
Abstract
Lipids in the cytoplasm membrane fulfill numerous functions. We focus on how lipid asymmetry is generated and its physiological and pathophysiological mission. The role of phosphatidylserine (PS), a prominent phospholipid that gets exposed during cell death, in health and disease as well as in the clearance process will be outlined in detail. Attraction signals, bridging molecules, and danger signals being involved in the PS-dependent clearance of apoptotic and necrotic cells and in subsequent immune modulation are presented. Furthermore, modulations of immune responses by PS-exposing cells, organisms, microparticles, and by the PS-binding protein annexin A5 are discussed. Interference with PS-dependent clearance of apoptotic tumor cells by macrophages fosters uptake and presentation of cancer antigens by dendritic cells and thereby induces specific anti-tumor immunity. The lipid composition of microvesicles is also depicted. Tumor microvesicles are often rich in PS and thereby contribute to tumor escape mechanisms. Understanding the role of PS in membranes of dying cells and microvesicles will help to develop novel drugs and treatment options for controlling immune-mediated diseases like chronic autoimmunity and cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The structure and the functions of cellular membranes are linked to each other. They determine cell’s live and physiology. The plasma membrane of the cells is in the opinion of many scientists the most important cellular membrane because its solidity is a matter of life or death for the cell. It is at first the barrier between the internal space (cytoplasm) and the environment. Either the release or the uptake of substances to/from the environment may be toxic for the environment or the cell itself. The understanding of the membranes of the cells was growing since the early nineteenth century, when Groter and Grendel proposed a bilayer matrix of lipids, which surrounds each single cell [21, 31]. During the years, it became evident that the plasma membrane of the cells is not a crude bilayer of lipids that blocks the transfer of water and solubles between out- and inside of the cell; moreover, the membrane acts as a cellular organelle which has different duties and responsibilities for the cell itself, for the environment, and the whole organism which harbors the single cell [79]. The present understanding of cellular membranes was first established by Singer and Nicolson. They proposed in 1972 a fluid mosaic model of the plasma membrane [151]. Based on this model, many investigators advanced this model still based on the proposed lipid bilayer as backbone [37].
The initial function of mostly every biological membrane in eukaryotic cells is to serve as a barrier between the cytoplasmic and the extracellular (plasma membrane) or intra-organelle space (internal membrane), respectively. This function is performed by the lipid bilayers, which display a barrier with a high hydrophobic core and a high hydrophilic outer face. The bilayer is able to divide mostly aqueous compartments [79]. However, the membranes of eukaryotic cells are not only a motionless barrier between compartments built by a simple bilayer of several lipids. Nowadays, it has become clear that eukaryotic cells use about 5% of their genes to synthesize thousands of different lipids. The specific function of numerous lipids is well characterized, but a fully description of the whole lipid repertoire of eukaryotes (and that of other species) is still elusive. Generally, lipids are known to fulfill three general functions: (a) lipids are used for the storage of energy, (b) lipids can act as first or second messenger, and (c) polar lipids are able to form cellular membranes [159]. The last two functions of the lipids will be further highlighted in this review. In addition, we will focus on a distinct phospholipid of cellular membranes, namely phosphatidylserine (PS), and its role in diseases, clearance, and modulation of the immune system. The role of PS in membranes of microvesicles will also be outlined.
General composition of the cytoplasm membrane of eukaryotic cells
For the formation of the cytoplasm membrane, the eukaryotic cell uses different phospholipids (PL). These PL are derived either from glycerol, a three carbon alcohol, or sphingosine, a long-chain unsaturated amino alcohol [27, 36]. However, the main structural PL in biological membranes are the glycerophospholipids [159]. Glycerophospholipids consist of a glycerol backbone with two fatty acid chains, the diacylglycerol “tail”. The latter is esterified with various phosphorylated alcohol “heads”. A common scheme of the PL, which can be found in nearly any eukaryotic cytoplasm membrane, is depicted in Fig. 1a. The composition of the PL depends on the membrane type. The plasma membrane of an eukaryotic cell consists mainly of the aminophospholipids phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylserine (PS), and sphingomyelin (SPM; a phospholipid with ceramide as backbone instead of glycerol) in different amounts, as shown in Fig. 1b. Furthermore, the plasma membrane is enriched in cholesterol, which gives the bilayer of about 5 nm thickness a mechanical stability [79]. The membranes of the cellular organelles are mostly built with PC, and the other PL also show a different distribution compared to the cytoplasm membrane. Furthermore, the lipids of the membranes of organelles contain a different ration between cholesterol (CHOL) and PL (CHOL/PL < 1) than the cytoplasm membrane (CHOL/PL = 1) [79, 159].

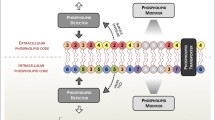
Membrane lipids of eukaryotic cells. a The structure of glycerophosphate-based lipids and sphingomyelin, the main lipids of the cytoplasm membrane of eukaryotic cells, is displayed. b Phospholipid composition of the cytoplasm membrane of a eukaryotic cell. The data are expressed as weight percent of total PL. The data are the mean values of PL composition analyses published by [36, 130, 131, 159]. PC phosphatidylcholine, PE phosphatidylethanolamine, PI phosphatidylinositol, PS phosphatidylserine, SPM sphingomyelin
Generation of lipid asymmetry
The composition of the membranes can be further characterized by analyzing the orientation of the lipids to the cytosol. Whereas the membrane of the endoplasmic reticulum (ER) displays a symmetric distribution of the PL between the inner and outer layer of the lipid bilayer, the membranes of Golgi apparatus and cytoplasmic endosomes as well as the cytoplasm membrane display strong asymmetric distribution of their membrane lipids [33, 159, 178]. The loss of the asymmetry, especially the appearance of PS on the outer membrane leaflet, will lead to many physiologic and pathologic phenomena (as discussed later in this article). Figure 2 shows schematically the asymmetric distribution of the lipids in eukaryotic cytoplasm membranes. Predominantly PE, PI, and PS are retained in the cytoplasmic leaflet, while PC and SPM are exposed to the extracellular space [91, 165, 180]. This asymmetry in PL has attracted the interest of many investigators. One could expect that the transport leading to asymmetry of the membrane lipids is a passive process, which is true for neutral lipids like cholesterol and charged lipids in a protonated form [33, 121]. However, all findings up to now point out that the cells invest a distinct amount of energy to induce and preserve their lipid asymmetry. The cells have developed concerted proteinaceous machinery, which controls the asymmetric distribution of the PL within the membranes. The passive process of lateral diffusion of PL within one leaflet of the bilayer is very fast (exchange rates of about 107 s−1), whereas the transverse diffusion (also called flip-flop) is very slow and has a half-time for exchange of hours to days, depending on the phospholipid [33]. However, several studies have shown that the transversal transport to the inner leaflet of the plasma membrane is achieved within minutes for labeled PS and within hours for labeled PE. Interestingly, PC and SPM stay nearly completely in the outer leaflet of the plasma membrane [33]. These observations lead to the hypothesis that different proteinaceous transporters (Fig. 3) will handle the transverse transport of the different lipid classes. At least three distinct activities will handle the asymmetry: two of them are energy requiring and the third is Ca2+-ion dependent.

Lipid asymmetry in the bilayer of the cytoplasm membrane of eukaryotic cells. The amount (percent) of each lipid found in the inner and outer leaflet of the plasma membrane of a viable cell is depicted. The amounts are the mean values of already described analyses of asymmetric distributions. The data were collected from [31, 33, 47, 131, 165, 178]. PC phosphatidylcholine, PE phosphatidylethanolamine, PI phosphatidylinositol, PS phosphatidylserine, SPM sphingomyelin

Schematic overview of the known phospholipid transporters in the cytoplasm membrane of eukaryotic cells. A schematic drawing of the transporter-controlled phospholipid exchange between the both leaflets of eukaryotic cytoplasm membrane is depicted. The unidirectional phospholipid transport is maintained by the flippase (inward transport of PS and PE) and floppase (outward transport of PC and SPM). Both transporters are ATP dependent and move phospholipids against their respective concentration gradient. Furthermore, the bidirectional scramblase is shown, which scrambles phospholipids between the two leaflets in an ATP independent manner. However, this transporter is highly Ca2+ dependent and does not work under normal physiologic concentrations of Ca2+. In addition, the mechanism of spontaneous diffusion of phospholipids which is neither ATP nor Ca2+ dependent and takes hours to days is displayed. The corresponding half-life (t 1/2) of the transportation time for phospholipids needed by each transporter is also shown. The drawing is a composition of figures of the following articles [121, 180, 181]. For detailed explanations, please refer to the main text
The aminophospholipid translocase (also known as flippase) processes the inward movement of aminophospholipids from the exoplasmic to the cytosolic leaflet of the membrane. The flippase may belong to the P-type ATPases family, a large family of transmembrane proteins. Furthermore, the flippase has been shown to be responsible for the selective movement of PS and PE (t 1/2 = 5–10 min for PS). The transport conducted by the flippase is strongly dependent upon ATP. Up to now, the aminophospholipid translocase has not been definitely identified, but it became evident that the flippase is a Mg2+ ATPase with a molecular mass of 115–120 kDa possibly associated with a 31-kDa protein at the endofacial membrane side. The flippase showed sensitivity to vanadate and to sulfhydryl oxidation. In red blood cells, it was evidenced that per molecule of transported aminophospholipid one molecule of ATP is hydrolyzed. Beyond the plasma membrane, the flippase activity can be detected in various intracellular membranes, to keep PS to the cytosolic leaflet [121, 180, 181].
Besides the location of PS and PE on the membrane inner leaflet, SPM and PC are mostly found on the outer leaflet of the cytoplasm membrane (Fig. 2). The so-called floppase, an ATP-dependent transporter, is responsible to transport cholin- and aminophospholipids from the inner to the outer leaflet of the bilayer. It works in a similar manner to the flippase. However, the transport is ten times slower than the transport realized with the flippase and has a t 1/2 = 1.5 h for PC [121, 180, 181]. The floppase belongs to the ATP-binding cassette (ABC) transporter which is encoded by the ABCC1 gene. This protein was formerly known as the multidrug resistance protein MRP1 and belongs to the superfamily of ABC transporters. The ABCC1 is a large integral membrane protein (M W ~180 kDa). Further experiments have indicated that the inhibition of the floppase results in a slow redistribution and furthermore a more random distribution of PC. However, the asymmetric distribution of PS and PE cannot be disturbed by floppase inhibition, pointing out that this transporter is highly substrate specific [27, 121, 181].
One could mention that the inhibition of both flippase and floppase, especially via ATP depletion, will simply raise the random distribution of the PL between the leaflets. This experiment was often conducted, resulting in a not rapidly distribution of PS on the outer membrane leaflet. However, as seen in activated platelets and apoptotic cells, the exposure of PS appears more rapidly [181]. This allows the conclusion that cells also have an ATP-independent mechanism to move PL via the membrane leaflets. The so-called scramblase is an ATP-independent but Ca2+-dependent mechanism to scramble PL bidirectional. The scrambling involves all major phospholipid classes with comparable scramble rates (t 1/2 = 10–20 min), expect SPM that is moved significantly slower [121, 180, 181]. The scramblase has been proposed to be a 37-kDa membrane protein, called scramblase 1 being responsible for the Ca2+-dependent lipid scrambling. However, this protein may serve only as one good candidate. Nevertheless, it was shown that scramblase 1 disturbs the asymmetry of membranes with PS exposure on apoptotic cells [121, 150, 181].
Disturbance of lipid asymmetry—exposure of phosphatidylserine
The disintegration of the phospholipid asymmetry, especially the exposure of PS on the outer leaflet of the membrane, has complex physiological impact on not only the cell itself but also on the whole tissue and the whole organism. Therefore, the necessity of the development of an efficient mechanism by the cells restricting PS to the inner leaflet of the plasma membrane bilayer becomes evident. Inappropriate exposure of PS on the cell surface may lead to the “destruction” of the cell but is also important in the case of apoptotic cell removal [169]. Apoptotic cells are not able to maintain the lipid asymmetry of their membranes for longer times. They expose PS and to a lower extend also PE on the outer surface of the membranes [44, 48]. The mechanism leading to the disturbance of the lipid asymmetry and consecutive to the exposure of PS on the outer surface appears to be elementary but displays a challenging and also highly regulated mechanism. Different theories exist how PS gets exposed on the cytoplasm membrane, but it is as sure as fate that apoptotic cells possess a complete loss of the asymmetric distribution of the plasma membrane PL [27, 44, 59, 69, 122, 150, 156]. Much knowledge about the transmembrane transporters was obtained by analyzing platelets, erythrocytes, or model membranes. However, the PS exposure on apoptotic cells seems to be different from the activation-induced PS exposure on platelets or other model systems [69].
Two main theories exist up to now, both very valid, but lacking the ultimate proof, how PS gets exposed on apoptotic cells. The first theory, which combines various findings of many researches, states as follows: The simple inhibition of aminophospholipid translocase (flippase) does not alone results in collapse of plasma membrane PL asymmetry, as long as normally low Ca2+ concentration is maintained. In addition, passive and spontaneous translocation of PL is very slow [150]. The increase in Ca2+ concentration in the cytosol yet can induce a rapid movement of PL between the plasma membrane leaflets [150, 169]. These experiments could be proven by the induction of apoptosis with thapsigargin. The latter is a non-competitive inhibitor of endoplasmic reticulum Ca2+ ATPases and raises cytosolic calcium concentration by blocking the pumping of calcium into the sarcoplasmic and endoplasmic reticula and causes depletion of these storages. At least prolonged exposure to thapsigargin can induce apoptosis. Furthermore, the PS exposure could be abolished by addition of zVAD-fmk, a cell-permeant pan caspase inhibitor which irreversibly binds to the catalytic site of nearly all caspases and therefore inhibits induction of apoptosis [69, 169]. It was further described that lymphocytes of patients with Scott syndrome, a rare bleeding disorder that reflects impaired expression of PS by activated platelets because of a mutation in the scramblase proteins, do not have any defects in apoptotic cell clearance; even the latter is triggered by PS exposure (further details are discussed later in this review) [45, 69, 150, 169]. Over and above experiments showed dependency as well as independency of caspases, BH3-only proteins (small apoptosis regulating proteins), and Ca2+ concentration on the exposure of PS on the surface of apoptotic cells [45, 169], indicating that multiple exposure pathways have to exist. Alterations in flippase and scramblase activity are not solely responsible for PS exposure on apoptotic cells [169].
Another main model for the exposure of PS by dying cells has been proposed by Tyurina and colleagues [156]. They observed that oxidized PS (oxPS) is crucial for uptake of apoptotic cells and therefore hypothesized that oxPS, formed during apoptosis, stimulates exposure of PS via increased rates of PS and/or oxPS transmembrane diffusion [83]. Examination of the mechanisms by which the loss of membrane asymmetry is regulated and induced is a fertile future research field. Interestingly, the mechanisms of PS exposure on apoptotic cells are similar to the effects exerted by distinct forms of dying cells (inflammation or anti-inflammation), namely it depends on the death stimulus [95, 169].
Information on the mechanisms how cells dying not exclusively via apoptosis expose PS are very scarce. Cells dying by autophagic cell death (autophagy fails and induces apoptosis) seem to behave similar to normal apoptotic cells [94]. Necrotic cells should be divided into secondary and primary necrotic cells (for extensive review, see [63]). Necrotic cells are characterized by disturbed membrane integrity. Because secondary necrotic cells are proposed to have passed an apoptotic program, the exposed PS on those kinds of necrotic cells may have had a similar fate like that in apoptotic cells. Primary necrotic cells display a high binding of annexin A5 (AnxA5), a protein that binds with high specificity and affinity in a Ca2+-dependent manner to PS. Due to the disrupted plasma membrane of necrotic cells, PS gets accessible to AnxA5 binding on the inner and on the outer leaflet of the membrane, but until now no investigations have been undertaken to characterize the exposure of PS on the outer leaflet of necrotic cells. Nevertheless, the clearance of necrotic cells is also mediated by PS and various other surface molecules that play roles in apoptotic cell clearance [19]. The procedure of necrotic cell removal has to be further specified in the future and to be adapted to cell death forms similar to necrosis, like necroptosis [13].
The exposure of PS on the outer leaflet of the plasma membrane is mostly an early event of apoptosis and may under certain circumstances also be reversible. PS exposure also occurs under physiological circumstances that are not connected to cell death [94]. The initial function of the PS exposure on viable cells is manifold and somehow open-ended. Table 1 gives examples for situations where PS is exposed on membranes of non-dying cells. The PS exposure of cells that do not show any signs of apoptosis (e.g., caspase activation) is even thought to avoid clearance. Detailed explanations about PS-dependent clearance mechanisms are outlined later in this review.
Phosphatidylserine and microparticles
The description of microparticles (MP) can be found in the literature years before apoptotic bodies were mentioned. The first known report of MP, also called microvesicles, emerged from 1967, when Wolf described the existence of fragments derived from platelets in human plasma [171]. Nowadays, it has become obvious that a variety of microparticles derived from the plasma membrane of platelets, leukocytes, endothelial cells, and several other cell types are present in human blood [35, 148]. A precise definition of the term MP is still missing and will be discussed in much more detail in other articles of in this issue. We define here MP as a heterogeneous population of small phospholipid bilayer coated vesicles with a diameter of 0.1–2.0 μm. The MP are derived from cell’s plasma membrane during activation or apoptosis of the cell. The type of stimulation can be correlated with the spectrum of released MP phenotypes. Stimulation causes the MP formation by membrane budding of the cell [35, 75, 125, 148]. First descriptions of MP suggested them to be dormant cell debris, released after distinct stimuli. Nowadays, it is well established that MP are able to serve as physiologically active effectors, playing important roles in inflammation, hemostasis, thrombosis, angiogenesis, cancer, and vascular reactivity [6]. The small size of the MP allows them to circulate easily in the vasculature. The distribution of the MP via the circulation allows not only a local but also a long-range signaling. MP bind to cells via surface ligands, enabling a long distance cell–cell interaction for cells typically dislodged from each other [6, 35, 125, 148].
MP are derived from the membrane of the ejecting cell. Furthermore, the release of the MP occurs by budding of the membrane in concerted action with the cytoskeleton. The detailed process of the budding and the interaction with the cytoskeleton are reviewed in this issue and elsewhere [6, 35, 75, 148]. We want to point out the following: During the budding process, the normal membrane lipid asymmetry of the cytoplasma membrane gets already lost [35, 75].
Characterizations of the surface of MP have been mostly performed by analyzing membrane surface proteins, which cause lot of the signaling function of the MP. In contrast, little is known about the lipid composition of the MP. When the MP were discovered, they were described as platelet dust because platelets shed a huge number of MP upon activation. These MP express PS on the outer leaflet, detected by staining of the MP with AnxA5 [75, 125, 148]. In Fig. 4a, the PL formations of platelet-derived MP are exemplarily displayed according to the experiments of Weerheim et al. [167]. Their analyses revealed that the main PL in MP ejected from platelets are PC and SPM [167]. These MP also show a prominent exposure of PS (detected by AnxA5 staining). MP are a potential trigger of the clotting cascade in combination with activated platelets. The latter also expose PS on their membrane surface [125]. However, if MP are extracted from the synovial fluid of inflamed joints, the composition of the membrane has a more equal composition, as shown in Fig. 4b. The source of the MP in the inflamed joint is not clearly worked out until now because most of inflammatory immune cells (e.g., neutrophils, macrophages, and lymphocytes) as well as endothelial/synovial cells can shed MP upon inflammatory activation. Information about the allocation of the PL between the two single layers of the MP membrane are elusive; however, PS exposure seems to be ubiquitous [6, 35, 80, 92, 125, 153]. Moreover, it was shown that the membrane composition of the MP correlate with the membrane composition of the releasing cell. The PL composition of the MP correlates strongly with the cell type and the stimulus leading to the budding and the release of the MP [35]. It has to be mentioned that MP are distinct from apoptotic bodies. The apoptotic program leads to chromatin condensation and cellular rearrangement resulting in formation of blebs filled with cytoplasmic material. These MP are released very early during the apoptotic process and are swiftly cleared by macrophages. If clearance of apoptotic cells is delayed or inhibited (for review, see [55, 62, 63]), the apoptotic cell collapses and fragments. The vesicles that are passively released in later phases of apoptosis are called apoptotic bodies. They differ from MP in size and composition, since the cytoplasm membrane has been substituted by intracellular membranes [35, 56]. Again, these apoptotic bodies are in contrast to MP (blebs). The latter are released early during the apoptotic process [35, 56]. MP are also different from exosomes, which are derived from late endosomes and released by the cells upon activation. Compared to MP, exosomes form vesicles and are stored intracellularly in multivesicular bodies. The latter fuse with the cell membrane and secrete the exosomes [6, 35]. Because the membrane composition of endosomes differs from the membrane composition of the plasma membrane, the exosome’s membrane composition may differ from MP membrane composition [154, 159].

Lipid composition of microparticle membrane bilayer. The composition of the microparticle membrane bilayer of eukaryotic cells is depicted. a The composition of the lipids which can be found in the bilayer of microparticles shed by platelets after activation. b In comparison to microparticles of activated platelets, the microparticles found in the synovial fluid of an inflamed joint have a different composition of membrane lipids. Note: The data are expressed as weight percent of total PL. The amounts are the mean values of already described analyses of asymmetric distributions. The data were collected from [53, 167]. PC phosphatidylcholine, PE phosphatidylethanolamine, PI phosphatidylinositol, PS phosphatidylserine, SPM sphingomyelin, L lyso
Diseases linked to loss of lipid asymmetry
Scott syndrome
The Scott syndrome is a rare, moderately severe, bleeding disorder, which is characterized by impaired blood coagulation of the patient’s platelets. It is known that the patients have an ineffective scramblase resulting in an impaired scrambling of the PL after activation, leading to disturbances in the clotting cascade. Apart from that, the platelets show normal behavior after activation. However, it was shown that these cells are also not able to shed PS-expressing MP. Up to now, the molecular basis of the Scott syndrome is still elusive. Most of the studies claim that a deletion or mutation in multiple hematological lineages may lead either to an ineffective lipid scramblase activity or avoids a correct Ca2+-induced activation pathway [159, 180, 181].
Antiphospholipid syndrome
Patients with the antiphospholipid syndrome (APL) demonstrate circulating “antiphospholipid” antibodies accompanied with severe arterial and venous thrombosis, recurrent abortions, and thrombocytopenia. The antiphospholipid syndrome is mostly seen in patients who suffer from systemic lupus erythematosus (SLE). While this chronic autoimmune disease can trigger an antiphospholipid syndrome, antiphospholipid antibodies can be detected in a variety of diseases that are accompanied by loss of membrane asymmetry and cell surface exposure of PS (e.g., sickle cell anemia, thalassemia, malaria, uremia, diabetes, pre-eclampsia, cancer, and diseases/conditions with elevated levels of circulating microvesicles). First it was thought that the antiphospholipid antibodies directly interact with PL. However, it has become evident that those antibodies are directed against plasma proteins like β-2-glycoprotein-1 that interact with anionic PL. While the APL is mostly accompanied by autoimmune disorders, the patients do not show a bleeding tendency but have an increased risk for thrombosis [159, 180, 181].
Sickle cell anemia
The sickle cell disease results from a point mutation in the β-chain of hemoglobin, leading to hemolytic anemia and vaso-occlusive episodes. The mutation is responsible for hemoglobin polymerization and sickling of erythrocytes when deoxygenized conditions exist. Furthermore, it leads to loss of the lipid asymmetry and exposure of PS on the surface of sickled cells and sickle cell-derived microvesicles [159, 180, 181].
Kidney stone disease
It was shown that an abnormal exposure of PS on membranes of renal epithelial cells has an emerging role in the formation of kidney stones. If cultured renal epithelial cells are treated with oxalate, they switch to apoptotic cell like phenotype and expose PS on the outer membrane. The exposed PS promotes the binding of calcium oxalate crystals and furthermore the growth of the renal stones. The mechanism of the oxalate-induced PS exposure remains unclear. However, a direct physical interaction between the oxalate and the membrane lipids of the epithelial cells is discussed [159, 180, 181].
Phosphatidylserine and clearance
“Find-me” signals of apoptotic cells
As outlined above, the exposure of PS is a key event in the apoptotic program. This anionic phospholipid acts as major “eat-me” signal that ensures efficient recognition and phagocytosis of dying cells and nuclei by phagocytes [177]. However, a prerequisite for recognition of apoptotic cells is the attraction of the professional or semi-professional phagocytes by their “prey”. Since phagocytes are mostly not located in the immediate neighborhood of apoptotic cells, the secretion of chemotactic factors attracting monocytes and macrophages is very likely. Even our knowledge about the soluble factors released from apoptotic cells is rather limited, current research identified several attraction factors for phagocytes (summarized in [119]). Proteins as well as lipids have been identified to contribute to the attraction process. More than a decade ago, Horino and colleagues identified the dimer of the ribosomal protein S19 as attraction signals for apoptotic HL-60 cells [74]. Recently, CX3CL1/fractalkine, being a chemokine and intercellular adhesion molecule, has been identified to be released by apoptotic cells leading to the stimulation of macrophage chemotaxis [155]. A new class of dying cell-derived attraction signals was identified by Elliot and colleagues [42]. The selective attraction of macrophages has been shown to be mediated by the nucleotides ATP and UTP. In addition, an apoptotic cell-derived lipid acting as “find-me” signal was identified by Lauber and co-workers [97]. The phospholipid lysophosphatidylcholine, but not its metabolic derivates or related lysophospholipids, stimulates chemotaxis of macrophages [118]. Importantly, not only single molecules but also apoptotic cell-derived microparticles are capable to attract phagocytes [144]. PS and membrane-bound TGF-beta exposed by blebs or microparticles may further contribute to anti-inflammatory processes exerted by a timely clearance of apoptotic cells by macrophages [175].
Danger signals of necrotic cells
A delayed or improper recognition and uptake of apoptotic cells lead to further changes in the membrane composition of the dying cells and finally to the disruption of the cellular membrane integrity. The cells turn from apoptosis to secondary necrosis [147]. The latter form of cell death mainly results, like primary necrosis, in immune activation and inflammatory reactions [63]. Uric acid has been identified as a danger signal released from dying cells [146]. The amount of released uric acid crystals seems to be higher in the case of secondary necrosis compared to primary necrosis [119]. The monosodium salt of uric acid, namely monosodium urate (MSU), forms birefringent crystals inducing reactive oxygen species production and release of pro-inflammatory cytokines in monocytes, in contrast to monopotassium urate, indicating that the microenvironment at the site of crystal formation is important for the immunogenic and inflammatory potential of uric acid [141]. The high mobility group box 1 (HMGB1) protein is a prominent example for a protein being also released by necrotic cells. Inside the cell nucleus, HMGB1 stabilizes chromatin and contributes to DNA bending. Outside the cell, it is a potent mediator of inflammation [136]. Tissue damage induced “Damage associate molecular patterns (DAMP)” activate the innate and adaptive immune system. Secondary necrotic cells release HMGB1 in complexed forms, since the danger signal remains bound to nucleosomes of late apoptotic cells [157]. Danger signals released from primary to secondary necrotic cells overcome the anti-inflammatory mode of action of PS. The latter gets accessible to the immune system on the inner leaflet of necrotic cells. DAMP molecules, including HMGB-1, S100 proteins, hepatoma-derived growth factor (HDGF), uric acid, ATP, altered matrix proteins, and heat shock proteins (HSP), represent important danger signals that mediate inflammatory responses [52]. Even HSP, playing key roles in the recovery from stress to acting as chaperons mainly inside the cell, exert various extracellular functions leading to immune activation. Hsp70 has been shown to function as danger signal and is released within membranous structures from the cells [163]. The release of Hsp70 has been controversial discussed because this protein does not present a secretory signal. However, a co-expression of Hsp70 and PS on the cell surface after stress conditions has recently been described [139]. Furthermore, lipid profiling experiments demonstrated that Hsp70 membrane-positive cells differ from their membrane-negative counterparts by containing significantly higher amounts of the lipid globotriaosylceramide (Gb3) [66]. Taken together, Gb3 and PS enable anchorage of Hsp70 in the plasma membrane of cells. PS interacting with proteins like HSP therefore contributes to inflammatory reactions and, as outlined above, in its pure form or complexed with bridging molecules (see below) to anti-inflammation. This discrepancy is also observed for the nucleotide ATP that functions as attraction signal for macrophages in non- or even anti-inflammatory apoptotic cell removal [42] but in the context of tissue damage in higher concentrations as immune activating danger signal [9, 77].
PS-dependent phagocytosis of dying cells
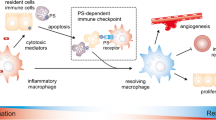
After attraction of the phagocytes, recognition and engulfment of the dying cells has to be managed. PS on the outer leaflet of the plasma membrane represents the key signal for triggering phagocytosis of both apoptotic as well as necrotic cells [19, 173]. How PS arrives at the plasma membrane outer leaflet during apoptosis is still fragmentarily understood [20], and the current knowledge is summarized in the top of this article. Shortly, the translocation of PS from the outer to the inner leaflet may fail due to inhibition of an aminophospholipid translocase, which is essential for the ATP-dependent maintenance of the asymmetric distribution of PL across the membrane bilayer. Alternatively or in addition, an activation of a not yet identified phospholipid scramblase, which rapidly flips PL from either side of the bilayer to the other, may contribute to PS exposure during apoptosis [169]. Though clear is that PS is recognized either directly by receptors or in combination with soluble adaptor proteins. The latter bear unique domains that bind to specialized receptors on the phagocyte. Integrins and receptor tyrosine kinases like Mer are examples that enable the transduction of the PS stimulus into various signaling outcomes [143]. Milk fat globule protein MFG-E8 [70], growth arrest-specific gene product GAS-6 (ligand for the receptor tyrosine kinase MerTK) [78], β-2-glycoprotein-1 [11], C-reactive protein (CRP) [111], serum-derived protein S [2], and annexin A1 [7] bind to PS and join apoptotic cells with the phagocyte. Changes of the glycoprotein composition of apoptotic cells often occur later than the exposure of PS [54]. They may therefore act as back-up mechanism for apoptotic cell clearance like the binding of complement components. Complement binding is an early event in necrosis and a rather late event in apoptosis/secondary necrosis [58]. Various molecules like complement, serum DNase I [60], and CRP act as back-up molecules in the clearance process (summarized in [63]). Complement activation in the innate immune response is besides due to mannan-binding lectin and deposition of C3 by the alternative pathway also due to natural IgM antibodies and CRP [137]. Notably, all those molecules are also involved in the clearance of dying and dead cells (Fig. 5).

Attraction signals, bridging molecules, and danger signals are involved in the clearance of apoptotic and necrotic cells by phagocytes and in consecutive immune modulation. Phosphatidylserine (filled gray circles) is the main recognition structure for apoptotic and necrotic cells. Apoptotic cells attract phagocytes, bind directly or via bridging molecules to receptors on phagocytes, and elicit no or anti-inflammatory immune responses. Necrotic cells release danger signals contributing to immune activation. For detailed explanation, please refer to the part of the main text dealing with “phosphatidylserine and clearance”. 1° primary, 2° secondary, AnxA1 annexin A1, ATP adenosine-5′-triphosphate, b2GP1 beta2-glycoprotein 1, BAI1 brain-specific angiogenesis inhibitor 1, CR complement receptor, CRP C-reactive protein, dRP19 dimer of the ribosomal protein S19, Gas-6 growth arrest-specific protein 6, HDGF hepatoma-derived growth factor, HMGB1 high mobility group 1 protein, HSP heat shock protein, LPC lysophosphatidylcholine, Mer receptor tyrosine kinase, MFG-E8 milk fat globule protein E8, MSU monosodium urate, P2 purinergic receptors, PS phosphatidylserine, R receptor, RAGE receptor for advanced glycation end-products, sdProS serum-derived protein S, SR scavenger receptors (e.g., CD68, CD36, SR-A), TIM T cell immunoglobulin and mucin-domain-containing molecule, TLR Toll-like receptor, UA uric acid, UTP uridine-5′-triphosphate, ? receptors to be identified
There exist examples in which viable cells transiently expose PS but are ignored by phagocytes [34, 40]. Exposed PS may also play a role in endocytic pathways for the internalization of annexins [84]. One big question remains: How can a phagocyte distinguish between PS on viable and on apoptotic cells? Binding studies with AnxA5, being a natural occurring ligand for PS, to apoptotic, necrotic, and viable cells, revealed that the restricted lateral mobility of PS may be one crucial point to distinguish life from death. We suggested that the PS density on membranes of viable cells is too low to induce the “eat-me” stimulus for phagocytes [5]. Kagan and co-workers suggested that oxidation of PS during apoptosis is mandatory for both the exposure of PS to the cell surface and for the “activation” of PS being an “eat-me” signal for phagocytes [82]. Another hint for this hypothesis is that oxidized PS binds adaptor proteins like MFG-E8 with higher affinity in comparison to non-oxidized PS [17]. Furthermore, a spontaneous but only ex vivo observable exposure of PS on viable B cells may be the cause of the proper function of PS as unique “eat-me” signal for dying cells [41]. Viable cells further expose a combination of “don’t-eat-me” signals such as CD47, CD31, and specific carbohydrate structures to prevent their uptake by phagocytes [124].
Several macrophage scavenger receptors may also interact directly with PS on the surface of the apoptotic cells [128]. Regardless of the receptors engaged on the phagocyte, ingestion does not occur in the absence of PS. Receptors exist which are involved in “tethering” (recognition and binding) of apoptotic cells versus those inducing their uptake and signal transduction, namely their “tickling” [72]. Monoclonal antibodies (mAb) against stimulated phagocytes were produced to identify receptors for PS (PSR). The antigenic target of the mAb 217 had hallmarks of a receptor for PS, namely it recognized PS and blocked the engulfment of apoptotic cells [49]. Phenotypes of PSR-deficient mice were described consistent with a role of the PSR in the removal of apoptotic cells [96, 100]. In another line of PSR-deficient mice, where the mAb 217 still stained positive, the PSR displayed essential functions during embryogenesis but not in apoptotic cell removal [18]. Also PSR-deficient cells were fully competent to recognize, engulf, and respond to apoptotic cells [108]. It became evident that this protein is not at the cell surface but in the nucleus [140]. But there is no doubt of PS itself and its putative receptor(s) playing an important role in the uptake process of apoptotic and necrotic cells. Using library of hamster monoclonal antibodies against mouse peritoneal macrophages Tim4 (T cell immunoglobulin and mucin-domain-containing molecule) has recently be identified as one receptor for PS [109]. Tim4 was found to be expressed on human and mouse macrophages and on dendritic cells and recognized PS via its immunoglobulin domain. Interestingly, among other Tim family members, only Tim1, but neither Tim2 nor Tim3, was found to bind PS. A metal-ion-dependent ligand binding site seems to be crucial for PS binding [135]. Just recently it was demonstrated that Tim3 is also a receptor for PS and binds in a pocket on the N-terminal IgV domain in coordination with a calcium ion. However, in contrast to fibroblastic cells, T or B cells expressing Tim3 formed conjugates with but failed to engulf apoptotic cells [32]. Another PS-specific membrane receptor that binds directly to the exposed PS and provides a tickling signal has been identified by Park and colleagues. Their experiments led to the conclusion that stabilin-2 is the first of the membrane PS receptors to provide tethering and tickling signals [117]. Stabilin-2, the hepatic hyaluronan receptor, has been shown to be essential for the clearance of hyaluronan from the lymph or the blood circulation [50]. It acts like a scavenger receptor, a number of which are already known to bind native and/or oxidized PS. The brain-specific angiogenesis inhibitor 1 (BAI1) has also been shown to be a receptor that can bind PS on apoptotic cells. The thrombospondin type 1 repeats within the extracellular region of BAI1 mediate direct binding to PS [116]. BAI1 functions as engulfment receptor for both recognition and subsequent internalization of dying cells.
A plethora of molecules are involved in PS recognition and binding (Fig. 5). A typical receptor that triggers internalization of apoptotic cells is Mer, originally named for its distribution in myeloid cells, epithelial cells, and reproductive tissue and responsible for the downregulation of TNF-alpha in macrophages [25]. Ligands for Mer are GAS-6 and protein S that bridge PS to Mer. Activation of Mer induces Src-dependent phosphorylation of FAK on Tyr861 finally leading to Rac1 activation and internalization of bound apoptotic cells (summarized in [173]). The activation of the small GTPase Rac leads to cytoskeletal reorganization of the phagocyte membrane allowing internalization of the PS-exposing corpse. In general, TAM receptor protein tyrosine kinases, TYRO3, AXL, and Mer, have been shown, besides fostering the phagocytosis of apoptotic cells and membranous organelles, to inhibit inflammation in both macrophages and dendritic cells most likely by suppression of Toll-like receptor (TLR) signaling [99].
The receptor for advanced glycation end-products and TLR mediate the signals of HMGB1 to the immune system [149]. The danger signal ATP activates purinergic P2RX7 receptors on DC leading to activation of the inflammasome. The latter drives the secretion of IL-1beta resulting in full differentiation of IFNgamma polarized CD8+ T cells during the priming of anti-tumor immune responses [9]. However, very recently, Bles and colleagues demonstrated that ATP released from tumor cells might also exert a tumorigenic action by stimulating the secretion of amphiregulin from DC [14]. An engagement of the P2 receptors also influences the ability of macrophages to bind apoptotic bodies. After binding of nucleotides to P2 receptor, the ability of macrophages to internalize and present antigens from the apoptotic and necrotic cells was enhanced [105]. Triggering of P2Y receptors induces the maturation of DC and leads to increased intracellular levels of cAMP [168]. Scavenger receptors and CD91 have been implicated in binding of the danger signal Hsp70 and may direct Hsp70–peptide complexes into the MHC class II presentation pathway [51]. The receptors for HDGF and MSU are not known by now. MSU seems to modulate the release of other danger signals since eosinophils exposed to MSU crystals within minutes of exposure released ATP into the extracellular milieu [90]. The identification of receptors specifically mediating the uptake of secondary necrotic cells has to be performed in future research. Uptake via complement receptors of complement opsonized secondary necrotic cells certainly contributes as back-up clearance mechanism. The molecular mechanisms of late apoptotic/secondary necrotic cell clearance were recently summarized by Poon and co-workers [124]. The attraction signals, bridging molecules, and danger signals for and of apoptotic and necrotic cells and the respective receptors on phagocytes outlined in the part “phosphatidylserine and clearance” are summarized in Fig. 5.
The PS-binding protein annexin A5
Switching from the physiological role of PS to its diagnostic application, it has to be stressed that PS has become a promising target to visualize dying and dead cells in vivo. AnxA5 is the most prominent molecular imaging agent to detect PS-bearing membranes in the body [86]. Being a natural occurring ligand for PS, AnxA5 belongs to a huge family of evolutionary related annexin proteins detected in most eukaryotic phyla. The core domain of annexins consists of four similar repeats approximately 70 amino acids long with the exception of AnxA6, which has eight repeats. The core domain is responsible for the binding of Ca2+ ions and phospholipid. AnxA5 is present in both the intracellular and extracellular milieu. The concentration in the circulation is low and about 1.5 nM. However, annexins are considered to be only cytosolic proteins since they lack a 5′-leader sequence in their messenger RNA. Shedding of plasma membrane-derived microparticles might be one possibility to shuffle AnxA5 in the extracellular space like it has been described for interleukin-1beta [126]. Complex formation with other proteins and binding to PS on the inner membrane leaflet may also lead to externalization of AnxA5 [158]. Once present in the extracellular space, AnxA5 is capable to inhibit the release of microparticles by apoptotic cells but can also foster endocytosis [67]. Proteins present in the extracellular environment are endocytosed via receptor-mediated internalization or macropinocytosis. Ligands can be taken up by multiple lipid raft-mediated pathways. Just recently, it has been demonstrated that magnetic nanoparticles can be endocytosed efficiently by conjugation to a specific anti-polysulfated heparan sulfate antibody. The studies from Wittrup and colleagues revealed the chaperone glucose-regulated protein 75 as a functional constituent of a defined endocytic pathway [170]. Nevertheless, Kenis and colleagues demonstrated that AnxA5 internalization by apoptotic and viable PS-expressing tumor cells is independent from the described endocytic pathways [84]. The major prerequisite for internalization seems to be the binding of AnxA5 to PS. This pathway opens novel aspects to exploit AnxA5 as a targeting agent for delivery compounds into PS-expressing cells [142]. It was further suggested that AnxA5 blocks via this internalization mechanism the phagocytosis of dying cells by macrophages by delivering PS back to the inner leaflet of the cell membrane [85].
Modulation of inflammation by PS-exposing cells and organisms
Under healthy conditions, apoptotic cells are poorly immunogenic [123] and their clearance is non- or even anti-inflammatory. In contrast to the uptake of pathogens or FcR-mediated phagocytosis [134], the engulfment of apoptotic cells does not induce inflammatory cytokine production. Voll and colleagues demonstrated more than a decade ago that activated macrophages secrete more IL-10 and TGF-beta and less inflammatory cytokines like TNF-alpha, IL-1beta, and IL-12 after phagocytosis of apoptotic cells [166]. TGF-beta released by stimulated macrophages that ingest apoptotic cells was shown to be a central player in mediating those anti-inflammatory responses [103]. The production of TGF-beta is regulated at both transcriptional and translational steps. Transcription required p38 MAPK, ERK, and JNK whereas translation was dependent on Rho GTPase, PI3K, and Akt [174]. Cells like human monomyelocytes that do not express PS during apoptosis failed to induce TGF-beta. However, PS liposomes transferred onto the cell surface membranes were capable to restore the induction of this anti-inflammatory cytokine suggesting an important role for PS as immune suppressor [76].
PS in exosomes further contributes to an anti-inflammatory environment. Exosomes are vesicles of endocytic origin, and dendritic cells as well as macrophages are capable to spontaneously secret them. MFG-E8 being an adaptor protein for PS is one of the major exosomal proteins. It targets exosomes to other immune cells [164] or possibly to apoptotic cells. The binding of both exosomes and apoptotic cells is feasible, since MFG-E8, also called lactadherin, has two lipid binding domains [1]. The uptake of the dying cells together with exosomes might strongly contribute to the anti-inflammatory responses exerted by PS-exposing particles like apoptotic cells [173]. Ectosomes derived from polymorphonuclear neutrophils expose PS and induce immune suppression by inhibiting the maturation of DC and by inducing an increased release of TGF-beta1 [39]. The clearance of apoptotic cells is involved in the resolution of inflammation, and the regulation of pro- and anti-inflammatory cytokine production during the ingestion of dying and PS-exposing cells has been identified as one key regulatory mechanism. Kim and colleagues suggested that the pH value of the microenvironment where phagocytosis events take place further contributes to this regulation. The PS-binding activity of stabilin-2, being one receptor for PS, is enhanced in acidic areas, suggesting that low pH might act as a warning signal to foster stabilin-2-mediated phagocytosis to resolve inflammation [89].
Malignant cells and some virus and parasites are taking advantage of this ubiquitous PS regulated mechanism. PS is expressed on monocytes as part of their differentiation program to macrophages [23]. Since HIV and many other viruses cause extensive apoptosis, the infected cells express elevated levels of PS which, consecutively, can also be found in the outer membrane of the enveloped retrovirus [24]. The latter therefore enter macrophages “silently”, like apoptotic cells possibly contributing to the impaired immune response accompanying these infections. We also analyzed the influence of AnxA5 on HIV-I replication in infected phagocytes and found that the virus replication in macrophages was significantly reduced after treatment with a single dose of AnxA5 [112]. An opsonin for dying cells, namely complement component C1q, was further identified to mediate the binding of HIV to erythrocytes, playing a crucial role in the progression of the primary infection [73]. In addition, ectosomes derived from erythrocytes bear immune suppressive properties [133]. Recent research revealed that cell death in protozoan parasites also occurs in a programmed fashion [162]. The obligate intracellular pathogen Leishmania major survives and multiplies in professional phagocytes. The survival of the parasites inside macrophages was shown to be dependent on the presence of apoptotic parasites exposing PS. Apoptotic promastigotes from Leishmania parasites induced release of TGF-beta by neutrophils, suggesting that the presence of apoptotic parasites provides survival advantage for the viable parasites fostering disease development [161]. Apoptotic neutrophils with Leishmania parasites inside further act as “Trojan horse” to deliver the parasites into macrophages by inducing even anti-inflammation [160]. Vaccinia virus also uses apoptotic mimicry to infect host cells. It enters the cells via macropinocytosis after inducing the extrusion of blebs. All entry events were strictly dependent on the presence of PS in the viral membrane [106]. To block the PS-dependent “silent” uptake of tumor cells, virus, and parasites is a promising target to induce immunity against those “invaders”. Figure 6 schematically displays how interference with PS-dependent clearance processes leads to immune modulation.

PS-exposing particles enter silently in macrophages while blocking their clearance induces specific immunity. Apoptotic cells and microparticles expose PS leading to an anti-inflammatory clearance by macrophages. Clearance defects for naturally in the body occurring cells lead to the accumulation of apoptotic cells which finally proceed to secondary necrosis. Secondarily necrotic cell-derived antigens are taken up and presented by DC assumed that co-stimulation is provided by danger signals released from the dead cells. The presented antigens lead to activation of T cells, which provide survival signals for B cells leading to chronic autoimmunity. In the case of cancer cells, a conscious blocking of their clearance by, e.g., the PS-binding protein AnxA5 can lead by the same mechanism to specific anti-tumor immunity. This blocking strategy is also instrumental to induce immune reactions against PS-exposing virus or Leishmania parasites. Both membrane wrapped virus and Leishmania parasites use the PS exposure as a tool to assure their survival by fooling the immune system. Ag antigen, DC dendritic cell, PS phosphatidylserine
Modulation of PS-dependent clearance—implications in disease
An efficient clearance of PS-exposing apoptotic cells is strongly linked to homeostasis in healthy tissues. One of the most intensively examined relationship between disturbed dying cell clearance and disease pathogenesis represents chronic autoimmunity. The clearance failure of apoptotic cells leads to the accumulation of secondarily necrotic cells. The latter have lost their membrane integrity resulting in the release of intracellular antigens, danger signals, DNA, and noxious contents. Under those circumstances, dendritic cells get the chance to take up dead cell-derived antigens, and they receive further co-stimulation by danger signals released from the secondarily necrotic cells. The presentation of antigens, which is also fostered by danger signals, leads to activation of T cells, which provide survival signals for B cells leading to specific immune reactions against dead cell-derived antigens (Fig. 6). SLE is a multifactorial systemic autoimmune disease characterized by a chronic and antigen-driven autoimmune response against antigens that are cleaved, phosphorylated, and/or originated from dying or dead cells. A clustering and concentration of these molecules in the surface blebs of apoptotic cells is often observed [132]. In about half of the patients with SLE, deficiencies in the recognition and engulfment of apoptotic cells have been reported to be one main alteration [64]. Early mechanistic studies revealed that an excess of apoptotic cells leads to autoantibody production [102, 107]. When lower amounts of apoptotic cells are present but the PS on the dying cell surface is masked, autoantibody production is also induced, most likely due to impaired clearance of the apoptotic cells by macrophages [8]. Mice mutant for TAM receptor protein tyrosine kinases also bear signs of chronic autoimmunity [99]. Various mouse models displaying defects in the PS mediated clearance have confirmed that impairment in apoptotic cell removal can lead to chronic and systemic autoimmunity (summarized in [61]). Examples for deficiencies of the following molecules in mice with clearance failures are C1q, DNase I, Mer, MFG-E8, secreted IgM, serum amyloid P component, and surfactant protein-D. All of those adaptor or chromatin processing molecules are involved in the clearance process of apoptotic cells [98]. Just recently, it has been described that in Tim4-deficient mice peritoneal macrophages and B-1 cells do not efficiently clear apoptotic thymocytes in vivo [129], emphasizing that defects in receptors for PS also contribute to chronic autoimmunity.
As already mentioned, Rac inside the phagocyte is mandatory for cytoskeletal rearrangement during internalization of apoptotic cells. Rac can be antagonized by the small GTPase RhoA resulting in impaired engulfment [113]. Those mechanisms may play pivotal roles in the diseased lung. Increased levels of apoptotic cells have been observed in lung tissue of various respiratory diseases. The impact of an impaired clearance on the pathogenesis of those diseases as well as on neurological diseases and on atherosclerosis was just recently reviewed very detailed by Elliott and Ravichandran [43]. Mice deficient in typical apoptotic cell adaptor and bridging molecules develop atheriosclerotic plaques. MFG-E8 seems to be the prominent adaptor protein for clearance of apoptotic cells by microglia in the brain. Decreased levels of MFG-E8 have been shown to be associated with Alzheimer’s disease [15].
The MFG-E8-dependent clearance may be beneficial in the case of removal of tumor cells. The induction of anti-inflammatory mediators could neutralize the inflammatory environment often fostering tumorigenesis [127]. However, the immune suppressive effects resulting from clearance processes could also suppress specific anti-tumor immune responses. Increased MFG-E8 levels in the tumor microenvironment have been found to foster tumor cell survival, invasion, and angiogenesis by contributing to local immune suppression. The expression of other molecules also involved in engulfment processes is upregulated in certain tumor entities [22]. Blockade of MFG-E8 cooperated with cytotoxic chemotherapy, molecularly targeted therapy, and radiation therapy to induce destruction of already established mouse tumors [81]. It has to be stressed that the concentration of MFG-E8 strongly determines the immunological outcome. An excess of MFG-E8 has an inverse effect on the engulfment of apoptotic cells, meaning that apoptotic cell removal is blocked instead of fostered [176]. High MFG-E8 concentrations may therefore have similar immune effects like AnxA5 (Fig. 6). Besides concentration, the timely regulated release of immune modulating molecules is essential. The temporarily release of immune activating danger signals like HMGB1 by necrotic cells, resulting among others from impaired clearance, provide co-stimulatory signals for dendritic cells. Such danger signals act as early warning system to activate innate and adaptive immune responses. Recently, it was demonstrated that activation of a tumor antigen-specific T cell immunity involves secretion of HMGB1 protein by dying and dead tumor cells [4]. Besides HMGB1, ATP, uric acid, or extracellular HSP can act as danger signals and interact with several receptors leading to the induction of the secretion of pro-inflammatory cytokines by immune cells. Combinations of ionizing irradiation with hyperthermia resulted in increased amounts of necrotic tumor cells and extracellular HMGB1 in comparison to single treatments [104, 138]. The release of HMGB1 by tumor cells leads to cross-presentation of tumor cell-derived antigens by DC while on the tumor cell surface exposed and normally intracellular located proteins may foster the uptake of tumor cells by DC. Anthracyclines have been discovered by Dr. Obeid and co-workers to elicit immunogenic tumor cell death. The latter correlates with the exposure of the ER-resident protein calreticulin (CRT) on the tumor cell surface in very early phases of the apoptotic program [114]. Cell death-related molecules like CRT and HMGB1 have to compile a spatiotemporal code to translate cell death into a specific anti-tumor immune response [179]. It has to be stressed that the general view that necrosis always does induce inflammation and apoptosis anti-inflammation is an oversimplification. One main important step in the induction of anti-tumor immunity is to block the uptake of dying tumor cells by macrophages and to shift it toward DC [57]. We apply the PS-binding protein AnxA5 to block the uptake of apoptotic tumor cells by macrophages (Fig. 6). AnxA5 decreased apoptotic cell uptake by macrophages and concomitantly increased their uptake by DC in vitro and in vivo. Furthermore, activated macrophages secreted higher amounts of TNF-alpha and IL-1beta and lower amounts of TGF-beta after contact with dying tumor cells in the presence of AnxA5 in comparison to solely dying tumor cells [16, 57]. Vaccination and cure assays of tumor-bearing mice proceeded significantly better when AnxA5 was present to mask PS exposed by irradiated apoptotic tumor cells [16]. PS also becomes exposed on the luminal surface of vascular endothelium in tumors. Targeting of PS with antibodies in addition to irradiation led to damage of tumor blood vessels in rats and to the induction of tumor immunity, even in glioblastoma [71]. Intervention with molecules acting as “find-me” signals may also be a promising target in anti-cancer therapies [43]. IL-10 activated macrophages showing an enhanced apoptotic cell clearance capacity sustain an anti-inflammatory environment thereby contributing to the suppression of anti-tumor immunity [115]. The “find-me” signal fractalkine (CX3CL1) seems to be involved in the recruitment of such immune suppressive macrophages [155]. Small amounts of ATP could also foster anti-inflammatory clearance processes [42], while present in high local concentrations, it acts as inflammatory danger signal.
Microvesicles as modulators of the tumor microenvironment
The cell biology of microvesicles and their influence on inflammatory processes was recently summarized in detail by Cocucci and colleagues [30]. Microvesicles often consist of mixed vesicle populations containing shedding vesicles and exosomes. An anti-inflammatory microenvironment is sustained by tumor-derived microvesicles by multiple mechanisms (Fig. 7). Macrophages secreted high amounts of TGF-beta after engulfment of tumor microvesicles being rich in PS. Pre-clinical models revealed that the metastatic potential of melanoma was enhanced in the presence of such PS-exposing microvesicles. Notably, the effects were reversed by adding AnxA5 [101]. An accumulation of cytotoxic drug and their discharge in shed vesicles is another mechanism by which tumor cells escape from therapy [145]. Inhibition of drug export in microvesicles may therefore offer new potential for reverting multidrug resistance of cancer cells [38]. The transfer of microparticles is often dependent on bridging molecules like already outlined for the uptake of apoptotic cells by macrophages. After binding of complement, microparticles have been shown to be taken up by immune cells. They thereby reduce the activation of B cells and skip the cytokine secretion profile of monocytes toward anti-inflammation [93]. Tumor-derived microvesicles significantly modulate the biological activity of monocytes resembling very closely the effects of tumor cells by themselves on immune cells [10]. Growing tumor cells shed microvesicles. The rate of shedded vesicles is often increased in malignant tumors. Analyses of the specific proteome of such microvesicles identified annexins to be involved in their biogenesis [29]. The binding of annexins to PL is strictly dependent on Ca2+ ions, like it was identified to be for the release of ectosomes [120]. Besides microvesicles, cancer cells also shed soluble PS [88] contributing to active immune suppression that is also sustained by secretion of TGF-beta. TGF-beta1 secretion and the exposure of PS on the surface of ectosomes both down-modulate cellular activation in macrophages [65]. The extracellular matrix is also modulated by microvesicles. Metalloproteinases and cathepsin B are main modulators of extracellular matrix and are transferred in the tumor microenvironment via microvesicles [68]. A disintegrin and metalloproteinases are expressed in various human tumors and promote cell growth as well as tumor spreading [110]. Microvesicles contain mRNA, microRNA, and angiogenic proteins and are thereby capable to deliver genetic information and proteins to recipient cells in the tumor microenvironment [152]. Another possibility for tumor progression and cancer cell immune escape exerted by microvesicles is to induce killing of immune cells. Microvesicles containing FasL induced loss of mitochondrial membrane potential, cytochrome c release, and caspase-3 cleavage leading to T cell apoptosis [87]. Taken together, the release of extracellular microvesicles is a highly important process in tumorigenesis [172].

The role of microvesicles in tumor escape. PS-exposing microvesicles interact with various immune cells, vessels, tumor cells, and the extracellular matrix leading to an anti-inflammatory microenvironment fostering tumor escape. For detailed explanation, please refer to the part of the main text dealing with “microvesicles as modulators of the tumor microenvironment”. PS phosphatidylserine
Microvesicles further compete for macrophages with apoptotic cells. The production of PS-exposing plasma membrane-derived vesicles is increased during apoptosis, the so-called blebbing of the dying cell. The blebs may lead to workload of macrophages resulting in an accumulation of apoptotic cells. The latter can sustain the plasma membrane integrity only for a certain time before loosing it. The resulting secondarily necrotic cells foster inflammatory reactions and may contribute to the etiopathogenesis of chronic autoimmune diseases like SLE [3]. Blebs are further known for long time to be, besides their anti-inflammatory properties, responsible for spread of antigen in SLE [26]. Figure 7 summarizes how microvesicles contribute to the escape of tumors. The isolation and analysis of microvesicles from blood samples of autoimmune or tumor patients have the potential to provide information about state and progression of diseases and could ascent as innovative biomarkers [28].
Concluding remark
Resolving further pathophysiological roles of PS will be prerequisites for the development of novel drugs and in exploiting immunogenic signals of apoptotic and necrotic cells and microvesicles for controlling diseases like chronic autoimmunity and cancer.
References
Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT (2000) Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry 39:6200–6206
Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E (2003) Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat Immunol 4:87–91
Antwi-Baffour S, Kholia S, Aryee YK, Ansa-Addo EA, Stratton D, Lange S, Inal JM (2010) Human plasma membrane-derived vesicles inhibit the phagocytosis of apoptotic cells—possible role in SLE. Biochem Biophys Res Commun 398:278–283
Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S, Andre F, Tursz T, Kroemer G, Zitvogel L (2007) The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 220:47–59
Appelt U, Sheriff A, Gaipl US, Kalden JR, Voll RE, Herrmann M (2005) Viable, apoptotic and necrotic monocytes expose phosphatidylserine: cooperative binding of the ligand annexin V to dying but not viable cells and implications for PS-dependent clearance. Cell Death Differ 12:194–196
Ardoin SP, Shanahan JC, Pisetsky DS (2007) The role of microparticles in inflammation and thrombosis. Scand J Immunol 66:159–165
Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK (2003) Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev Cell 4:587–598
Asano K, Miwa M, Miwa K, Hanayama R, Nagase H, Nagata S, Tanaka M (2004) Masking of phosphatidylserine inhibits apoptotic cell engulfment and induces autoantibody production in mice. J Exp Med 200:459–467
Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, Smyth MJ, Zitvogel L (2010) Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res 70:855–858
Baj-Krzyworzeka M, Szatanek R, Weglarczyk K, Baran J, Zembala M (2007) Tumour-derived microvesicles modulate biological activity of human monocytes. Immunol Lett 113:76–82
Balasubramanian K, Chandra J, Schroit AJ (1997) Immune clearance of phosphatidylserine-expressing cells by phagocytes. The role of beta2-glycoprotein I in macrophage recognition. J Biol Chem 272:31113–31117
Balasubramanian K, Schroit AJ (2003) Aminophospholipid asymmetry: a matter of life and death. Annu Rev Physiol 65:701–734
Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P (2010) Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 17:922–930
Bles N, Di Pietrantonio L, Boeynaems JM, Communi D (2010) ATP confers tumorigenic properties to dendritic cells by inducing amphiregulin secretion. Blood. doi:10.1182/blood-2010-01-265611
Boddaert J, Kinugawa K, Lambert JC, Boukhtouche F, Zoll J, Merval R, Blanc-Brude O, Mann D, Berr C, Vilar J, Garabedian B, Journiac N, Charue D, Silvestre JS, Duyckaerts C, Amouyel P, Mariani J, Tedgui A, Mallat Z (2007) Evidence of a role for lactadherin in Alzheimer’s disease. Am J Pathol 170:921–929
Bondanza A, Zimmermann VS, Rovere-Querini P, Turnay J, Dumitriu IE, Stach CM, Voll RE, Gaipl US, Bertling W, Poschl E, Kalden JR, Manfredi AA, Herrmann M (2004) Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med 200:1157–1165
Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B (2004) Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ 11:943–945
Bose J, Gruber AD, Helming L, Schiebe S, Wegener I, Hafner M, Beales M, Kontgen F, Lengeling A (2004) The phosphatidylserine receptor has essential functions during embryogenesis but not in apoptotic cell removal. J Biol 3:15
Bottcher A, Gaipl US, Furnrohr BG, Herrmann M, Girkontaite I, Kalden JR, Voll RE (2006) Involvement of phosphatidylserine, alphavbeta3, CD14, CD36, and complement C1q in the phagocytosis of primary necrotic lymphocytes by macrophages. Arthritis Rheum 54:927–938
Bratton DL, Henson PM (2008) Apoptotic cell recognition: will the real phosphatidylserine receptor(s) please stand up? Curr Biol 18:R76–R79
Bretscher MS, Raff MC (1975) Mammalian plasma membranes. Nature 258:43–49
Burvenich I, Schoonooghe S, Vervoort L, Dumolyn C, Coene E, Vanwalleghem L, Van Huysse J, Praet M, Cuvelier C, Mertens N, De Vos F, Slegers G (2008) Monoclonal antibody 14C5 targets integrin alphavbeta5. Mol Cancer Ther 7:3771–3779
Callahan MK, Halleck MS, Krahling S, Henderson AJ, Williamson P, Schlegel RA (2003) Phosphatidylserine expression and phagocytosis of apoptotic thymocytes during differentiation of monocytic cells. J Leukoc Biol 74:846–856
Callahan MK, Popernack PM, Tsutsui S, Truong L, Schlegel RA, Henderson AJ (2003) Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J Immunol 170:4840–4845
Camenisch TD, Koller BH, Earp HS, Matsushima GK (1999) A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol 162:3498–3503
Casciola-Rosen L, Rosen A, Petri M, Schlissel M (1996) Surface blebs on apoptotic cells are sites of enhanced procoagulant activity: implications for coagulation events and antigenic spread in systemic lupus erythematosus. Proc Natl Acad Sci USA 93:1624–1629
Chaurio RA, Janko C, Munoz LE, Frey B, Herrmann M, Gaipl US (2009) Phospholipids: key players in apoptosis and immune regulation. Molecules 14:4892–4914
Chen C, Skog J, Hsu CH, Lessard RT, Balaj L, Wurdinger T, Carter BS, Breakefield XO, Toner M, Irimia D (2010) Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip 10:505–511
Choi DS, Lee JM, Park GW, Lim HW, Bang JY, Kim YK, Kwon KH, Kwon HJ, Kim KP, Gho YS (2007) Proteomic analysis of microvesicles derived from human colorectal cancer cells. J Proteome Res 6:4646–4655
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51
Cullis PR, Fenske DB, Hope MJ (1996) Physical properties and functional roles of lipids in membranes. In: Vance DE, Vance JE (eds) Biochemistry of lipids, lipoproteins and membranes. Elsevier Science, New York, pp 1–32
DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, Karisola P, Pichavant M, Kaplan GG, Umetsu DT, Freeman GJ, Casasnovas JM (2010) T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol 184:1918–1930
Devaux PF (1991) Static and dynamic lipid asymmetry in cell membranes. Biochemistry 30:1163–1173
Dillon SR, Mancini M, Rosen A, Schlissel MS (2000) Annexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activation. J Immunol 164:1322–1332
Distler JH, Pisetsky DS, Huber LC, Kalden JR, Gay S, Distler O (2005) Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum 52:3337–3348
Dowhan W, Bogdanov M, Mileykovskaya E (2008) Functional roles of lipids in membranes. In: Vance DE, Vance JE (eds) Biochemistry of lipids, lipoproteins and membranes. Elsevier, Amsterdam, pp 1–38
Dowhan W, Bogdanov M, Mileykovskaya E (2008) Functional roles of lipids in membranes. In: Vance DE, Vance JE (eds) Biochemistry of lipids, lipoproteins and membranes. Elsevier, New York, pp 2–36
Dumitriu IE, Roedel F, Beyer TD, Gaipl US, Kalden JR, Herrmann M (2003) UV or X-irradiation increases the cytoplasmic accumulation of rhodamine 123 in various cancer cell lines. Strahlenther Onkol 179:564–570
Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA (2008) Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol 180:817–824
Elliott JI, Surprenant A, Marelli-Berg FM, Cooper JC, Cassady-Cain RL, Wooding C, Linton K, Alexander DR, Higgins CF (2005) Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol 7:808–816
Elliott JI, Sardini A, Cooper JC, Alexander DR, Davanture S, Chimini G, Higgins CF (2006) Phosphatidylserine exposure in B lymphocytes: a role for lipid packing. Blood 108:1611–1617
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS (2009) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461:282–286
Elliott MR, Ravichandran KS (2010) Clearance of apoptotic cells: implications in health and disease. J Cell Biol 189:1059–1070
Emoto K, Toyama-Sorimachi N, Karasuyama H, Inoue K, Umeda M (1997) Exposure of phosphatidylethanolamine on the surface of apoptotic cells. Exp Cell Res 232:430–434
Fadeel B (2004) Plasma membrane alterations during apoptosis: role in corpse clearance. Antioxid Redox Signal 6:269–275
Fadeel B, Xue D (2006) PS externalization: from corpse clearance to drug delivery. Cell Death Differ 13:360–362
Fadeel B, Xue D (2009) The ins and outs of phospholipid asymmetry in the plasma membrane: roles in health and disease. Crit Rev Biochem Mol Biol 44:264–277
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148:2207–2216
Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM (2000) A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405:85–90
Falkowski M, Schledzewski K, Hansen B, Goerdt S (2003) Expression of stabilin-2, a novel fasciclin-like hyaluronan receptor protein, in murine sinusoidal endothelia, avascular tissues, and at solid/liquid interfaces. Histochem Cell Biol 120:361–369
Fischer N, Haug M, Kwok WW, Kalbacher H, Wernet D, Dannecker GE, Holzer U (2010) Involvement of CD91 and scavenger receptors in Hsp70-facilitated activation of human antigen-specific CD4+ memory T cells. Eur J Immunol 40:986–997
Foell D, Wittkowski H, Roth J (2007) Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol 3:382–390
Fourcade O, Simon MF, Viode C, Rugani N, Leballe F, Ragab A, Fournie B, Sarda L, Chap H (1995) Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 80:919–927
Franz S, Frey B, Sheriff A, Gaipl US, Beer A, Voll RE, Kalden JR, Herrmann M (2006) Lectins detect changes of the glycosylation status of plasma membrane constituents during late apoptosis. Cytom A 69:230–239
Franz S, Gaipl US, Munoz LE, Sheriff A, Beer A, Kalden JR, Herrmann M (2006) Apoptosis and autoimmunity: when apoptotic cells break their silence. Curr Rheumatol Rep 8:245–247
Franz S, Herrmann K, Furnrohr BG, Sheriff A, Frey B, Gaipl US, Voll RE, Kalden JR, Jack HM, Herrmann M (2007) After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ 14:733–742
Frey B, Schildkopf P, Rodel F, Weiss EM, Munoz LE, Herrmann M, Fietkau R, Gaipl US (2009) AnnexinA5 renders dead tumor cells immunogenic—implications for multimodal cancer therapies. J Immunotoxicol 6:209–216
Gaipl US, Kuenkele S, Voll RE, Beyer TD, Kolowos W, Heyder P, Kalden JR, Herrmann M (2001) Complement binding is an early feature of necrotic and a rather late event during apoptotic cell death. Cell Death Differ 8:327–334
Gaipl US, Beyer TD, Baumann I, Voll RE, Stach CM, Heyder P, Kalden JR, Manfredi A, Herrmann M (2003) Exposure of anionic phospholipids serves as anti-inflammatory and immunosuppressive signal—implications for antiphospholipid syndrome and systemic lupus erythematosus. Immunobiology 207:73–81
Gaipl US, Beyer TD, Heyder P, Kuenkele S, Bottcher A, Voll RE, Kalden JR, Herrmann M (2004) Cooperation between C1q and DNase I in the clearance of necrotic cell-derived chromatin. Arthritis Rheum 50:640–649
Gaipl US, Voll RE, Sheriff A, Franz S, Kalden JR, Herrmann M (2005) Impaired clearance of dying cells in systemic lupus erythematosus. Autoimmun Rev 4:189–194
Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, Kalden JR, Herrmann M (2006) Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun 9:173–187
Gaipl US, Sheriff A, Franz S, Munoz LE, Voll RE, Kalden JR, Herrmann M (2006) Inefficient clearance of dying cells and autoreactivity. Curr Top Microbiol Immunol 305:161–176
Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, Herrmann M (2007) Clearance deficiency and systemic lupus erythematosus (SLE). J Autoimmun 28:114–121
Gasser O, Schifferli JA (2004) Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104:2543–2548
Gehrmann M, Liebisch G, Schmitz G, Anderson R, Steinem C, De Maio A, Pockley G, Multhoff G (2008) Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 3:e1925
Gidon-Jeangirard C, Hugel B, Holl V, Toti F, Laplanche JL, Meyer D, Freyssinet JM (1999) Annexin V delays apoptosis while exerting an external constraint preventing the release of CD4+ and PrPc+ membrane particles in a human T lymphocyte model. J Immunol 162:5712–5718
Giusti I, D'Ascenzo S, Millimaggi D, Taraboletti G, Carta G, Franceschini N, Pavan A, Dolo V (2008) Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia 10:481–488
Hampton MB, Vanags DM, Porn-Ares MI, Orrenius S (1996) Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett 399:277–282
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417:182–187
He J, Yin Y, Luster TA, Watkins L, Thorpe PE (2009) Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res 15:6871–6880
Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM (2001) Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J Cell Biol 155:649–659
Horakova E, Gasser O, Sadallah S, Inal JM, Bourgeois G, Ziekau I, Klimkait T, Schifferli JA (2004) Complement mediates the binding of HIV to erythrocytes. J Immunol 173:4236–4241
Horino K, Nishiura H, Ohsako T, Shibuya Y, Hiraoka T, Kitamura N, Yamamoto T (1998) A monocyte chemotactic factor, S19 ribosomal protein dimer, in phagocytic clearance of apoptotic cells. Lab Invest 78:603–617
Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM (2005) Membrane microparticles: two sides of the coin. Physiol (Bethesda) 20:22–27
Huynh ML, Fadok VA, Henson PM (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109:41–50
Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC Jr, Lambrecht BN (2007) Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 13:913–919
Ishimoto Y, Ohashi K, Mizuno K, Nakano T (2000) Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem 127:411–417
Janmey PA, Kinnunen PK (2006) Biophysical properties of lipids and dynamic membranes. Trends Cell Biol 16:538–546
Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS (2003) Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res 109:175–180
Jinushi M, Sato M, Kanamoto A, Itoh A, Nagai S, Koyasu S, Dranoff G, Tahara H (2009) Milk fat globule epidermal growth factor-8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J Exp Med 206:1317–1326
Kagan VE, Borisenko GG, Serinkan BF, Tyurina YY, Tyurin VA, Jiang J, Liu SX, Shvedova AA, Fabisiak JP, Uthaisang W, Fadeel B (2003) Appetizing rancidity of apoptotic cells for macrophages: oxidation, externalization, and recognition of phosphatidylserine. Am J Physiol Lung Cell Mol Physiol 285:L1–L17
Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato AA, Fujii Y (2004) Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med 37:1963–1985
Kenis H, van Genderen H, Bennaghmouch A, Rinia HA, Frederik P, Narula J, Hofstra L, Reutelingsperger CP (2004) Cell surface-expressed phosphatidylserine and annexin A5 open a novel portal of cell entry. J Biol Chem 279:52623–52629
Kenis H, van Genderen H, Deckers NM, Lux PA, Hofstra L, Narula J, Reutelingsperger CP (2006) Annexin A5 inhibits engulfment through internalization of PS-expressing cell membrane patches. Exp Cell Res 312:719–726
Kenis H, Zandbergen HR, Hofstra L, Petrov AD, Dumont EA, Blankenberg FD, Haider N, Bitsch N, Gijbels M, Verjans JW, Narula N, Narula J, Reutelingsperger CP (2010) Annexin A5 uptake in ischemic myocardium: demonstration of reversible phosphatidylserine externalization and feasibility of radionuclide imaging. J Nucl Med 51:259–267
Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL (2005) Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res 11:1010–1020
Kim R, Emi M, Tanabe K (2005) Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol Ther 4:924–933
Kim S, Bae DJ, Hong M, Park SY, Kim IS (2010) The conserved histidine in epidermal growth factor-like domains of stabilin-2 modulates pH-dependent recognition of phosphatidylserine in apoptotic cells. Int J Biochem Cell Biol 42:1154–1163
Kobayashi T, Kouzaki H, Kita H (2010) Human eosinophils recognize endogenous danger signal crystalline uric acid and produce proinflammatory cytokines mediated by autocrine ATP. J Immunol 184:6350–6358
Kol MA, de Kroon AI, Killian JA, de Kruijff B (2004) Transbilayer movement of phospholipids in biogenic membranes. Biochemistry 43:2673–2681
Kolowos W, Gaipl US, Sheriff A, Voll RE, Heyder P, Kern P, Kalden JR, Herrmann M (2005) Microparticles shed from different antigen-presenting cells display an individual pattern of surface molecules and a distinct potential of allogeneic T-cell activation. Scand J Immunol 61:226–233
Koppler B, Cohen C, Schlondorff D, Mack M (2006) Differential mechanisms of microparticle transfer toB cells and monocytes: anti-inflammatory properties of microparticles. Eur J Immunol 36:648–660
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G (2009) Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ 16:3–11
Krysko DV, D'Herde K, Vandenabeele P (2006) Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis 11:1709–1726
Kunisaki Y, Masuko S, Noda M, Inayoshi A, Sanui T, Harada M, Sasazuki T, Fukui Y (2004) Defective fetal liver erythropoiesis and T lymphopoiesis in mice lacking the phosphatidylserine receptor. Blood 103:3362–3364
Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S (2003) Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113:717–730
Lauber K, Blumenthal SG, Waibel M, Wesselborg S (2004) Clearance of apoptotic cells: getting rid of the corpses. Mol Cell 14:277–287
Lemke G, Rothlin CV (2008) Immunobiology of the TAM receptors. Nat Rev Immunol 8:327–336
Li MO, Sarkisian MR, Mehal WZ, Rakic P, Flavell RA (2003) Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 302:1560–1563
Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA (2009) Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett 283:168–175
Lorenz HM, Herrmann M, Winkler T, Gaipl U, Kalden JR (2000) Role of apoptosis in autoimmunity. Apoptosis 5:443–449
Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M, Silverstein R, Savill J, Lacy-Hulbert A (2006) Requirements for apoptotic cell contact in regulation of macrophage responses. J Immunol 177:4047–4054
Mantel F, Frey B, Haslinger S, Schildkopf P, Sieber R, Ott OJ, Lödermann B, Rödel F, Sauer R, Fietkau R, Gaipl US (2010) Combination of ionising irradiation and hyperthermia activates programmed apoptotic and necrotic cell death pathways in human colorectal carcinoma cells. Strahlenther Onkol (in press)
Marques-da-Silva C, Burnstock G, Ojcius DM, Coutinho-Silva R (2010) Purinergic receptor agonists modulate phagocytosis and clearance of apoptotic cells in macrophages. Immunobiology. doi:10.1016/j.imbio.2010.03.010
Mercer J, Helenius A (2008) Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535
Mevorach D, Zhou JL, Song X, Elkon KB (1998) Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med 188:387–392
Mitchell JE, Cvetanovic M, Tibrewal N, Patel V, Colamonici OR, Li MO, Flavell RA, Levine JS, Birge RB, Ucker DS (2006) The presumptive phosphatidylserine receptor is dispensable for innate anti-inflammatory recognition and clearance of apoptotic cells. J Biol Chem 281:5718–5725
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450:435–439
Mochizuki S, Okada Y (2007) ADAMs in cancer cell proliferation and progression. Cancer Sci 98:621–628
Mortensen RF, Osmand AP, Lint TF, Gewurz H (1976) Interaction of C-reactive protein with lymphocytes and monocytes: complement-dependent adherence and phagocytosis. J Immunol 117:774–781
Munoz LE, Franz S, Pausch F, Furnrohr B, Sheriff A, Vogt B, Kern PM, Baum W, Stach C, von Laer D, Brachvogel B, Poschl E, Herrmann M, Gaipl US (2007) The influence on the immunomodulatory effects of dying and dead cells of Annexin V. J Leukoc Biol 81:6–14
Nakaya M, Tanaka M, Okabe Y, Hanayama R, Nagata S (2006) Opposite effects of rho family GTPases on engulfment of apoptotic cells by macrophages. J Biol Chem 281:8836–8842
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13:54–61
Ogden CA, Pound JD, Batth BK, Owens S, Johannessen I, Wood K, Gregory CD (2005) Enhanced apoptotic cell clearance capacity and B cell survival factor production by IL-10-activated macrophages: implications for Burkitt’s lymphoma. J Immunol 174:3015–3023
Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450:430–434
Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS (2008) Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ 15:192–201
Peter C, Waibel M, Radu CG, Yang LV, Witte ON, Schulze-Osthoff K, Wesselborg S, Lauber K (2008) Migration to apoptotic “find-me” signals is mediated via the phagocyte receptor G2A. J Biol Chem 283:5296–5305
Peter C, Wesselborg S, Herrmann M, Lauber K (2010) Dangerous attraction: phagocyte recruitment and danger signals of apoptotic and necrotic cells. Apoptosis 15:1007–1028
Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z (2005) Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol 27:375–387
Pomorski T, Hrafnsdottir S, Devaux PF, van Meer G (2001) Lipid distribution and transport across cellular membranes. Semin Cell Dev Biol 12:139–148
Pomorski T, Holthuis JC, Herrmann A, van Meer G (2004) Tracking down lipid flippases and their biological functions. J Cell Sci 117:805–813
Ponner BB, Stach C, Zoller O, Hagenhofer M, Voll R, Kalden JR, Herrmann M (1998) Induction of apoptosis reduces immunogenicity of human T-cell lines in mice. Scand J Immunol 47:343–347
Poon IK, Hulett MD, Parish CR (2010) Molecular mechanisms of late apoptotic/necrotic cell clearance. Cell Death Differ 17:381–397
Puddu P, Puddu GM, Cravero E, Muscari S, Muscari A (2010) The involvement of circulating microparticles in inflammation, coagulation and cardiovascular diseases. Can J Cardiol 26:140–145
Qu Y, Franchi L, Nunez G, Dubyak GR (2007) Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 179:1913–1925
Rakoff-Nahoum S (2006) Why cancer and inflammation? Yale J Biol Med 79:123–130
Rigotti A, Acton SL, Krieger M (1995) The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J Biol Chem 270:16221–16224
Rodriguez-Manzanet R, Sanjuan MA, Wu HY, Quintana FJ, Xiao S, Anderson AC, Weiner HL, Green DR, Kuchroo VK (2010) T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci USA 107:8706–8711
Roelofsen B, van Meer G, Op den Kamp JA (1981) The lipids of red cell membranes. Compositional, structural and functional aspects. Scand J Clin Lab Invest Suppl 156:111–115
Roelofsen B (1991) Molecular architecture and dynamics of the plasma membrane lipid bilayer: the red blood cell as a model. Infection 19 Suppl 4:S206–S209
Rosen A, Casciola-Rosen L (1999) Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ 6:6–12
Sadallah S, Eken C, Schifferli JA (2008) Erythrocyte-derived ectosomes have immunosuppressive properties. J Leukoc Biol 84:1316–1325
Sanchez-Mejorada G, Rosales C (1998) Signal transduction by immunoglobulin Fc receptors. J Leukoc Biol 63:521–533
Santiago C, Ballesteros A, Martinez-Munoz L, Mellado M, Kaplan GG, Freeman GJ, Casasnovas JM (2007) Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27:941–951
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191–195
Schifferli JA (2005) Complement: a member of the innate immune system. Springer Semin Immunopathol 27:273–275
Schildkopf P, Frey B, Mantel F, Ott OJ, Weiss EM, Sieber R, Janko C, Sauer R, Fietkau R, Gaipl US (2010) Application of hyperthermia in addition to ionizing irradiation fosters necrotic cell death and HMGB1 release of colorectal tumor cells. Biochem Biophys Res Commun 391:1014–1020
Schilling D, Gehrmann M, Steinem C, De Maio A, Pockley AG, Abend M, Molls M, Multhoff G (2009) Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J 23:2467–2477
Schlegel RA, Williamson P (2007) P.S. to PS (phosphatidylserine)—pertinent proteins in apoptotic cell clearance. Sci STKE 2007:pe57
Schorn C, Janko C, Munoz L, Schulze C, Strysio M, Schett G, Herrmann M (2009) Sodium and potassium urate crystals differ in their inflammatory potential. Autoimmunity 42:314–316
Schutters K, Reutelingsperger C (2010) Phosphatidylserine targeting for diagnosis and treatment of human diseases. Apoptosis 15:1072–1082
Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411:207–211
Segundo C, Medina F, Rodriguez C, Martinez-Palencia R, Leyva-Cobian F, Brieva JA (1999) Surface molecule loss and bleb formation by human germinal center B cells undergoing apoptosis: role of apoptotic blebs in monocyte chemotaxis. Blood 94:1012–1020
Shedden K, Xie XT, Chandaroy P, Chang YT, Rosania GR (2003) Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res 63:4331–4337
Shi Y, Evans JE, Rock KL (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425:516–521
Silva MT, do Vale A, dos Santos NM (2008) Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis 13:463–482
Simak J, Gelderman MP (2006) Cell membrane microparticles in blood and blood products: potentially pathogenic agents and diagnostic markers. Transfus Med Rev 20:1–26
Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ (2010) HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 28:367–388
Sims PJ, Wiedmer T (2001) Unraveling the mysteries of phospholipid scrambling. Thromb Haemost 86:266–275
Singer SJ, Nicolson GL (1972) The fluid mosaic model of the structure of cell membranes. Science 175:720–731
Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10:1470–1476
Stadler K, Frey B, Munoz LE, Finzel S, Rech J, Fietkau R, Herrmann M, Hueber A, Gaipl US (2009) Photopheresis with UV-A light and 8-methoxypsoralen leads to cell death and to release of blebs with anti-inflammatory phenotype in activated and non-activated lymphocytes. Biochem Biophys Res Commun 386:71–76
Thery C, Zitvogel L, Amigorena S (2002) Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579
Truman LA, Ford CA, Pasikowska M, Pound JD, Wilkinson SJ, Dumitriu IE, Melville L, Melrose LA, Ogden CA, Nibbs R, Graham G, Combadiere C, Gregory CD (2008) CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112:5026–5036
Tyurina YY, Tyurin VA, Zhao Q, Djukic M, Quinn PJ, Pitt BR, Kagan VE (2004) Oxidation of phosphatidylserine: a mechanism for plasma membrane phospholipid scrambling during apoptosis? Biochem Biophys Res Commun 324:1059–1064
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE (2008) Induction of inflammatory and immune responses by HMGB1–nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med 205:3007–3018
van Genderen HO, Kenis H, Hofstra L, Narula J, Reutelingsperger CP (2008) Extracellular annexin A5: functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim Biophys Acta 1783:953–963
van Meer G, Voelker DR, Feigenson GW (2008) Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol 9:112–124
van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T (2004) Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol 173:6521–6525
van Zandbergen G, Bollinger A, Wenzel A, Kamhawi S, Voll R, Klinger M, Muller A, Holscher C, Herrmann M, Sacks D, Solbach W, Laskay T (2006) Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proc Natl Acad Sci USA 103:13837–13842
van Zandbergen G, Luder CG, Heussler V, Duszenko M (2010) Programmed cell death in unicellular parasites: a prerequisite for sustained infection? Trends Parasitol 26:477–483
Vega VL, Rodriguez-Silva M, Frey T, Gehrmann M, Diaz JC, Steinem C, Multhoff G, Arispe N, De Maio A (2008) Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol 180:4299–4307
Veron P, Segura E, Sugano G, Amigorena S, Thery C (2005) Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol Dis 35:81–88
Voelker DR (1996) Lipid assembly into cell membranes. In: Vance DE, Vance JE (eds) Biochemistry of lipids, lipoproteins and membranes. Elsevier Science, New York, pp 391–423
Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I (1997) Immunosuppressive effects of apoptotic cells. Nature 390:350–351
Weerheim AM, Kolb AM, Sturk A, Nieuwland R (2002) Phospholipid composition of cell-derived microparticles determined by one-dimensional high-performance thin-layer chromatography. Anal Biochem 302:191–198
Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B (2001) The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol 166:7172–7177
Williamson P, Schlegel RA (2002) Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim Biophys Acta 1585:53–63
Wittrup A, Zhang SH, Svensson KJ, Kucharzewska P, Johansson MC, Morgelin M, Belting M (2010) Magnetic nanoparticle-based isolation of endocytic vesicles reveals a role of the heat shock protein GRP75 in macromolecular delivery. Proc Natl Acad Sci U S A 107:13342–13347
Wolf P (1967) The nature and significance of platelet products in human plasma. Br J Haematol 13:269–288
Wright PK (2008) Targeting vesicle trafficking: an important approach to cancer chemotherapy. Recent Pat Anticancer Drug Discov 3:137–147
Wu Y, Tibrewal N, Birge RB (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol 16:189–197
Xiao YQ, Freire-de-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM (2008) Transcriptional and translational regulation of TGF-beta production in response to apoptotic cells. J Immunol 181:3575–3585
Xie Y, Bai O, Yuan J, Chibbar R, Slattery K, Wei Y, Deng Y, Xiang J (2009) Tumor apoptotic bodies inhibit CTL responses and antitumor immunity via membrane-bound transforming growth factor-beta1 inducing CD8+ T-cell anergy and CD4+ Tr1 cell responses. Cancer Res 69:7756–7766
Yamaguchi H, Takagi J, Miyamae T, Yokota S, Fujimoto T, Nakamura S, Ohshima S, Naka T, Nagata S (2008) Milk fat globule EGF factor 8 in the serum of human patients of systemic lupus erythematosus. J Leukoc Biol 83:1300–1307
Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S (2005) Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437:754–758
Zachowski A (1993) Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J 294(Pt 1):1–14
Zitvogel L, Kepp O, Kroemer G (2010) Decoding cell death signals in inflammation and immunity. Cell 140:798–804
Zwaal RF, Schroit AJ (1997) Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89:1121–1132
Zwaal RF, Comfurius P, Bevers EM (2005) Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci 62:971–988
Acknowledgments
This work was supported by the Doktor Robert Pfleger Foundation, by the German Research Foundation (GA 1507/1-1), and by the Bundesministerium für Bildung und Forschung (BMBF; m4 Cluster, 01EX1021R).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Frey, B., Gaipl, U.S. The immune functions of phosphatidylserine in membranes of dying cells and microvesicles. Semin Immunopathol 33, 497–516 (2011). https://doi.org/10.1007/s00281-010-0228-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0228-6