
Abstract
Whether paroxysmal nocturnal hemoglobinuria (PNH) clone in aplastic anemia (AA) is a prognostic factor to immunosuppressive therapy is a subject of debate. We evaluated hematological responses to immunosuppressive therapy (IST) in severe AA (SAA) patients with or without the presence of a PNH clone. In 97 SAA patients who received first-line IST between January and December 2011, 24 (24.7 %) had a PNH clone prior to treatment, with a median clone size of 7.82 % (range 1.19–45.46 %). The response rates to IST for patients with or without a PNH clone were 66.7 and 50.7 % (P < 0.172), 79.2 and 57.5 % (P < 0.057), and 79.2 and 67.1 % (P < 0.264) at 3, 6, and 12 months, respectively. Combined rate of complete and good partial responses differed between patients with or without a PNH clone: insignificantly at 3 months (41.7 vs. 21.9 %, P < 0.058), but significantly at 6 (66.7 vs. 31.5 %, P < 0.002) and 12 (75.0 vs. 46.6 %, P < 0.015) months. Multivariate analysis revealed that a pretreatment neutrophil count of >0.2 × 109/L is indicative of a better response, while the presence of a PNH clone is predictive to a higher combined rate of complete and good partial responses. This study demonstrated that the presence of a PNH clone could predict a better hematological response instead of a higher response rate in patients with SAA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Data from peripheral blood flow cytometry analysis had shown that approximately 15–68 % of aplastic anemia (AA) patients were glycosylphosphatidylinositol-anchored membrane protein-deficient (GPI-AP−), which is characteristic to paroxysmal nocturnal hemoglobinuria (PNH)-type cells without evidence of hemolysis [1–7]. The percentage of affected red cells is always smaller than the percentages of affected neutrophils and monocytes; the percentages of PNH-type cells in severe aplastic anemia (SAA) and very severe aplastic anemia (VSAA) patients are almost always smaller than that in classical PNH [8–10]. There were reports showing that the presence of a PNH clone in AA is indicative of an immunopathogenesis and may affect the response to immunosuppressive therapy (IST), although results varied [1–7]. The present study enrolled 97 SAA patients with PNH clone pre-tested to evaluate the impact of PNH clone on the response rate and response quality to IST.
Patients and methods
Patients
Ninety-seven SAA patients who received IST in the Blood Diseases Hospital, Tianjin, China, between January and December 2011, were analyzed. Diagnosis of AA was in accordance with the criteria of the International Agranulocytosis and Aplastic Anemia Study Group [11]. AA severity was evaluated according to Camitta’s and Bacigalupo’s criteria. All patients were negative on the Ham’s test and had no clinical or laboratory sign of hemolysis. Patients were treated with rabbit antithymocyte globulin (rATG, Genzyme, Cambridge, MA, USA) at 3.75 mg/kg/day, or with porcine antihuman T lymphocyte immunoglobulin (pATG, Wuhan Institute of Biological Products, Wuhan, China) at 20 mg/kg/day, for 5 days, plus cyclosporine (CsA) at 3 mg/kg/day. The dose of CsA was adjusted to maintain the trough and peak blood levels at 150∼250 and 700∼1000 ng/ml, respectively.
PNH-type cell test
PNH clone was analyzed on red blood cells, granulocytes and monocytes using a FACSCalibur (Becton Dickinson, Franklin Lakes, NJ, USA) flow cytometer with the CellQuest software. For the analysis of granulocytes and monocytes, 5–10 × 105 nucleated cells were incubated with an antibody mixture containing CD45-PerCP (Becton Dickinson), CD33-APC (Becton Dickinson), CD24-PE (Beckman Coulter, Miami, FL, USA), CD14-PE (Beckman Coulter), and FLAER (Protox Biotech, Victoria, BC, Canada). Five thousand cells were acquired for each sample after debris were excluded by FSC/SSC gating. Granulocytes and monocytes were gated based on CD45/CD33 staining. PNH clones were defined as FLAER/CD24 double negative for granulocytes and FLAER/CD14 double negative for monocytes. For the analysis of red blood cells, diluted blood suspensions were stained with antibodies including IgG1-FITC, IgG1-PE, CD235a-FITC (Beckman Coulter), and CD59-PE (Invitrogen, Carlsbad, CA, USA). A total of 1 × 104 RBCs were acquired for each sample in which the PNH clone was defined by the CD235a+CD59− phenotype. All samples were analyzed within 24 h after blood collection. The presence of a PNH clone was defined by more than 1 % of neutrophils, monocytes or red cells carrying the PNH phenotype (absence of GPI-anchored surface proteins) . The size of the PNH clone was defined by the highest percentage of neutrophils, monocytes or red cells lacking GPI-AP.
Response evaluation and follow-up
Responses to treatment were divided into the following groups: complete response (CR): hemoglobin level (Hb) >100 g/L, absolute neutrophil count (ANC) >1.5 × 109/L, and platelet (PLT) >100 × 109/L; good partial response (GPR): Hb >80 g/dL, ANC >1.0 × 109/L, and PLT >50 × 109/L; partial response (PR): blood cell counts above SAA criteria but below GPR criteria with patient being independent of blood transfusion; no response (NR): blood counts within the range of SAA criteria and/or patient being transfusion dependent. Overall response rate was defined as the sum of CR, GPR, and PR rates. Patients were followed up to 12 months after IST.
Statistical analysis
Differences in the baseline characteristics were assessed using the χ 2 test for categorical variables and the Mann-Whitney U test for continuous variables. Logistic regression was used to assess independent predictors of response in multivariate analyses using a stepwise selection algorithm of important covariates. Univariate and multivariate analyses were conducted at 3, 6, and 12 months after IST. P value less than 0.05 was considered statistically significant.
Results
Patient characteristics
Ninety-seven AA patients, 52 males and 45 females, were at 2 to 65 years of age (median 22 years) when enrolled in the study. Seventy-one patients were diagnosed as SAA of which 68 were categorized as idiopathic and three as hepatitis-associated AA. Twenty-six patients were diagnosed as VSAA of which 23 were idiopathic and three were hepatitis associated. Patient geographic and hematologic characteristics were shown in Table 1. There was no significant difference in age, gender, time from diagnosis to IST, hemoglobin, reticulocytes, lymphocytes, neutrophils, soluble transferrin receptor (sTfR), and ATG preparations between PNH+ and PNH− patient groups (P > 0.05).
PNH clone
PNH cell clone (GPI-AP−) was found in 24/97 (24.7 %) SAA/VSAA patients before IST. The PNH clone size ranged from 1.19 to 45.46 % with a median of 7.82 % involving single or multiple cell lineages: granulocytes in 19/24 patients (PNH clone size range 1.09–28.90 %, median 6.87 %); monocytes in 16/24 patients (1.32–45.46 %, 7.975 %); and red cells in 8/24 patients (1.10–20.70 %, 5.45 %). Patients with GPI-AP− were defined as the PNH+ group, and those with normal GPI-AP were defined as the PNH− group.
Hematologic responses
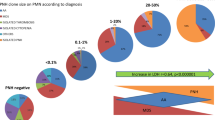
As shown in Table 2, the total hematologic response rates at 3, 6, and 12 months were 54.6, 62.9, and 70.1 %, and the combined CR + GPR rates were 26.8, 40.2, and 53.6 %, respectively. Three patients died within 3 months after IST (two died from infection, and one died from intracranial hemorrhage and septicemia) without any improvement of blood cell counts and were classified as NR. No difference in total hematologic response rate was noted between PNH+ and PNH− patients (66.7 vs. 50.7 % at 3 months, 79.2 vs. 57.5 % at 6 months, and 79.2 vs. 67.1 % at 12 months). All hematologic response in the PNH+ group occurred within 6 months, whereas 85.7 % (42/49) of hematologic response in the PNH− group happened within 6 months. Although the CR + GPR rates were similar in the PNH+ and PNH− groups at 3 months (41.7 vs. 21.9 %, P < 0.058), more patients achieved CR and GPR in the PNH+ group than in the PNH− group at 6 (66.7 vs. 31.5 %, P < 0.002) and 12 (75.0 vs. 46.6 %, P < 0.015) months after IST, respectively, as shown in Fig. 1.

The response rate in severe aplastic anemia. The total response rate (a) and combined complete response and good partial response rate (CR + GPR rate) (b) of severe aplastic anemia are shown. No difference in total hematologic response rate was noted between PNH+ and PNH− patients. The CR + GPR rates were similar in the PNH+ and PNH− groups at 3 months, but more patients achieved CR and GPR in the PNH+ group than in the PNH− group at 6 and 12 months
Three SAA and three VSAA patients were hepatitis associated, of which none had a PNH clone. Two of the six patients responded to IST within 6 months, and another two responded by 12 months, with a total response rate of 66.7 % at 12 months.
According to the disease severity, as shown in Table 2, the total response rate of SAA patients was higher than that of VSAA patients at 3 months after IST (P < 0.017). A more detailed analysis of these patients according to PNH clone showed no significant difference in IST response rate between PNH+ and PNH− patient in either the SAA patients or VSAA patient groups. However, more PNH+ SAA patients achieved CR and GPR at 6 and 12 months in comparison to PNH− SAA patients.
Risk factors for hematologic responses
Univariate analysis was performed to analyze the relationships between response rate and age, sex, time from diagnosis to IST, hemoglobin level, reticulocyte count, lymphocyte count, neutrophil count, sTfR, ATG variety and the presence of PNH clone. The results showed that neutrophil count >0.2 × 109/L indicated a higher response rate at 3 months (as shown above). The PNH clone indicated a higher CR + GPR rate at 6 (P < 0.002) and 12 (P < 0.015) months.
Epidemiological and hematologic parameters were analyzed by logistic regression, and the results showed that neutrophil count >0.2 × 109/L indicated a better response rate at 3 months (P < 0.019; OR = 3.078; 95 % CI 1.203–7.875), and that the PNH clone indicated a higher CR + GPR rate at 6 (P < 0.003; OR = 4.756; 95 % CI 1.717–13.174) and 12 (P < 0.019; OR = 3.441; 95 % CI 1.226–9.659) months. pATG indicated a higher CR + GPR rate at 6 months (P < 0.033; OR = 2.624; 95 % CI 1.082–6.366).
PNH clone evolution after IST
Three patients in PNH+ group had their PNH clone reexamined only once after IST at 3 months, two (1 NR, 1 GPR) presented enlarged, and one (CR) showed decreased clone size. Three patients refused PNH clone test for financial reason. The other 18 PNH+ AA patients had their PNH clone monitored regularly every 3 months during follow-up, and the median population size was 6.575 % (range 0–33.70 %) at 12 months after IST with five (27.8 %) enlarged clone size compared with pretreatment (NR 1, CR 4) and 13 (72.2 %) decreased size (CR 9, GPR 3, PR 1). Eight patients with pretreatment PNH clones (range 1.09–28.9 %; median 1.73 %) were undetectable for PNH clone at 12 months. Four of 69 (5.8 %, CR2, PR1, NR1) pre-IST PNH− patients developed a small PNH clone (range 1.55–5.56 %) without evidence of hemolysis. The PNH clone evolution correlate with neither IST response rate nor CR + GPR rate.
Relapse and survival
All patients were followed up to at least 12 months after IST. None of the patients in the PNH+ group relapsed or died. A total of six patients died in the PNH− group, including the three patients who died within 3 months. For the other three patients who died later, two NR died from infection and transplant preparation regimen, whereas one GPR patient relapsed and died from infection.
Discussion
Whether the presence of the PNH clone can predict response to IST in AA patients remains controversial. In 1995, Schrezenmeier [1] reported that the response rate to standard IST was significantly higher in AA patients with normal PIG-AP than those with PIG-AP− cells in at least one cell lineage. A small prospective study from Italy reached the same conclusion [12]. However, the results of De Lord [2], Scheinberg [3], and Yoshida [6] were inconsistent with those conclusions. They reported no difference in response rate to IST between AA patients with a GPI-AP− and those with normal GPI-AP expression. Moreover, several studies [4, 5, 7, 13] reported that the presence of PNH clones was associated with a higher response rate to IST and better prognosis. The reasons for different conclusions drawn from different studies are not all clear. Differences in the definition of GPI-AP− cell percentage and the small sizes of patient cohort might be contributing factors as we envision. That enlightened us to conduct this study based on a large cohort of patients recently diagnosed with SAA/VSAA. We demonstrated that existence of a small population of PNH-type cells predicts a higher CR + GPR rate but not a higher response rate to IST for patients with SAA/VSAA.
In this study, patients with or without PNH clone achieved the same hematologic response rate in either of SAA and VSAA group. However, 95 % of the responders in PNH + group achieved complete hematologic response or good partial response by 12 months after IST, higher than that of PNH− patients. We postulate that the higher CR + GPR rate of PNH+ AA patients may be attributed to the following factors: (1) Presence of a larger number of residual hematopoietic stem cells in PNH+ AA patients. Although neutrophil, lymphocyte, reticulocyte count, and sTfR that had been reported indicating residual hemopoiesis [13–15] were comparable between the PNH+ and PNH− AA patients, all these measurements may likely reflect only later hemopoiesis. (2) The abnormal immune reaction of PNH+ AA patients were more sensitive to IST. (3) Homogeneity of the immune mechanism was present in PNH+ AA patients. Both PNH+ AA and PNH− AA patients may develop immune-mediated suppression of hemopoiesis since more than two thirds of those patients responded well to IST. However, PNH+ AA patients might have a higher probability of an immune pathogenesis while some of the PNH− AA patients might develop the disease through a nonimmune mechanism.
It appears that AA patients with a PNH clone developed a faster onset of response, because all of the responses in PNH+ AA occurred within 6 months, whereas nearly 10 % of PNH− patients achieved response after 6 months. Such a timing inconsistency helps to make a strategic decision about the second IST. In most instances, a lack of response at 8 months should be coded as a failure of IST [16]. The British Committee for Standards in Haematology [17] recommends a 4-month interval between the first and second courses of ATG, because it usually takes approximately 3 months for a response to occur. In this study, seven patients without a PNH clone responded between 6 and 12 months and accounted for a quarter of nonresponders within the PNH− AA patient cohort at 6 months. Thus, we suggest that even if PNH− AA patients did not respond to IST at 4 months, the salvage therapy could be postponed, while CsA is used regularly and consistently.
Recently, Kulagin [5] reported that PNH+ AA patients had a better response to IST than PNH− patients with a defined threshold of ≥0.01 % GPI/FLAER-deficient cells in granulocytes and RBCs as a criterion for the presence of PNH-type cells, and suggested that the definitive PNH clone threshold of 1 % did not have predictive value. This proposal could partially explain the lack of prognostic value of PNH positivity in the NIH group study [18] and was supported by Sugimori’s report [4]. However, it does not support the results reported by Afable et al. [13]. By using the threshold of ≥0.01 % GPI− cells as PNH+ (data not shown), patients in this study were regrouped into 76 (78.3 %) PNH+ and 21 (21.7 %) PNH− patients. A higher response rate in PNH+ group was found at 3 months (60.5 vs 33.3 %, P < 0.027), but not at 6 and 12 months. Existence of a PNH clone, based on the 0.01 % threshold, still predicted a higher CR + GPR rate at 3 (31.6 vs. 9.5 %, P < 0.043), 6 (46.1 vs. 19 %, P < 0.025), and 12 (59.2 vs. 33.3 %, P < 0.035) months, respectively. On a practical note, the lower PNH threshold would enable the identification of more patients carrying PNH clone. SAA/VSAA patients are extremely low in granulocyte and monocyte counts, it is not easy to harvest enough cells to achieve 104 cells for a clinical PNH clone test. But the usage of more than 1 % PNH cells as the threshold would lead to erroneously low detection rate of PNH clone and would fail to identify true PNH− cases, as some patients with <1 % PNH cells may not be truly PNH negative.
Previous studies had reported that the higher response rate of PNH+ AA to IST could be attributed to an authentic type of immune-mediated marrow failure, because patients with PNH clone often have a specific HLA-DR allele (HLA-DR15) and antigen-driven T cell proliferation in the bone marrow [19, 20]. Several measurements, such as increased ratio of activated T cells, increased interferon-γ expression in bone marrow and peripheral blood T cells, and increased expression of heat-shock protein 70, have been proposed as good markers reflecting the immune pathogenesis of AA. Although these markers are useful in predicting responses to IST, none of them have been practically applied in the clinic at this stage [4]. Cytokines are soluble low-molecular-weight proteins that mediate inflammatory responses and regulate hematopoiesis by modulating bone marrow microenvironment [21]. While cytokine levels may reflect the body immune status, no difference in cytokine was found between PNH+ and PNH− AA patients. Sugimori [4] reported that in most PNH+ patients, successful IST resulted in increases of both PNH-type and normal-type granulocytes. It was postulated that an immune attack against hematopoietic stem cells at the onset of AA that allow PNH-type stem cells to survive does not contribute to the subsequent progression of bone marrow failure. The development of bone marrow failure might be caused by different immune mechanisms targeting epitopes other than those that induce disease. In our study, comparison of PNH clone size before and after IST showed no association with IST response rate. Nonresponders could have their PNH clone enlarged after IST, while some PNH− AA patients could evolve into PNH+. It is possible that immune-mediated suppression of hemopoiesis occur in both GPI-AP+ and GPI-AP− AA patients, but the initiation factors for bone marrow failure might be different with GPI-AP− cells being more susceptible to IST.
References
Schrezenmeier H, Hertenstein B, Wagner B, Raghavachar A, Heimpel H (1995) A pathogenetic link between aplastic anemia and paroxysmal nocturnal hemoglobinuria is suggested by a high frequency of aplastic anemia patients with a deficiency of phosphatidylinositol glycan anchored proteins. Exp Hematol 23:81–87
De Lord C, Tooze JA, Saso R, Marsh JC, Gordon-Smith EC (1998) Deficiency of glycosylphosphatidylinositol-anchored proteins in patients with aplastic anaemia does not affect response to immunosuppressive therapy. Br J Haematol 101:90–93
Scheinberg P, Marte M, Nunez O, Young NS (2010) Paroxysmal nocturnal hemoglobinuria clones in severe aplastic anemia patients treated with horse anti-thymocyte globulin plus cyclosporine. Haematologica 95:1075–1080
Sugimori C, Chuhjo T, Feng X et al (2006) Minor population of CD55-CD59-blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood 107:1308–1314
Kulagin A, Lisukov I, Ivanova M et al (2014) Prognostic value of paroxysmal nocturnal haemoglobinuria clone presence in aplastic anaemia patients treated with combined immunosuppression: results of two-centre prospective study. Br J Haematol 164:546–554
Yoshida N, Yagasaki H, Takahashi Y et al (2008) Clinical impact of HLA-DR15, a minor population of paroxysmal nocturnal haemoglobinuria-type cells, and an aplastic anaemia-associated autoantibody in children with acquired aplastic anaemia. Br J Haematol 142:427–435
Yamazaki H, Saito C, Sugimori N et al (2011) Thymoglobuline is as effective as lymphoglobuline in Japanese patients with aplastic anemia possessing increased glycosylphosphatidylinositol-anchored protein (GPI-AP) deficient cells. Blood (ASH Ann Meet Abstr) 118:1339
Socié G, Rosenfeld S, Frickhofen N, Gluckman E, Tichelli A (2000) Late clonal diseases of treated aplastic anemia. Semin Hematol 37:91–101
Azenishi Y, Ueda E, Machii T et al (1999) CD59-deficient blood cells and PIG-A gene abnormalities in Japanese patients with aplastic anaemia. Br J Haematol 104:523–529
Dunn DE, Tanawattanacharoen P, Boccuni P et al (1999) Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med 131:401–408
International Agranulocytosis and Aplastic Anaemia Study (1987) Incidence of aplastic anaemia: relevance of diagnosis criteria. Blood 70:1718–1721
Timeus F, Crescenzio N, Lorenzati A et al (2010) Paroxysmal nocturnal haemoglobinuria clones in children with acquired aplastic anaemia: a prospective single centre study. Br J Haematol 150:483–485
Afable MG 2nd, Shaik M, Sugimoto Y et al (2011) Efficacy of rabbit anti-thymocyte globulin in severe aplastic anemia. Haematologica 96:1269–1275
Chang MH, Kim KH, Kim HS et al (2010) Predictors of response to immunosuppressive therapy with antithymocyte globulin and cyclosporine and prognostic factors for survival in patients with severe aplastic anemia. Eur J Haematol 84:154–159
Yang WR, Xiong YY, Zhang L et al (2013) Predicting early response to immunosuppressive therapy in severe aplastic anemia by soluble transferrin receptor assay. Zhonghua Xue Ye Xue Za Zhi 34:709–713
Camitta BM (2000) What is the definition of cure for aplastic anemia? Acta Haematol 103:16–18
Marsh JC, Ball SE, Cavenagh J et al (2009) Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol 147:43–70
Scheinberg P, Wu CO, Nunez O, Young NS (2009) Predicting response to immunosuppressive therapy and survival in severe aplastic anaemia. Br J Haematol 144:206–216
Maciejewski JP, Follmann D, Nakamura R et al (2001) Increased frequency of HLA-DR2 in patients with paroxysmal nocturnal hemoglobinuria and the PNH/aplastic anemia syndrome. Blood 98:3513–3519
Zeng W, Nakao S, Takamatsu H et al (1999) Characterization of T-cell repertoire of the bone marrow in immune-mediated aplastic anemia: evidence for the involvement of antigen-driven T-cell response in cyclosporine-dependent aplastic anemia. Blood 93:3008–3016
Feng X, Scheinberg P, Wu CO et al (2011) Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica 96:602–606
Acknowledgments
The authors would like to thank the National Public Health Benefit Research Foundation of China (Grant No. 201202017), the Important National Science and Technology Specific Projects of China (Grant No. 2011ZX09302-007-04), and the Natural Science Foundation of Tianjin (Grant No. 11JCYBJC10500). The authors would also like to acknowledge Dr. Huijun Wang for carefully conducting the PNH clone testing technique.
Authors’ contributions
FZ served as the principal investigator for this study. XZ wrote the paper and contributed to patient recruitment and statistical analysis. LZ, LJ, KZ, YL, GP, LY, YL, JL, HF, LS, and WY contributed to patient recruitment and treatment.
Conflict of interest
The authors declare no competing financial interests.
Ethical statement
This study was approved by the ethics committee of Blood Diseases Hospital, CAMS, and PUMC. All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2008. Patients provided written informed consent for data exploitation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhao, X., Zhang, L., Jing, L. et al. The role of paroxysmal nocturnal hemoglobinuria clones in response to immunosuppressive therapy of patients with severe aplastic anemia. Ann Hematol 94, 1105–1110 (2015). https://doi.org/10.1007/s00277-015-2348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-015-2348-5