. It is important to consider that 1,25(OH)2D3 generated by DCs could also act on other cells that express the VDR, such as T cells [225]. This would have important implications during the adaptive immune response by downregulating T cell activation and promoting generation of Tregs. In addition, it could be important for maintaining tolerance. 1,25(OH)2D3 has a direct effect on T cells in vitro, characterized by an upregulation of Foxp3 and CTLA-4 and inhibition of pro-inflammatory cytokine production [253]. In contrast, the addition of 25-hydroxyvitamin D3, the precursor of the active metabolite, only had an effect on T cell activation when DCs were present, supporting the idea that DCs can regulate the activation of nearby T cells by metabolizing the VitD precursor into its active form [252]. A study by Sigmundsdottir et al. claimed that VitD conversion by DCs might play an important role in the skin. 1,25(OH)2D3 induced the chemokine receptor CCR10 in T cells, thus promoting their migration and retention in the epidermis once they enter the skin [254]. Furthermore, this effect was further enhanced by IL-12. These findings led the authors to hypothesize that T cell recruitment to the epidermis could have emerged as a response to the epidermal damage caused by sun exposure. Nevertheless, the implications of this and whether this process functions as a tissue-repair mechanism need to be further investigated. Interestingly, the ImmGen database shows high VDR expression in LCs [33], suggesting that 1,25(OH)2D3 synthesized in the skin could have an effect on this cell subset. Analysis of human ex vivo dermal DCs and LCs revealed that 1,25(OH)2D3 diminished the immunogenicity of both subpopulations, but it only endowed LCs with Treg-inducing ability [255].
Current studies estimate that 1 billion people worldwide suffer from VitD deficiency [256]. A growing body of evidence supports the notion that low VitD levels correlate with higher occurrence of autoimmune diseases, including MS, type 1 diabetes, SLE and RA. In many of these cases, the information available suggests that VitD supplementation could prevent/delay the onset of disease or ameliorate its outcome. Epidemiological studies point towards a connection between geographical latitude and prevalence of MS; regions located closer to the equator have a lower incidence of the disease [257]. This gives room for speculation regarding the role of sun exposure, and therefore, VitD, in the development of MS. Supporting this hypothesis, the amount of VitD in serum from MS patients negatively correlate with disease severity [258]. Moreover, a recent clinical trial revealed that high-dose VitD supplementation in MS patients is able to downregulate IL-17 production by CD4+ T cells, as well as the frequencies of effector CD4+ T cells [259]. Another link between VitD and MS incidence was provided by several genome-wide association studies which implicated the genes coding for the VitD-metabolizing enzymes (CYP27B1, CYP24A1) in the pathogenesis of MS, showing that specific alleles correlate with greater risk of developing the disease [260, 261]. Exon sequencing of CYP27B1 from individuals belonging to families with history of MS identified a number of loss-of-function variants which were associated with higher incidence of MS [262]. There is also evidence of an association between specific polymorphisms in the VDR gene and predisposition to the disease [263, 264]. Although the polymorphisms implicated do not result in changes to the protein structure, they might affect the stability of the mRNA or its translation efficiency.
VitD has also been implicated in IBD. IBD patients display deficient VitD levels [265, 266], while Vdr −/− mice display higher susceptibility to different models of colitis [267, 268, 269]. These findings suggest a link between VitD levels and gut homeostasis. A study of the gut microbiome of Vdr −/− and Cyp27b1 −/− mice, which cannot synthesize 1,25(OH)2D3, revealed that these mutations result in alteration of the composition of intestinal flora, supporting a role for VitD in regulating colonization by different bacterial families. Together, it appears that reduced VitD levels can give rise to reduced tolerogenic DC frequencies, leading to insufficient Treg numbers and break of tolerance.
Concluding remarks
The field of metabolites exerting effects on DCs, as well as other immune cell populations, is ever expanding. Other classes of metabolites which were not included in this review due to space limitations include tryptophan derivatives and aryl hydrocarbons (reviewed in [270]), cholesterol derivatives and bile acids (reviewed in [271]), lipoxins and resolvins (reviewed in [272]), and others (reviewed in [141]).
Although the first studies ascribing immunomodulatory properties to the metabolites listed in this review were published over three decades ago, their role as a means of communication between DCs and the environment has only recently begun to be deciphered. This is due in part to major advances in the development of tools to study DC biology and ontogeny that took place in the last few years. A significant proportion of the information available so far derives from studies using GM-CSF-derived DCs which present several limitations. Therefore, readdressing some of the initial questions with more physiological culture systems is highly important. Together with the use of mice that allow targeting specific DC subsets in vivo, this will shed more light on how metabolites can influence DC function in the context of immune-mediated diseases.
References
Mayer CT, Berod L, Sparwasser T (2012) Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol 3:183. doi:10.3389/fimmu.2012.00183
Kim JM, Rasmussen JP, Rudensky AY (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8(2):191–197. doi:10.1038/ni1428
Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T (2007) Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 204(1):57–63. doi:10.1084/jem.20061852
Sakaguchi S (2004) Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol 22:531–562. doi:10.1146/annurev.immunol.21.120601.141122
Swee LK, Bosco N, Malissen B, Ceredig R, Rolink A (2009) Expansion of peripheral naturally occurring T regulatory cells by fms-like tyrosine kinase 3 ligand treatment. Blood 113(25):6277–6287. doi:10.1182/blood-2008-06-161026
Collins CB, Aherne CM, McNamee EN, Lebsack MD, Eltzschig H, Jedlicka P, Rivera-Nieves J (2012) Flt3 ligand expands CD103(+) dendritic cells and FoxP3 (+) T regulatory cells, and attenuates Crohn's-like murine ileitis. Gut 61(8):1154–1162. doi:10.1136/gutjnl-2011-300820
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F (2007) A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764. doi:10.1084/jem.20070590
Yamazaki S, Dudziak D, Heidkamp GF, Fiorese C, Bonito AJ, Inaba K, Nussenzweig MC, Steinman RM (2008) CD8+ CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J Immunol 181(10):6923–6933
Darrasse-Jeze G, Deroubaix S, Mouquet H, Victora GD, Eisenreich T, Yao KH, Masilamani RF, Dustin ML, Rudensky A, Liu K, Nussenzweig MC (2009) Feedback control of regulatory T cell homeostasis by dendritic cells in vivo. J Exp Med 206(9):1853–1862. doi:10.1084/jem.20090746
Ohnmacht C, Pullner A, King SB, Drexler I, Meier S, Brocker T, Voehringer D (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206(3):549–559. doi:10.1084/jem.20082394
Collin M, Bigley V, Haniffa M, Hambleton S (2011) Human dendritic cell deficiency: the missing ID? Nat Rev Immunol 11(9):575–583. doi:10.1038/nri3046
Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, Jardine L, Pagan S, Dimmick I, Chua I, Wallis J, Lordan J, Morgan C, Kumararatne DS, Doffinger R, van der Burg M, van Dongen J, Cant A, Dick JE, Hambleton S, Collin M (2011) The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med 208(2):227–234. doi:10.1084/jem.20101459
Esterhazy D, Loschko J, London M, Jove V, Oliveira TY, Mucida D (2016) Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat Immunol 17(5):545–555. doi:10.1038/ni.3408
Amodio G, Gregori S (2012) Dendritic cells a double-edge sword in autoimmune responses. Front Immunol 3:233. doi:10.3389/fimmu.2012.00233
Pearce EJ, Everts B (2015) Dendritic cell metabolism. Nat Rev Immunol 15(1):18–29. doi:10.1038/nri3771
Luckey TD (1972) Introduction to intestinal microecology. Am J Clin Nutr 25(12):1292–1294
Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod LP, Harris NL, Marsland BJ (2014) Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med 20(2):159–166. doi:10.1038/nm.3444
Graf D, Di Cagno R, Fak F, Flint HJ, Nyman M, Saarela M, Watzl B (2015) Contribution of diet to the composition of the human gut microbiota. Microb Ecol Health Dis 26:26164. doi:10.3402/mehd.v26.26164
Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M (2014) Role of "western diet" in inflammatory autoimmune diseases. Current allergy and asthma reports 14(1):404. doi:10.1007/s11882-013-0404-6
Leslie RD, Hawa M (1994) Twin studies in auto-immune disease. Acta Genet Med Gemellol 43(1–2):71–81
Strachan DP (1989) Hay fever, hygiene, and household size. BMJ 299(6710):1259–1260
Walk ST, Blum AM, Ewing SA, Weinstock JV, Young VB (2010) Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm Bowel Dis 16(11):1841–1849. doi:10.1002/ibd.21299
Rausch S, Held J, Fischer A, Heimesaat MM, Kuhl AA, Bereswill S, Hartmann S (2013) Small intestinal nematode infection of mice is associated with increased enterobacterial loads alongside the intestinal tract. PLoS One 8(9):e74026. doi:10.1371/journal.pone.0074026
Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, Valdez Y, Yebra MJ, Finlay BB, Maizels RM (2014) Commensal-pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes 5(4):522–532. doi:10.4161/gmic.32155
Holm JB, Sorobetea D, Kiilerich P, Ramayo-Caldas Y, Estelle J, Ma T, Madsen L, Kristiansen K, Svensson-Frej M (2015) Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of lactobacilli. PLoS One 10(5):e0125495. doi:10.1371/journal.pone.0125495
Houlden A, Hayes KS, Bancroft AJ, Worthington JJ, Wang P, Grencis RK, Roberts IS (2015) Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: effects reversed by pathogen clearance. PLoS One 10(5):e0125945. doi:10.1371/journal.pone.0125945
Kay GL, Millard A, Sergeant MJ, Midzi N, Gwisai R, Mduluza T, Ivens A, Nausch N, Mutapi F, Pallen M (2015) Differences in the Faecal Microbiome in Schistosoma haematobium Infected Children vs. Uninfected Children. PLoS neglected tropical diseases 9(6):e0003861. doi:10.1371/journal.pntd.0003861
Lee SC, Tang MS, Lim YA, Choy SH, Kurtz ZD, Cox LM, Gundra UM, Cho I, Bonneau R, Blaser MJ, Chua KH, Loke P (2014) Helminth colonization is associated with increased diversity of the gut microbiota. PLoS Negl Trop Dis 8(5):e2880. doi:10.1371/journal.pntd.0002880
Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM (1990) The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. J Exp Med 171(5):1753–1771
Homann D, Jahreis A, Wolfe T, Hughes A, Coon B, van Stipdonk MJ, Prilliman KR, Schoenberger SP, von Herrath MG (2002) CD40L blockade prevents autoimmune diabetes by induction of bitypic NK/DC regulatory cells. Immunity 16(3):403–415
Lin Y, Roberts TJ, Sriram V, Cho S, Brutkiewicz RR (2003) Myeloid marker expression on antiviral CD8+ T cells following an acute virus infection. Eur J Immunol 33(10):2736–2743. doi:10.1002/eji.200324087
Huleatt JW, Lefrancois L (1995) Antigen-driven induction of CD11c on intestinal intraepithelial lymphocytes and CD8+ T cells in vivo. J Immunol 154(11):5684–5693
Heng TS, Painter MW, Immunological Genome Project C (2008) The immunological genome project: networks of gene expression in immune cells. Nat Immunol 9(10):1091–1094. doi:10.1038/ni1008-1091
Arnold-Schrauf C, Berod L, Sparwasser T (2015) Dendritic cell specific targeting of MyD88 signalling pathways in vivo. Eur J Immunol 45(1):32–39. doi:10.1002/eji.201444747
Dudek M, Puttur F, Arnold-Schrauf C, Kuhl AA, Holzmann B, Henriques-Normark B, Berod L, Sparwasser T (2015) Lung epithelium and myeloid cells cooperate to clear acute pneumococcal infection. Mucosal immunology. doi:10.1038/mi.2015.128
Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM (2012) Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209(6):1135–1152. doi:10.1084/jem.20120030
Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC (2012) Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med 209(6):1153–1165. doi:10.1084/jem.20112675
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14(8):571–578. doi:10.1038/nri3712
Martin-Fontecha A, Lanzavecchia A, Sallusto F (2009) Dendritic cell migration to peripheral lymph nodes. Handb Exp Pharmacol 188:31–49. doi:10.1007/978-3-540-71029-5_2
Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, Salama A, Movassaghi K, Opitz C, Mages HW, Henn V, Kloetzel PM, Gurka S, Kroczek RA (2010) Superior antigen cross-presentation and XCR1 expression define human CD11c + CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med 207(6):1273–1281. doi:10.1084/jem.20100348
Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B, Larbi A, Tan P, Zhao H, Poidinger M, Pagan S, Cookson S, Dickinson R, Dimmick I, Jarrett RF, Renia L, Tam J, Song C, Connolly J, Chan JK, Gehring A, Bertoletti A, Collin M, Ginhoux F (2012) Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity 37(1):60–73. doi:10.1016/j.immuni.2012.04.012
Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, Chen CJ, Dunbar PR, Wadley RB, Jeet V, Vulink AJ, Hart DN, Radford KJ (2010) Human CD141+ (BDCA-3) + dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 207(6):1247–1260. doi:10.1084/jem.20092140
Yu CI, Becker C, Wang Y, Marches F, Helft J, Leboeuf M, Anguiano E, Pourpe S, Goller K, Pascual V, Banchereau J, Merad M, Palucka K (2013) Human CD1c + dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity 38(4):818–830. doi:10.1016/j.immuni.2013.03.004
Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre E, Vu Manh TP, Baranek T, Storset AK, Marvel J, Boudinot P, Hosmalin A, Schwartz-Cornil I, Dalod M (2010) The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha + dendritic cells. J Exp Med 207(6):1283–1292. doi:10.1084/jem.20100223
Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le Moine A, Faure F, Donckier V, Sancho D, Cerundolo V, Bonnet D, Reis-e Sousa C (2010) Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha + dendritic cells. J Exp Med 207(6):1261–1271. doi:10.1084/jem.20092618
den Haan JM, Lehar SM, Bevan MJ (2000) CD8(+) but not CD8(−) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192(12):1685–1696
Pooley JL, Heath WR, Shortman K (2001) Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8- dendritic cells, but cross-presented to CD8 T cells by CD8+ dendritic cells. J Immunol 166(9):5327–5330
Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y, Takahara K, Steinman RM, Inaba K (2002) The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 195(10):1289–1302
Schulz O, Reis e Sousa C (2002) Cross-presentation of cell-associated antigens by CD8alpha + dendritic cells is attributable to their ability to internalize dead cells. Immunology 107(2):183–189
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha + dendritic cells in cytotoxic T cell immunity. Science 322(5904):1097–1100. doi:10.1126/science.1164206
Belz GT, Smith CM, Eichner D, Shortman K, Karupiah G, Carbone FR, Heath WR (2004) Cutting edge: conventional CD8 alpha + dendritic cells are generally involved in priming CTL immunity to viruses. J Immunol 172(4):1996–2000
Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, Ploegh HL, Murphy TL, Sibley LD, Murphy KM (2011) CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by toxoplasma gondii tachyzoites. Immunity 35(2):249–259. doi:10.1016/j.immuni.2011.08.008
Hopp AK, Rupp A, Lukacs-Kornek V (2014) Self-antigen presentation by dendritic cells in autoimmunity. Front Immunol 5:55. doi:10.3389/fimmu.2014.00055
Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M (2001) Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J Immunol 166(9):5448–5455
Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, Bhattacharya D, Stappenbeck TS, Holtzman MJ, Sung SS, Murphy TL, Hildner K, Murphy KM (2010) Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha + conventional dendritic cells. J Exp Med 207(4):823–836. doi:10.1084/jem.20091627
Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AW, See P, Shin A, Wasan PS, Hoeffel G, Malleret B, Heiseke A, Chew S, Jardine L, Purvis HA, Hilkens CM, Tam J, Poidinger M, Stanley ER, Krug AB, Renia L, Sivasankar B, Ng LG, Collin M, Ricciardi-Castagnoli P, Honda K, Haniffa M, Ginhoux F (2013) IRF4 transcription factor-dependent CD11b + dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity 38(5):970–983. doi:10.1016/j.immuni.2013.04.011
Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, Nussenzweig MC (2007) Differential antigen processing by dendritic cell subsets in vivo. Science 315(5808):107–111. doi:10.1126/science.1136080
Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, Ivanov II, Liu K, Merad M, Reizis B (2011) Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35(5):780–791. doi:10.1016/j.immuni.2011.08.013
Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, Woltman AM, Reyal Y, Bonnet D, Sichien D, Bain CC, Mowat AM, Reis e Sousa C, Poulin LF, Malissen B, Guilliams M (2012) CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol 42(12):3150–3166. doi:10.1002/eji.201242847
Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, Vanhoutte L, Neyt K, Killeen N, Malissen B, Hammad H, Lambrecht BN (2013) Conventional and monocyte-derived CD11b (+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38(2):322–335. doi:10.1016/j.immuni.2012.10.016
Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, Mucida D, Merad M, Steinman RM Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest 123(2):844–854. doi:10.1172/JCI65260
Kim TS, Gorski SA, Hahn S, Murphy KM, Braciale TJ (2014) Distinct dendritic cell subsets dictate the fate decision between effector and memory CD8 (+) T cell differentiation by a CD24-dependent mechanism. Immunity 40(3):400–413. doi:10.1016/j.immuni.2014.02.004
Cerovic V, Bain CC, Mowat AM, Milling SW (2014) Intestinal macrophages and dendritic cells: what's the difference? Trends Immunol 35(6):270–277. doi:10.1016/j.it.2014.04.003
Merad M, Sathe P, Helft J, Miller J, Mortha A (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604. doi:10.1146/annurev-immunol-020711-074950
Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, Pollet E, Ardouin L, Luche H, Sanchez C, Dalod M, Malissen B, Henri S (2013) Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39(5):925–938. doi:10.1016/j.immuni.2013.10.004
Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, Kissenpfennig A, Barbaroux JB, Groves R, Geissmann F (2009) Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med 206(13):3089–3100. doi:10.1084/jem.20091586
Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, Choy SH, Grisotto M, Renia L, Conway SJ, Stanley ER, Chan JK, Ng LG, Samokhvalov IM, Merad M, Ginhoux F (2012) Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209(6):1167–1181. doi:10.1084/jem.20120340
Malissen B, Tamoutounour S, Henri S (2014) The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol 14(6):417–428. doi:10.1038/nri3683
Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176(6):1693–1702
Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G (1999) An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223(1):77–92
Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C (2015) GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c (+) MHCII (+) macrophages and dendritic cells. Immunity 42(6):1197–1211. doi:10.1016/j.immuni.2015.05.018
Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, O'Keeffe M, Shortman K (2006) Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol 7(6):663–671. doi:10.1038/ni1340
Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stuve P, Puttur F, Merad M, Sparwasser T (2014) Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood 124(20):3081–3091. doi:10.1182/blood-2013-12-545772
Proietto AI, Mittag D, Roberts AW, Sprigg N, Wu L (2012) The equivalents of human blood and spleen dendritic cell subtypes can be generated in vitro from human CD34 (+) stem cells in the presence of fms-like tyrosine kinase 3 ligand and thrombopoietin. Cellular & molecular immunology 9(6):446–454. doi:10.1038/cmi.2012.48
Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP (2014) Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol 12(1):e1001759. doi:10.1371/journal.pbio.1001759
Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ (2010) Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115(23):4742–4749. doi:10.1182/blood-2009-10-249540
Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol 15(4):323–332. doi:10.1038/ni.2833
Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, Pearce EL, Pearce EJ (2012) Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120(7):1422–1431. doi:10.1182/blood-2012-03-419747
Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Glasner J, Warnecke C, Wiesener MS, Eckardt KU, Steinkasserer A, Hensel M, Willam C (2008) Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol 180(7):4697–4705
Bhandari T, Olson J, Johnson RS, Nizet V (2013) HIF-1alpha influences myeloid cell antigen presentation and response to subcutaneous OVA vaccination. J Mol Med 91(10):1199–1205. doi:10.1007/s00109-013-1052-y
Berod L, Sparwasser T (2016) pDCs take a deep breath to fight viruses. Immunity 44(6):1246–1248. doi:10.1016/j.immuni.2016.06.005
Wu D, Sanin DE, Everts B, Chen Q, Qiu J, Buck MD, Patterson A, Smith AM, Chang CH, Liu Z, Artyomov MN, Pearce EL, Cella M, Pearce EJ (2016) Type 1 interferons induce changes in Core metabolism that are critical for immune function. Immunity 44(6):1325–1336. doi:10.1016/j.immuni.2016.06.006
O'Neill LA, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213(1):15–23. doi:10.1084/jem.20151570
Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwarzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, Carballido JM (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9(11):1261–1269. doi:10.1038/ni.1657
Wani R, Nagata A, Murray BW (2014) Protein redox chemistry: post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front Pharmacol 5:224. doi:10.3389/fphar.2014.00224
Rooks MG, Garrett WS (2016) Gut microbiota, metabolites and host immunity. Nat Rev Immunol 16(6):341–352. doi:10.1038/nri.2016.42
Passalacqua KD, Charbonneau ME, O'Riordan MX (2016) Bacterial metabolism shapes the host-pathogen Interface. Microbiology spectrum 4(3). doi:10.1128/microbiolspec.VMBF-0027-2015
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461(7261):282–286. doi:10.1038/nature08296
Homolya L, Steinberg TH, Boucher RC (2000) Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. J Cell Biol 150(6):1349–1360
Pakdeechote P, Rummery NM, Ralevic V, Dunn WR (2007) Raised tone reveals purinergic-mediated responses to sympathetic nerve stimulation in the rat perfused mesenteric vascular bed. Eur J Pharmacol 563(1–3):180–186. doi:10.1016/j.ejphar.2007.02.011
Deaglio S, Robson SC (2011) Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv Pharmacol 61:301–332. doi:10.1016/B978-0-12-385526-8.00010-2
Fredholm BB, Irenius E, Kull B, Schulte G (2001) Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol 61(4):443–448
Cabrita MA, Baldwin SA, Young JD, Cass CE (2002) Molecular biology and regulation of nucleoside and nucleobase transporter proteins in eukaryotes and prokaryotes. Biochemistry and cell biology = Biochimie et biologie cellulaire 80(5):623–638
Pastor-Anglada M, Casado FJ, Valdes R, Mata J, Garcia-Manteiga J, Molina M (2001) Complex regulation of nucleoside transporter expression in epithelial and immune system cells. Mol Membr Biol 18(1):81–85
Burnstock G, Kennedy C (1985) Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol 16(5):433–440
Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-Haerter PJ, Mrowietz U, Dichmann S, Norgauer J (2001) Expression and function of adenosine receptors in human dendritic cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 15(11):1963–1970. doi:10.1096/fj.01-0169com
Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, Girolomoni G, Norgauer J (2003) Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 101(10):3985–3990. doi:10.1182/blood-2002-07-2113
Challier J, Bruniquel D, Sewell AK, Laugel B (2013) Adenosine and cAMP signalling skew human dendritic cell differentiation towards a tolerogenic phenotype with defective CD8 (+) T-cell priming capacity. Immunology 138(4):402–410. doi:10.1111/imm.12053
Ben Addi A, Lefort A, Hua X, Libert F, Communi D, Ledent C, Macours P, Tilley SL, Boeynaems JM, Robaye B (2008) Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the a(2B) receptor. Eur J Immunol 38(6):1610–1620. doi:10.1002/eji.200737781
Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, Linden J, Ernst PB (2009) The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol 182(8):4616–4623. doi:10.4049/jimmunol.0801279
Ring S, Pushkarevskaya A, Schild H, Probst HC, Jendrossek V, Wirsdorfer F, Ledent C, Robson SC, Enk AH, Mahnke K (2015) Regulatory T cell-derived adenosine induces dendritic cell migration through the Epac-Rap1 pathway. J Immunol 194(8):3735–3744. doi:10.4049/jimmunol.1401434
Takenaka MC, Robson S, Quintana FJ (2016) Regulation of the T cell response by CD39. Trends Immunol 37(7):427–439. doi:10.1016/j.it.2016.04.009
Molle C, Goldman M, Goriely S (2010) Critical role of the IFN-stimulated gene factor 3 complex in TLR-mediated IL-27p28 gene expression revealing a two-step activation process. J Immunol 184(4):1784–1792. doi:10.4049/jimmunol.0902005
Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL (2007) A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol 8(12):1380–1389. doi:10.1038/ni1541
Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N (2006) Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol 7(9):929–936. doi:10.1038/ni1375
Fitzgerald DC, Zhang GX, El-Behi M, Fonseca-Kelly Z, Li H, Yu S, Saris CJ, Gran B, Ciric B, Rostami A (2007) Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol 8(12):1372–1379. doi:10.1038/ni1540
Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O'Shea JJ, Hennighausen L, Ernst M, Hunter CA (2006) Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol 7(9):937–945. doi:10.1038/ni1376
Karakhanova S, Bedke T, Enk AH, Mahnke K (2011) IL-27 renders DC immunosuppressive by induction of B7-H1. J Leukoc Biol 89(6):837–845. doi:10.1189/jlb.1209788
Wang S, Miyazaki Y, Shinozaki Y, Yoshida H (2007) Augmentation of antigen-presenting and Th1-promoting functions of dendritic cells by WSX-1 (IL-27R) deficiency. J Immunol 179(10):6421–6428
Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, Kuchroo VK, Robson SC, Quintana FJ (2013) IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol 14(10):1054–1063. doi:10.1038/ni.2695
Ghaemi Oskouie F, Shameli A, Yang A, Desrosiers MD, Mucsi AD, Blackburn MR, Yang Y, Santamaria P, Shi Y (2011) High levels of adenosine deaminase on dendritic cells promote autoreactive T cell activation and diabetes in nonobese diabetic mice. J Immunol 186(12):6798–6806. doi:10.4049/jimmunol.1004222
Pacheco R, Martinez-Navio JM, Lejeune M, Climent N, Oliva H, Gatell JM, Gallart T, Mallol J, Lluis C, Franco R (2005) CD26, adenosine deaminase, and adenosine receptors mediate costimulatory signals in the immunological synapse. Proc Natl Acad Sci U S A 102(27):9583–9588. doi:10.1073/pnas.0501050102
Desrosiers MD, Cembrola KM, Fakir MJ, Stephens LA, Jama FM, Shameli A, Mehal WZ, Santamaria P, Shi Y (2007) Adenosine deamination sustains dendritic cell activation in inflammation. J Immunol 179(3):1884–1892
Casanova V, Naval-Macabuhay I, Massanella M, Rodriguez-Garcia M, Blanco J, Gatell JM, Garcia F, Gallart T, Lluis C, Mallol J, Franco R, Climent N, McCormick PJ (2012) Adenosine deaminase enhances the immunogenicity of human dendritic cells from healthy and HIV-infected individuals. PLoS One 7(12):e51287. doi:10.1371/journal.pone.0051287
Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP (2009) Contribution of adenosine A2B receptors to inflammatory parameters of experimental colitis. J Immunol 182(8):4957–4964. doi:10.4049/jimmunol.0801324
Friedman DJ, Kunzli BM, YI, AR, Sevigny J, Berberat PO, Enjyoji K, Csizmadia E, Friess H, Robson SC (2009) From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A 106 (39):16788–16793. doi:10.1073/pnas.0902869106
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57(13):2602–2605
Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z (2011) Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol 187(2):676–683. doi:10.4049/jimmunol.1003884
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, Dikov MM (2008) Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112(5):1822–1831. doi:10.1182/blood-2008-02-136325
Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J (2012) Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol 188(1):198–205. doi:10.4049/jimmunol.1101845
Leone RD, Lo YC, Powell JD (2015) A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Computational and structural biotechnology journal 13:265–272. doi:10.1016/j.csbj.2015.03.008
Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF (1993) The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc Natl Acad Sci U S A 90(1):312–316
Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS (2010) Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature 467(7317):863–867. doi:10.1038/nature09413
Schwiebert EM, Egan ME, Hwang TH, Fulmer SB, Allen SS, Cutting GR, Guggino WB (1995) CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell 81(7):1063–1073
Idzko M, Dichmann S, Ferrari D, Di Virgilio F, la Sala A, Girolomoni G, Panther E, Norgauer J (2002) Nucleotides induce chemotaxis and actin polymerization in immature but not mature human dendritic cells via activation of pertussis toxin-sensitive P2y receptors. Blood 100(3):925–932
Liu QH, Bohlen H, Titzer S, Christensen O, Diehl V, Hescheler J, Fleischmann BK (1999) Expression and a role of functionally coupled P2Y receptors in human dendritic cells. FEBS Lett 445(2–3):402–408
Coutinho-Silva R, Persechini PM, Bisaggio RD, Perfettini JL, Neto AC, Kanellopoulos JM, Motta-Ly I, Dautry-Varsat A, Ojcius DM (1999) P2Z/P2X7 receptor-dependent apoptosis of dendritic cells. Am J Phys 276(5 Pt 1):C1139–C1147
Ferrari D, La Sala A, Chiozzi P, Morelli A, Falzoni S, Girolomoni G, Idzko M, Dichmann S, Norgauer J, Di Virgilio F (2000) The P2 purinergic receptors of human dendritic cells: identification and coupling to cytokine release. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 14(15):2466–2476. doi:10.1096/fj.00-0031com
Nihei OK, de Carvalho AC, Savino W, Alves LA (2000) Pharmacologic properties of P (2Z)/P2X (7 )receptor characterized in murine dendritic cells: role on the induction of apoptosis. Blood 96(3):996–1005
Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA (2001) Altered cytokine production in mice lacking P2X (7) receptors. J Biol Chem 276(1):125–132. doi:10.1074/jbc.M006781200
Perregaux D, Gabel CA (1994) Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem 269(21):15195–15203
Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440(7081):228–232. doi:10.1038/nature04515
Greenberg S, Di Virgilio F, Steinberg TH, Silverstein SC (1988) Extracellular nucleotides mediate Ca2+ fluxes in J774 macrophages by two distinct mechanisms. J Biol Chem 263(21):10337–10343
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Genin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, Andre F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15(10):1170–1178. doi:10.1038/nm.2028
la Sala A, Ferrari D, Corinti S, Cavani A, Di Virgilio F, Girolomoni G (2001) Extracellular ATP induces a distorted maturation of dendritic cells and inhibits their capacity to initiate Th1 responses. J Immunol 166(3):1611–1617
Ivanova EP, Alexeeva YV, Pham DK, Wright JP, Nicolau DV (2006) ATP level variations in heterotrophic bacteria during attachment on hydrophilic and hydrophobic surfaces. International microbiology : the official journal of the Spanish Society for Microbiology 9(1):37–46
Mempin R, Tran H, Chen C, Gong H, Kim Ho K, Lu S (2013) Release of extracellular ATP by bacteria during growth. BMC Microbiol 13:301. doi:10.1186/1471-2180-13-301
Hironaka I, Iwase T, Sugimoto S, Okuda K, Tajima A, Yanaga K, Mizunoe Y (2013) Glucose triggers ATP secretion from bacteria in a growth-phase-dependent manner. Appl Environ Microbiol 79(7):2328–2335. doi:10.1128/AEM.03871-12
Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K (2008) ATP drives lamina propria T (H) 17 cell differentiation. Nature 455(7214):808–812. doi:10.1038/nature07240
Neves AR, Castelo-Branco MT, Figliuolo VR, Bernardazzi C, Buongusto F, Yoshimoto A, Nanini HF, Coutinho CM, Carneiro AJ, Coutinho-Silva R, de Souza HS (2014) Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn's disease. Inflamm Bowel Dis 20(3):444–457. doi:10.1097/01.MIB.0000441201.10454.06
Castro CN, Freitag J, Berod L, Lochner M, Sparwasser T (2015) Microbe-associated immunomodulatory metabolites: Influence on T cell fate and function. Molecular immunology 68(2 Pt C):575–584. doi:10.1016/j.molimm.2015.07.025
Macfarlane S, Macfarlane GT (2003) Regulation of short-chain fatty acid production. Proc Nutr Soc 62(1):67–72. doi:10.1079/PNS2002207
Roediger WE (1980) Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 21(9):793–798
Cummings JH (1984) Colonic absorption: the importance of short chain fatty acids in man. Scand J Gastroenterol Suppl 93:89–99
Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT (1987) Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28(10):1221–1227
Cummings JH, Macfarlane GT (1991) The control and consequences of bacterial fermentation in the human colon. The Journal of applied bacteriology 70(6):443–459
Ganapathy V, Thangaraju M, Prasad PD, Martin PM, Singh N (2013) Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol 13(6):869–874. doi:10.1016/j.coph.2013.08.006
Frankel WL, Zhang W, Singh A, Klurfeld DM, Don S, Sakata T, Modlin I, Rombeau JL (1994) Mediation of the trophic effects of short-chain fatty acids on the rat jejunum and colon. Gastroenterology 106(2):375–380
Wolever TM, Brighenti F, Royall D, Jenkins AL, Jenkins DJ (1989) Effect of rectal infusion of short chain fatty acids in human subjects. Am J Gastroenterol 84(9):1027–1033
Canfora EE, Jocken JW, Blaak EE (2015) Short-chain fatty acids in control of body weight and insulin sensitivity. Nat Rev Endocrinol 11(10):577–591. doi:10.1038/nrendo.2015.128
De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, Backhed F, Mithieux G (2014) Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 156(1–2):84–96. doi:10.1016/j.cell.2013.12.016
Davie JR (2003) Inhibition of histone deacetylase activity by butyrate. J Nutr 133(7 Suppl):2485S–2493S
Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, Muir AI, Wigglesworth MJ, Kinghorn I, Fraser NJ, Pike NB, Strum JC, Steplewski KM, Murdock PR, Holder JC, Marshall FH, Szekeres PG, Wilson S, Ignar DM, Foord SM, Wise A, Dowell SJ (2003) The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem 278(13):11312–11319. doi:10.1074/jbc.M211609200
Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, Mellinger JD, Smith SB, Digby GJ, Lambert NA, Prasad PD, Ganapathy V (2009) GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res 69(7):2826–2832. doi:10.1158/0008-5472.CAN-08-4466
Le Poul E, Loison C, Struyf S, Springael JY, Lannoy V, Decobecq ME, Brezillon S, Dupriez V, Vassart G, Van Damme J, Parmentier M, Detheux M (2003) Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem 278(28):25481–25489. doi:10.1074/jbc.M301403200
Nilsson NE, Kotarsky K, Owman C, Olde B (2003) Identification of a free fatty acid receptor, FFA2R, expressed on leukocytes and activated by short-chain fatty acids. Biochem Biophys Res Commun 303(4):1047–1052
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR (2007) Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104(34):13780–13785. doi:10.1073/pnas.0706625104
Treem WR, Ahsan N, Shoup M, Hyams JS (1994) Fecal short-chain fatty acids in children with inflammatory bowel disease. J Pediatr Gastroenterol Nutr 18(2):159–164
Mazmanian SK, Round JL, Kasper DL (2008) A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453(7195):620–625. doi:10.1038/nature07008
Round JL, Mazmanian SK (2010) Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107(27):12204–12209. doi:10.1073/pnas.0909122107
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR (2009) Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461(7268):1282–U1119. doi:10.1038/nature08530
Sina C, Gavrilova O, Forster M, Till A, Derer S, Hildebrand F, Raabe B, Chalaris A, Scheller J, Rehmann A, Franke A, Ott S, Hasler R, Nikolaus S, Folsch UR, Rose-John S, Jiang HP, Li J, Schreiber S, Rosenstiel P (2009) G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol 183(11):7514–7522. doi:10.4049/jimmunol.0900063
Fournier BM, Parkos CA (2012) The role of neutrophils during intestinal inflammation. Mucosal immunology 5(4):354–366. doi:10.1038/mi.2012.24
Vinolo MA, Hatanaka E, Lambertucci RH, Newsholme P, Curi R (2009) Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem Funct 27(1):48–55. doi:10.1002/cbf.1533
Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R (2009) Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci 117(9):331–338. doi:10.1042/CS20080642
Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly YM, Stephens L, Hawkins PT, Curi R (2011) SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One 6(6):e21205. doi:10.1371/journal.pone.0021205
Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M (2014) Dextran sulfate sodium (DSS) induced colitis in mice. Curr Protoc Immunol 104:Unit 15 25. doi:10.1002/0471142735.im1525s104
Chang PV, Hao L, Offermanns S, Medzhitov R (2014) The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A 111(6):2247–2252. doi:10.1073/pnas.1322269111
Nastasi C, Candela M, Bonefeld CM, Geisler C, Hansen M, Krejsgaard T, Biagi E, Andersen MH, Brigidi P, Odum N, Litman T, Woetmann A (2015) The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci Rep 5:16148. doi:10.1038/srep16148
Iraporda C, Errea A, Romanin DE, Cayet D, Pereyra E, Pignataro O, Sirard JC, Garrote GL, Abraham AG, Rumbo M (2015) Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 220(10):1161–1169. doi:10.1016/j.imbio.2015.06.004
Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, Bassi EJ, Moraes-Vieira PM, Hiyane MI, Rodas AC, Peron JP, Aguiar CF, Reis MA, Ribeiro WR, Valduga CJ, Curi R, Vinolo MA, Ferreira CM, Camara NO (2015) Gut bacteria products prevent AKI induced by ischemia-reperfusion. Journal of the American Society of Nephrology : JASN 26(8):1877–1888. doi:10.1681/ASN.2014030288
Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY (2013) Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504(7480):451–455. doi:10.1038/nature12726
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40(1):128–139. doi:10.1016/j.immuni.2013.12.007
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504(7480):446–450. doi:10.1038/nature12721
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341(6145):569–573. doi:10.1126/science.1241165
Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW (2007) Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med 13(11):1299–1307. doi:10.1038/nm1652
de Zoeten EF, Wang L, Sai H, Dillmann WH, Hancock WW (2010) Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 138(2):583–594. doi:10.1053/j.gastro.2009.10.037
Wang LQ, de Zoeten EF, Greene MI, Hancock WW (2009) Immunomodulatory effects of deacetylase inhibitors: therapeutic targeting of FOXP3 (+) regulatory T cells. Nat Rev Drug Discov 8(12):969–981. doi:10.1038/nrd3031
Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW (2012) Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Science signaling 5(229):ra45. doi:10.1126/scisignal.2002873
Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, Kim CH (2015) Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6 K pathway. Mucosal immunology 8(1):80–93. doi:10.1038/mi.2014.44
Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, Schwierzeck V, Utermohlen O, Chun E, Garrett WS, McCoy KD, Diefenbach A, Staeheli P, Stecher B, Amit I, Prinz M (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 18(7):965–977. doi:10.1038/nn.4030
Vinolo MA, Rodrigues HG, Nachbar RT, Curi R (2011) Regulation of inflammation by short chain fatty acids. Nutrients 3(10):858–876. doi:10.3390/nu3100858
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142(5):687–698. doi:10.1016/j.cell.2010.07.041
Talukdar S, Olefsky JM, Osborn O (2011) Targeting GPR120 and other fatty acid-sensing GPCRs ameliorates insulin resistance and inflammatory diseases. Trends Pharmacol Sci 32(9):543–550. doi:10.1016/j.tips.2011.04.004
Fujita T, Matsuoka T, Honda T, Kabashima K, Hirata T, Narumiya S (2011) A GPR40 agonist GW9508 suppresses CCL5, CCL17, and CXCL10 induction in keratinocytes and attenuates cutaneous immune inflammation. The Journal of investigative dermatology 131(8):1660–1667. doi:10.1038/jid.2011.123
Suzuki M, Takaishi S, Nagasaki M, Onozawa Y, Iino I, Maeda H, Komai T, Oda T (2013) Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J Biol Chem 288(15):10684–10691. doi:10.1074/jbc.M112.420042
Blomhoff R, Blomhoff HK (2006) Overview of retinoid metabolism and function. J Neurobiol 66(7):606–630. doi:10.1002/neu.20242
Giguere V, Ong ES, Segui P, Evans RM (1987) Identification of a receptor for the morphogen retinoic acid. Nature 330(6149):624–629. doi:10.1038/330624a0
Petkovich M, Brand NJ, Krust A, Chambon P (1987) A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 330(6147):444–450. doi:10.1038/330444a0
Balmer JE, Blomhoff R (2002) Gene expression regulation by retinoic acid. J Lipid Res 43:1773–808. doi:10.1194/jlr.R100015-JLR200
Beijer MR, Kraal G, den Haan JM (2014) Vitamin A and dendritic cell differentiation. Immunology 142(1):39–45
Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, Ferreyra GA, Leonardi AJ, Borman ZA, Wang J, Palmer DC, Wilhelm C, Cai R, Sun J, Napoli JL, Danner RL, Gattinoni L, Belkaid Y, Restifo NP (2013) Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med 210(10):1961–1976. doi:10.1084/jem.20122508
Zeng R, Bscheider M, Lahl K, Lee M, Butcher EC (2016) Generation and transcriptional programming of intestinal dendritic cells: essential role of retinoic acid. Mucosal immunology 9(1):183–193. doi:10.1038/mi.2015.50
Haileselassie Y, Navis M, Vu N, Qazi KR, Rethi B, Sverremark-Ekstrom E (2016) Postbiotic modulation of retinoic acid imprinted mucosal-like dendritic cells by probiotic lactobacillus reuteri 17938 in vitro. Front Immunol 7:96. doi:10.3389/fimmu.2016.00096
Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC (2015) Retinoic acid primes human dendritic cells to induce gut-homing, IL-10-producing regulatory T cells. Mucosal immunology 8(2):265–278. doi:10.1038/mi.2014.64
Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, Song SY (2004) Retinoic acid imprints gut-homing specificity on T cells. Immunity 21(4):527–538. doi:10.1016/j.immuni.2004.08.011
Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, Rosenthal KM, Evavold BD, Pulendran B (2009) Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med 15(4):401–409. doi:10.1038/nm.1925
Wang S, Villablanca EJ, De Calisto J, Gomes DC, Nguyen DD, Mizoguchi E, Kagan JC, Reinecker HC, Hacohen N, Nagler C, Xavier RJ, Rossi-Bergmann B, Chen YB, Blomhoff R, Snapper SB, Mora JR (2011) MyD88-dependent TLR1/2 signals educate dendritic cells with gut-specific imprinting properties. J Immunol 187(1):141–150. doi:10.4049/jimmunol.1003740
Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ (2007) All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med 204(8):1765–1774. doi:10.1084/jem.20070719
Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH (2007) Vitamin a metabolites induce gut-homing FoxP3+ regulatory T cells. J Immunol 179(6):3724–3733
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785. doi:10.1084/jem.20070602
Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O'Shea JJ (2008) Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a stat-3/stat-5 independent signaling pathway. Blood 111(3):1013–1020. doi:10.1182/blood-2007-06-096438
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835):256–260. doi:10.1126/science.1145697
Schambach F, Schupp M, Lazar MA, Reiner SL (2007) Activation of retinoic acid receptor-alpha favours regulatory T cell induction at the expense of IL-17-secreting T helper cell differentiation. Eur J Immunol 37(9):2396–2399. doi:10.1002/eji.200737621
De Leenheer AP, Lambert WE, Claeys I (1982) All-trans-retinoic acid: measurement of reference values in human serum by high performance liquid chromatography. J Lipid Res 23(9):1362–1367
Uematsu S, Fujimoto K, Jang MH, Yang BG, Jung YJ, Nishiyama M, Sato S, Tsujimura T, Yamamoto M, Yokota Y, Kiyono H, Miyasaka M, Ishii KJ, Akira S (2008) Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing toll-like receptor 5. Nat Immunol 9(7):769–776. doi:10.1038/ni.1622
Cha HR, Chang SY, Chang JH, Kim JO, Yang JY, Kim CH, Kweon MN (2010) Downregulation of Th17 cells in the small intestine by disruption of gut flora in the absence of retinoic acid. J Immunol 184(12):6799–6806. doi:10.4049/jimmunol.0902944
Wang C, Kang SG, HogenEsch H, Love PE, Kim CH (2010) Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J Immunol 184(10):5519–5526. doi:10.4049/jimmunol.0903942
Pino-Lagos K, Guo Y, Brown C, Alexander MP, Elgueta R, Bennett KA, De Vries V, Nowak E, Blomhoff R, Sockanathan S, Chandraratna RA, Dmitrovsky E, Noelle RJ (2011) A retinoic acid-dependent checkpoint in the development of CD4+ T cell-mediated immunity. J Exp Med 208(9):1767–1775. doi:10.1084/jem.20102358
Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, Belkaid Y (2011) Essential role for retinoic acid in the promotion of CD4 (+) T cell effector responses via retinoic acid receptor alpha. Immunity 34(3):435–447. doi:10.1016/j.immuni.2011.03.003
Sommer A, Tarwotjo I, Hussaini G, Susanto D (1983) Increased mortality in children with mild vitamin a deficiency. Lancet 2(8350):585–588
Stephens D, Jackson PL, Gutierrez Y (1996) Subclinical vitamin a deficiency: a potentially unrecognized problem in the United States. Pediatr Nurs 22(5):377–389 456
Sommer A, Tarwotjo I, Djunaedi E, West KP Jr, Loeden AA, Tilden R, Mele L (1986) Impact of vitamin a supplementation on childhood mortality. A randomised controlled community trial. Lancet 1(8491):1169–1173
Imdad A, Yakoob MY, Sudfeld C, Haider BA, Black RE, Bhutta ZA (2011) Impact of vitamin A supplementation on infant and childhood mortality. BMC public health 11(Suppl 3):S20. doi:10.1186/1471-2458-11-S3-S20
Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, Rajewsky K, Adams DH, von Andrian UH (2006) Generation of gut-homing IgA-secreting B cells by intestinal dendritic cells. Science 314(5802):1157–1160. doi:10.1126/science.1132742
Rudraraju R, Surman SL, Jones BG, Sealy R, Woodland DL, Hurwitz JL (2012) Reduced frequencies and heightened CD103 expression among virus-induced CD8(+) T cells in the respiratory tract airways of vitamin A-deficient mice. Clinical and vaccine immunology : CVI 19(5):757–765. doi:10.1128/CVI.05576-11
Surman SL, Rudraraju R, Sealy R, Jones B, Hurwitz JL (2012) Vitamin a deficiency disrupts vaccine-induced antibody-forming cells and the balance of IgA/IgG isotypes in the upper and lower respiratory tract. Viral Immunol 25(4):341–344. doi:10.1089/vim.2012.0023
Surman SL, Jones BG, Rudraraju R, Sealy RE, Hurwitz JL (2014) Intranasal administration of retinyl palmitate with a respiratory virus vaccine corrects impaired mucosal IgA response in the vitamin A-deficient host. Clinical and vaccine immunology : CVI 21(4):598–601. doi:10.1128/CVI.00757-13
Surman SL, Jones BG, Sealy RE, Rudraraju R, Hurwitz JL (2014) Oral retinyl palmitate or retinoic acid corrects mucosal IgA responses toward an intranasal influenza virus vaccine in vitamin a deficient mice. Vaccine 32(22):2521–2524. doi:10.1016/j.vaccine.2014.03.025
DeLuca HF (2004) Overview of general physiologic features and functions of vitamin D. Am J Clin Nutr 80(6 Suppl):1689S–1696S
Omdahl JL, Morris HA, May BK (2002) Hydroxylase enzymes of the vitamin D pathway: expression, function, and regulation. Annu Rev Nutr 22:139–166. doi:10.1146/annurev.nutr.22.120501.150216
Lemire JM, Adams JS, Sakai R, Jordan SC (1984) 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J Clin Invest 74(2):657–661. doi:10.1172/JCI111465
Rigby WF, Stacy T, Fanger MW (1984) Inhibition of T lymphocyte mitogenesis by 1,25-dihydroxyvitamin D3 (calcitriol). J Clin Invest 74(4):1451–1455. doi:10.1172/JCI111557
Bhalla AK, Amento EP, Krane SM (1986) Differential effects of 1,25-dihydroxyvitamin D3 on human lymphocytes and monocyte/macrophages: inhibition of interleukin-2 and augmentation of interleukin-1 production. Cell Immunol 98(2):311–322
Bhalla AK, Amento EP, Clemens TL, Holick MF, Krane SM (1983) Specific high-affinity receptors for 1,25-dihydroxyvitamin D3 in human peripheral blood mononuclear cells: presence in monocytes and induction in T lymphocytes following activation. J Clin Endocrinol Metab 57(6):1308–1310. doi:10.1210/jcem-57-6-1308
Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC (1983) 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science 221(4616):1181–1183
Lemire JM, Archer DC (1991) 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J Clin Invest 87(3):1103–1107. doi:10.1172/JCI115072
Cantorna MT, Hayes CE, DeLuca HF (1996) 1,25-dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc Natl Acad Sci U S A 93(15):7861–7864
Mathieu C, Laureys J, Sobis H, Vandeputte M, Waer M, Bouillon R (1992) 1,25-dihydroxyvitamin D3 prevents insulitis in NOD mice. Diabetes 41(11):1491–1495
Haussler MR, Haussler CA, Bartik L, Whitfield GK, Hsieh JC, Slater S, Jurutka PW (2008) Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev 66(10 Suppl 2):S98–112. doi:10.1111/j.1753-4887.2008.00093.x
Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R (2001) Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A 98(12):6800–6805. doi:10.1073/pnas.121172198
Penna G, Adorini L (2000) 1 alpha,25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol 164(5):2405–2411
Piemonti L, Monti P, Sironi M, Fraticelli P, Leone BE, Dal Cin E, Allavena P, Di Carlo V (2000) Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol 164(9):4443–4451
Canning MO, Grotenhuis K, de Wit H, Ruwhof C, Drexhage HA (2001) 1-alpha,25-dihydroxyvitamin D3 (1,25 (OH) (2)D (3)) hampers the maturation of fully active immature dendritic cells from monocytes. European journal of endocrinology / European Federation of Endocrine Societies 145(3):351–357
Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R (2000) Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochem Biophys Res Commun 270(3):701–708. doi:10.1006/bbrc.2000.2490
Berer A, Stockl J, Majdic O, Wagner T, Kollars M, Lechner K, Geissler K, Oehler L (2000) 1,25-dihydroxyvitamin D(3) inhibits dendritic cell differentiation and maturation in vitro. Exp Hematol 28(5):575–583
Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO (2009) Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol 39(11):3147–3159. doi:10.1002/eji.200839103
Ferreira GB, van Etten E, Verstuyf A, Waer M, Overbergh L, Gysemans C, Mathieu C (2011) 1,25-dihydroxyvitamin D3 alters murine dendritic cell behaviour in vitro and in vivo. Diabetes Metab Res Rev 27(8):933–941. doi:10.1002/dmrr.1275
Harant H, Wolff B, Lindley IJ (1998) 1Alpha,25-dihydroxyvitamin D3 decreases DNA binding of nuclear factor-kappaB in human fibroblasts. FEBS Lett 436(3):329–334
D'Ambrosio D, Cippitelli M, Cocciolo MG, Mazzeo D, Di Lucia P, Lang R, Sinigaglia F, Panina-Bordignon P (1998) Inhibition of IL-12 production by 1,25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest 101(1):252–262. doi:10.1172/JCI1050
Wu S, Liao AP, Xia Y, Li YC, Li JD, Sartor RB, Sun J (2010) Vitamin D receptor negatively regulates bacterial-stimulated NF-kappaB activity in intestine. Am J Pathol 177(2):686–697. doi:10.2353/ajpath.2010.090998
Szeto FL, Sun J, Kong J, Duan Y, Liao A, Madara JL, Li YC (2007) Involvement of the vitamin D receptor in the regulation of NF-kappaB activity in fibroblasts. J Steroid Biochem Mol Biol 103(3–5):563–566. doi:10.1016/j.jsbmb.2006.12.092
Chen Y, Zhang J, Ge X, Du J, Deb DK, Li YC (2013) Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase beta protein. J Biol Chem 288(27):19450–19458. doi:10.1074/jbc.M113.467670
Ferreira GB, Kleijwegt FS, Waelkens E, Lage K, Nikolic T, Hansen DA, Workman CT, Roep BO, Overbergh L, Mathieu C (2012) Differential protein pathways in 1,25-dihydroxyvitamin d(3) and dexamethasone modulated tolerogenic human dendritic cells. J Proteome Res 11(2):941–971. doi:10.1021/pr200724e
Ferreira GB, Vanherwegen AS, Eelen G, Gutierrez AC, Van Lommel L, Marchal K, Verlinden L, Verstuyf A, Nogueira T, Georgiadou M, Schuit F, Eizirik DL, Gysemans C, Carmeliet P, Overbergh L, Mathieu C (2015) Vitamin D3 induces tolerance in human dendritic cells by activation of intracellular metabolic pathways. Cell Rep. doi:10.1016/j.celrep.2015.01.013
Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, Fairhurst AM, Connolly JE (2015) High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol 194(11):5174–5186. doi:10.4049/jimmunol.1303316
Lips P (2007) Relative value of 25(OH)D and 1,25(OH)2D measurements. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 22(11):1668–1671. doi:10.1359/jbmr.070716
Reichel H, Koeffler HP, Norman AW (1987) Synthesis in vitro of 1,25-dihydroxyvitamin D3 and 24,25-dihydroxyvitamin D3 by interferon-gamma-stimulated normal human bone marrow and alveolar macrophages. J Biol Chem 262(23):10931–10937
Kreutz M, Andreesen R, Krause SW, Szabo A, Ritz E, Reichel H (1993) 1,25-dihydroxyvitamin D3 production and vitamin D3 receptor expression are developmentally regulated during differentiation of human monocytes into macrophages. Blood 82(4):1300–1307
Fritsche J, Mondal K, Ehrnsperger A, Andreesen R, Kreutz M (2003) Regulation of 25-hydroxyvitamin D3-1 alpha-hydroxylase and production of 1 alpha,25-dihydroxyvitamin D3 by human dendritic cells. Blood 102(9):3314–3316. doi:10.1182/blood-2002-11-3521
Hewison M, Freeman L, Hughes SV, Evans KN, Bland R, Eliopoulos AG, Kilby MD, Moss PA, Chakraverty R (2003) Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol 170(11):5382–5390
Jeffery LE, Wood AM, Qureshi OS, Hou TZ, Gardner D, Briggs Z, Kaur S, Raza K, Sansom DM (2012) Availability of 25-hydroxyvitamin D (3) to APCs controls the balance between regulatory and inflammatory T cell responses. J Immunol 189(11):5155–5164. doi:10.4049/jimmunol.1200786
Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M, Walker LS, Lammas DA, Raza K, Sansom DM (2009) 1,25-dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol 183(9):5458–5467. doi:10.4049/jimmunol.0803217
Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, Butcher EC (2007) DCs metabolize sunlight-induced vitamin D3 to 'program' T cell attraction to the epidermal chemokine CCL27. Nat Immunol 8(3):285–293. doi:10.1038/ni1433
van der Aar AM, Sibiryak DS, Bakdash G, van Capel TM, van der Kleij HP, Opstelten DJ, Teunissen MB, Kapsenberg ML, de Jong EC (2011) Vitamin D3 targets epidermal and dermal dendritic cells for induction of distinct regulatory T cells. The Journal of allergy and clinical immunology 127(6):1532–1540 . doi:10.1016/j.jaci.2011.01.068e1537
Holick MF, Chen TC (2008) Vitamin D deficiency: a worldwide problem with health consequences. Am J Clin Nutr 87(4):1080S–1086S
Koch-Henriksen N, Sorensen PS (2010) The changing demographic pattern of multiple sclerosis epidemiology. The Lancet Neurology 9(5):520–532. doi:10.1016/S1474-4422(10)70064-8
Ascherio A, Munger KL, White R, Kochert K, Simon KC, Polman CH, Freedman MS, Hartung HP, Miller DH, Montalban X, Edan G, Barkhof F, Pleimes D, Radu EW, Sandbrink R, Kappos L, Pohl C (2014) Vitamin D as an early predictor of multiple sclerosis activity and progression. JAMA neurology 71(3):306–314. doi:10.1001/jamaneurol.2013.5993
Sotirchos ES, Bhargava P, Eckstein C, Van Haren K, Baynes M, Ntranos A, Gocke A, Steinman L, Mowry EM, Calabresi PA (2016) Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology 86(4):382–390. doi:10.1212/WNL.0000000000002316
International Multiple Sclerosis Genetics C, Wellcome Trust Case Control C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A, Saarela J, Bellenguez C, Fontaine B, Gillman M, Hemmer B, Gwilliam R, Zipp F, Jayakumar A, Martin R, Leslie S, Hawkins S, Giannoulatou E, D'Alfonso S, Blackburn H, Martinelli Boneschi F, Liddle J, Harbo HF, Perez ML, Spurkland A, Waller MJ, Mycko MP, Ricketts M, Comabella M, Hammond N, Kockum I, McCann OT, Ban M, Whittaker P, Kemppinen A, Weston P, Hawkins C, Widaa S, Zajicek J, Dronov S, Robertson N, Bumpstead SJ, Barcellos LF, Ravindrarajah R, Abraham R, Alfredsson L, Ardlie K, Aubin C, Baker A, Baker K, Baranzini SE, Bergamaschi L, Bergamaschi R, Bernstein A, Berthele A, Boggild M, Bradfield JP, Brassat D, Broadley SA, Buck D, Butzkueven H, Capra R, Carroll WM, Cavalla P, Celius EG, Cepok S, Chiavacci R, Clerget-Darpoux F, Clysters K, Comi G, Cossburn M, Cournu-Rebeix I, Cox MB, Cozen W, Cree BA, Cross AH, Cusi D, Daly MJ, Davis E, de Bakker PI, Debouverie M, D'Hooghe MB, Dixon K, Dobosi R, Dubois B, Ellinghaus D, Elovaara I, Esposito F, Fontenille C, Foote S, Franke A, Galimberti D, Ghezzi A, Glessner J, Gomez R, Gout O, Graham C, Grant SF, Guerini FR, Hakonarson H, Hall P, Hamsten A, Hartung HP, Heard RN, Heath S, Hobart J, Hoshi M, Infante-Duarte C, Ingram G, Ingram W, Islam T, Jagodic M, Kabesch M, Kermode AG, Kilpatrick TJ, Kim C, Klopp N, Koivisto K, Larsson M, Lathrop M, Lechner-Scott JS, Leone MA, Leppa V, Liljedahl U, Bomfim IL, Lincoln RR, Link J, Liu J, Lorentzen AR, Lupoli S, Macciardi F, Mack T, Marriott M, Martinelli V, Mason D, McCauley JL, Mentch F, Mero IL, Mihalova T, Montalban X, Mottershead J, Myhr KM, Naldi P, Ollier W, Page A, Palotie A, Pelletier J, Piccio L, Pickersgill T, Piehl F, Pobywajlo S, Quach HL, Ramsay PP, Reunanen M, Reynolds R, Rioux JD, Rodegher M, Roesner S, Rubio JP, Ruckert IM, Salvetti M, Salvi E, Santaniello A, Schaefer CA, Schreiber S, Schulze C, Scott RJ, Sellebjerg F, Selmaj KW, Sexton D, Shen L, Simms-Acuna B, Skidmore S, Sleiman PM, Smestad C, Sorensen PS, Sondergaard HB, Stankovich J, Strange RC, Sulonen AM, Sundqvist E, Syvanen AC, Taddeo F, Taylor B, Blackwell JM, Tienari P, Bramon E, Tourbah A, Brown MA, Tronczynska E, Casas JP, Tubridy N, Corvin A, Vickery J, Jankowski J, Villoslada P, Markus HS, Wang K, Mathew CG, Wason J, Palmer CN, Wichmann HE, Plomin R, Willoughby E, Rautanen A, Winkelmann J, Wittig M, Trembath RC, Yaouanq J, Viswanathan AC, Zhang H, Wood NW, Zuvich R, Deloukas P, Langford C, Duncanson A, Oksenberg JR, Pericak-Vance MA, Haines JL, Olsson T, Hillert J, Ivinson AJ, De Jager PL, Peltonen L, Stewart GJ, Hafler DA, Hauser SL, McVean G, Donnelly P, Compston A (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476(7359):214–219. doi:10.1038/nature10251
Australia, New Zealand Multiple Sclerosis Genetics C (2009) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet 41(7):824–828. doi:10.1038/ng.396
Ramagopalan SV, Dyment DA, Cader MZ, Morrison KM, Disanto G, Morahan JM, Berlanga-Taylor AJ, Handel A, De Luca GC, Sadovnick AD, Lepage P, Montpetit A, Ebers GC (2011) Rare variants in the CYP27B1 gene are associated with multiple sclerosis. Ann Neurol 70(6):881–886. doi:10.1002/ana.22678
Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, Handunnetthi L, Handel AE, Disanto G, Orton SM, Watson CT, Morahan JM, Giovannoni G, Ponting CP, Ebers GC, Knight JC (2010) A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res 20(10):1352–1360. doi:10.1101/gr.107920.110
Tizaoui K, Kaabachi W, Hamzaoui A, Hamzaoui K (2015) Association between vitamin D receptor polymorphisms and multiple sclerosis: systematic review and meta-analysis of case-control studies. Cellular & molecular immunology 12(2):243–252. doi:10.1038/cmi.2014.47
Blanck S, Aberra F (2013) Vitamin d deficiency is associated with ulcerative colitis disease activity. Dig Dis Sci 58(6):1698–1702. doi:10.1007/s10620-012-2531-7
Ulitsky A, Ananthakrishnan AN, Naik A, Skaros S, Zadvornova Y, Binion DG, Issa M (2011) Vitamin D deficiency in patients with inflammatory bowel disease: association with disease activity and quality of life. JPEN J Parenter Enteral Nutr 35(3):308–316. doi:10.1177/0148607110381267
Froicu M, Cantorna MT (2007) Vitamin D and the vitamin D receptor are critical for control of the innate immune response to colonic injury. BMC Immunol 8:5. doi:10.1186/1471-2172-8-5
Ooi JH, Li Y, Rogers CJ, Cantorna MT (2013) Vitamin D regulates the gut microbiome and protects mice from dextran sodium sulfate-induced colitis. J Nutr 143(10):1679–1686. doi:10.3945/jn.113.180794
Froicu M, Zhu Z, Cantorna MT (2006) Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology 117:310–318. doi:10.1111/j.1365-2567.2005.02290.x
Quintana FJ, Sherr DH (2013) Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev 65(4):1148–1161. doi:10.1124/pr.113.007823
Cyster JG, Dang EV, Reboldi A, Yi T (2014) 25-hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol 14(11):731–743. doi:10.1038/nri3755
Basil MC, Levy BD (2016) Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 16(1):51–67. doi:10.1038/nri.2015.4
Acknowledgements
We would like to thank Maxine Swallow, Matthias Lochner and Marcela Françozo for critical reading of the manuscript. This work was funded by the Deutsche Forschungsgemeinschaft SFB-900 (Sonderforschungsbereich 900) to T.S. and HiLF to L.B. L.M. received a PhD fellowship from the Boehringer Ingelheim Fonds, Foundation for Basic Research in Medicine. L.B. was funded by the Ellen Schmidt Program (Medizinische Hochschule Hannover).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on Dendritic Cell Subsets and Immune-mediated Diseases - Guest Editor: Francisco Quintana
Rights and permissions
About this article

Cite this article
Minarrieta, L., Ghorbani, P., Sparwasser, T. et al. Metabolites: deciphering the molecular language between DCs and their environment. Semin Immunopathol 39, 177–198 (2017). https://doi.org/10.1007/s00281-016-0609-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-016-0609-6