
Abstract
The present study was designed to establish the incidence of cytogenetic evolution (CE), defined as the acquisition of chromosomal defects during the course of MDS, in order to correlate it with the WHO classification and IPSS score, and to assess its impact on overall survival (OS) and risk of MDS/AML evolution (progression-free interval, PFI) by means of Cox models for time-dependent covariates. Adjustments for known risk factors were achieved by performing a bivariable analysis. The study was carried out in 153 MDS patients who were followed for a median period of 45.2 months. Disease progression occurred in 42.4% of patients after a 65.2-month median PFI, while CE occurred in 30.7% of patients. Our study shows that (1) CE was more common in advanced than in early MDS, and advanced MDS presented secondary chromosomal defects distinct from those of early MDS; (2) CE significantly affected OS and PFI independently of other prognostic variables; (3) del(7)(q31q34) was the only secondary chromosomal defect which significantly affected PFI; trisomy 8 had only a moderate influence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Myelodysplastic syndromes (MDS) are heterogeneous clonal stem cell disorders characterized by a hypercellular marrow showing ineffective hematopoiesis which is responsible for one or more peripheral blood cytopenia(s) [1, 2]. Currently, MDS is diagnosed according to the World Health Organization (WHO) classification which identifies eight subtypes [3]. This last classification has been validated in a large number of patients [4–8] and is continually subject to improvement [9–14].
The WHO classification system provides relevant prognostic information for a disease with an extremely variable natural course [1–15]. MDS patients experience morbidity and mortality rates higher than those of age-matched normal subjects because of complications related to peripheral cytopenia [16] and AML progression which occurs in about 30% of cases [17]. In 1997, the International Prognostic Scoring System (IPSS) revealed that the number of peripheral blood cytopenias, cytogenetic pattern, and blast cell percentage are the most significant prognostic factors in MDS [18]. Additional studies have demonstrated that IPSS power may be improved by better defining the prognostic relevance of the chromosomal defects included within the intermediate IPSS cytogenetic category [19–22] and this assumption has been further validated [23]. Even more recently, the prognostic power of the cytogenetic pattern has been proven by the newly developed WHO classification-based prognostic system [24]. However, all these studies have analyzed the clinical relevance of chromosomal abnormalities revealed only at clinical diagnosis (primary defects) [25, 26]. In contrast, very few studies have evaluated the incidence and the prognostic significance of cytogenetic evolution (CE), defined as the acquisition of either an abnormal clone in a karyotypically normal patient or additional defects in patients with an already abnormal chromosomal pattern (secondary abnormalities) [27–36]. The majority of these studies, which include small numbers of patients with a short follow-up period, have simply reported that the acquisition of secondary defects occurred at the time of AML progression in approximately 60% of MDS patients, but none have assessed how many patients experience CE before clinical progression and whether this CE is predictive of overall survival (OS) and progression-free interval (PFI) independently of WHO classification, primary defects, and IPSS cytogenetic categories.
These are the goals of the present study, which included 153 patients, observed for a sufficiently long follow-up period, in whom the significant effect of primary cytogenetic defects on disease outcome has already been revealed [21]. An additional goal was to establish which secondary defect significantly predicts leukemic transformation.
Materials and methods
Patients
All 153 de novo MDS patients analyzed in the present study were included in a previous series and were diagnosed at the Division of Hematology, Foundation IRCCS Policlinico San Matteo, Pavia between January 1990 and December 2004. All patients were classified according to WHO classification. Our study does not include any patient with either secondary MDS or with a white blood cell (WBC) count above 12 × 109/L. The diagnostic procedures, performed at clinical diagnosis and during follow-up, and clinical monitoring were carried out as already reported [22]. Moreover, since the IPSS was developed for patients undergoing treatments not affecting the natural course of MDS and since the MDS natural course may be significantly changed by intensive chemotherapy and allogeneic bone marrow transplantation, patients submitted to these therapeutic procedures were evaluated only until the time of such treatments.
Cytogenetic studies
Chromosome studies were performed as previously reported [21]. Chromosome identification and karyotype description were performed according to the International System for Chromosome Nomenclature [37]. Karyotypes were defined as complex when they included ≥3 chromosomal abnormalities.
Statistical analysis
Data were described as the mean and standard deviation (SD) or median and 25th–75th percentiles (IQR) if continuous and described as a count and a percentage if categorical. The role of CE on survival and MDS/AML evolution-free survival was assessed by Cox models for time-dependent covariates. A bivariable analysis was also performed in order to adjust for known risk factors. In a predefined subgroup analysis, the following risk factors were investigated: cytopenias, blast cell percentage, WHO classification, karyotype pattern at clinical diagnosis, and IPSS cytogenetic categories. Time to event or to censoring was computed from diagnosis. The role of specific secondary defects on survival and on MDL/AML evolution-free survival after CE was assessed by Cox regression, with the time to event or censoring computed from CE. Hazard ratios and their 95% confidence intervals (CI) were estimated. The Kaplan Meier method was used to compute the cumulative event-free survival (PFI) and its 95% CI at 2 and 5 years from diagnosis (for all patients) or CE onset (for patients with CE).
Stata 10.1 (StataCorp, College Station, TX, USA) was used for computation. A two-sided p value <0.05 was considered statistically significant.
Results
Patients
Patients’ clinical and hematological features at clinical diagnosis are listed in Table 1. Considering all 153 patients, the median follow-up was 45.2 months \( \left( {{\hbox{IQR}} = {23}.8 - 75.{7}} \right) \), whereas the median survival was not reached \( \left( {{\hbox{IQR}} = {34}.{3} - {\hbox{not reached}}} \right) \). At the time of analysis, 107 (69.9%) patients had survived, and 46 (30%) had died. Disease progression occurred in a total of 65 (42.4%) patients: 11 evolved to a more advanced MDS subtype and 54 to AML. The median PFI was 65.2 months (IQR 16.60–not reached).
Treatment was as follows: 128 patients received supportive care (transfusions and hematopoietic growth factors); three differentiation-inducing agents; two immunosuppressive agents; five low-dose chemotherapy aimed at reducing WBC counts; and 15 various regimens of intensive chemotherapy. Twelve patients were submitted to allogeneic bone marrow transplantation (allo-BMT): 11 had previously received supportive treatment only, whereas one had been submitted to intensive chemotherapy which induced a partial remission. The decision to administer intensive chemotherapy or allo-BMT was exclusively based on either the patients’ age or the fact that progression to a more advanced MDS or to AML had just occurred.
Since our patients were not uniformly treated, in order to avoid any effect of intensive chemotherapy or allo-BMT on our results, the 15 and 12 patients submitted to the respective procedures were censored at the start of both treatments. So, we are quite confident that by excluding these patients, we did not affect the impact of CE on OS or PFI.
Cytogenetic results
At diagnosis, all 153 patients had successful cytogenetic analyses (Table 1). Cytogenetic evolution occurred in a total of 47 (30.7%) patients. However, since six patients (one 5q− syndrome, one RA, one RCMD, and three RAEB-2) developed secondary abnormalities after disease evolution, they were considered chromosomally stable when assessing whether CE was predictive of OS or PFI. Thus, a total of 40 (26.1%) patients acquired secondary abnormalities during the course of the disease and before disease progression. Their WHO subtype and blast cell percentage at clinical diagnosis and at cytogenetic reevaluation are listed in Table 2. In these patients, the median time from clinical diagnosis to the acquisition of secondary defects was 12.9 months \( \left( {{\hbox{IQR}} = {6}.6 - 27.{3}} \right) \). The secondary abnormalities included 5q− in four patients, monosomy 7 in three, del(7)(q31q35) in eight, trisomy 8 in five, del(17)(p13) in four, and various defects (single and double abnormalities) in 16. The interstitial long-arm deletion of chromosome 5 more commonly occurred in patients who did not experience disease progression, instead −7, del(7)(q31q35), and del(17p) more frequently developed in patients with disease progression. Patients surviving with or without CE according to various parameters and those with either a stable disease or evolving to more advanced MDS/AML with or without CE according to the same parameters are reported in Table 3.
Prognostic relevance of cytogenetic evolution
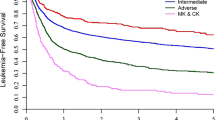
The significant impact of cytogenetic evolution on OS and MDS/AML progression was analyzed by time-dependent Cox regression. Patients who experienced CE during the follow-up period increased their risk of dying by seven-fold and their risk of disease progression (PFI) by 36-fold with respect to patients who remained karyotypically stable (p < 0.001 in both cases; Table 4). Two- and 5-year survivals were, respectively, 40% (95% CI, 19–60) and 10% (95% CI, 0.7–35) in patients with CE, versus 93% (95% CI, 86–96) and 70% (95% CI, 58–70) in patients without CE. After adjusting the effect of CE for other known risk factors (as listed in the methods section) in a bivariable analysis, the prognostic power of CE was proven in all cases for both death and PFI (Table 4). Furthermore, CE had a statistically relevant influence on the OS of patients who at clinical diagnosis were either chromosomally normal or abnormal (p = 0.004 and p < 0.001, respectively). In addition, when the impact of CE on the risk of MDS/AML evolution was adjusted for the presence of primary defects, which significantly affected PFI when analyzed as a single variable (\( {\hbox{Hazard ratio}} = {2}.{3}\left[ {{85}\% \,{\hbox{CI}} = {1}.0{7} - {4}.{94}} \right] \), p = 0.02), CE maintained its statistical power (p < 0.001), whereas primary defects lost their statistical power (p = 0.501; data not shown).
Finally, the OS and the risk of MDS/AML evolution for each of the most common single chromosomal defects acquired during the follow-up period before disease evolution were compared to those of the remaining set of cases with secondary defects. This analysis showed that no single secondary defect affected OS. In contrast, del(7)(q31q34) was the only chromosomal abnormality that significantly affected the risk of MDS/AML progression (p = 0.002), while trisomy 8 had only a moderate effect (p = 0.052).
Discussion
To the best of our knowledge, this is one of the few studies which evaluates the incidence of secondary chromosomal defects (CE) in an adequate number of MDS patients with a sufficient follow-up period of 45.2 months \( \left( {{\hbox{IQR}} = {23}.{8} - {75}.{7}} \right) \). These patients have been cytogenetically analyzed at least twice before MDS/AML evolution. Former studies, which often included small numbers of patients with short follow-up periods, have established that CE occurs during the course of the disease in about 14–46% of MDS patients, more commonly in those with a sudden evolution to AML [27–35]. In a more recent study, 24% of MDS patients developed disease progression which, in the eight patients cytogenetically examined during transformation, was associated with CE [36]. None of these studies has evaluated the incidence of secondary defects in relation to other biological and clinical parameters. In the present series, 47 patients (30.7%) acquired additional chromosomal defects during the follow-up period, 40 of them (25.4%) before disease progression (Table 2). The frequency of this event increased with the worsening of the MDS clinical stage (Table 3). All these data confirm current evidence which suggest that MDS evolution is accompanied by a steady increase in genetic instability and by an expansion of genomic alterations [17, 38, 39].
This is the first study which reveals that the acquisition of secondary chromosomal defects during the follow-up period before disease evolution predicts OS and PFI in MDS patients, an issue never addressed by previously published follow-up studies [27–36]. However, the clinical relevance of CE could be affected by the fact that the decision of performing a new bone marrow aspiration along with a cytogenetic reevaluation might have been dictated by other clinical indicators of disease progression. These last include a worsening of anemia [16], neutropenia [40], thrombocytopenia [41], a sudden need for red blood cell (RBC) transfusions [24], and an abrupt increase in lactic dehydrogenase levels [42]. Recently, a study has evaluated the impact of cytopenias on MDS outcomes [43]. This study has established that in IPSS intermediate-1 and intermediate-2 categories, the hemoglobin value only has an additive prognostic value regarding IPSS for evaluation of OS, but not for time to AML evolution. Another retrospective analysis has pointed out that in low-risk MDS, the total number of RBC transfusions is an independent negative prognostic factor for OS and PFI [24]. In the present study, the 40 patients who experienced CE before disease progression underwent a cytogenetic reassessment because of a reduction in peripheral blood values, especially hemoglobin values. Thus, most of our patients presented a worsening of anemia that could be due either to an increase in bone marrow apoptosis, the feature dissociating MDS from AML [17, 39], or to an increase in marrow blast cell percentage. However, this last event was excluded. In fact, at the time of CE, no patient changed the subtype of MDS diagnosed at the onset of the disease (Table 2).
In our series, patients who experienced CE presented a risk of dying seven-fold superior and a risk of transformation to advanced MDS and AML 36-fold superior than those of chromosomally stable patients (Table 4). However, since many studies have demonstrated that primary chromosomal defects and other clinical–biological factors significantly affect MDS natural history [18–23], it was questioned whether after adjusting for these last variables the statistical power of cytogenetic evolution was maintained. In a first step, the bivariable analysis applied revealed that the impact of CE on OS was independent of the number and type of peripheral blood cytopenias, the blast cell percentage, the WHO classification, the initial chromosomal pattern, and the IPSS cytogenetic categories.
In the subsequent bivariable analysis, it was revealed that after adjusting for all other prognostic variables, the relevance of CE on PFI was still significant (Table 4). In particular, the acquisition of secondary defects remained significant even after adjusting for the prognostic influence of the initial chromosomal pattern (p < 0.001 in patients with either a normal or an abnormal karyotype at clinical diagnosis), which impact on PFI was lost after performing this analysis. We speculate that this effect could be due to the fact that 28 of the 94 patients (30%) with an abnormal karyotype at clinical diagnosis acquired additional defects during the follow-up period (Table 2).
As far as leukemic evolution is concerned, until now, no study has established which numerical or structural abnormality is required for such an event. It has been proposed that some chromosomal alterations may be responsible for each step in the evolution of the disease, but since the pathway of evolution is not unique, it is very difficult to define which genetic alteration comes first [36]. However, this assumption seems to be disproved by a recent array comparative genomic hybridization study which revealed that in low-risk MDS, CD34 negative cells harbor genetic alterations in addition to those present in CD34 positive cells [44]. Thus, disease progression might be caused by the outgrowth of a clonal CD34 negative cell population already present at clinical diagnosis and by the type of genetic lesion present in these dysplastic cells. This last suggestion is also strengthened by our study. In fact, in our series, the majority of secondary defects revealed in early/low-risk MDS were distinct from those revealed in advanced MDS: 5q− and del(11)(q14q23) prevailed in patients with early disease, −7, del(7)(q31q34), and del(17p) in those with advanced disease. Trisomy 8 was equally distributed. Thus, it is not surprising that del(7)(q31q34) was the only secondary defect significantly affecting the risk of MDS/AML evolution.
In conclusion, (1) CE occurs in 30.7% of patients, particularly in those with more advanced MDS; (2) it has a significant influence on OS and PFI independently of other variables with well-known prognostic relevance; (3) del(7)(q31q34) is the only single secondary chromosomal defect which significantly effects PFI, whereas trisomy 8 has a moderate influence.
References
Heaney ML, Golde DW (1999) Myelodysplasia. N Engl J Med 340:1649–1660
Nimer SD (2008) Myelodysplastic syndromes. Blood 11:4841–4851
Jaffe ES, Harris NL, Stein H, Vardiman JW (eds) (2008) Tumours of haematopoietic and lymphoid tissues. World health organization classification of tumours. IARC, Lyon
Germing U, Gattermann N, Strupp C, Aivado M, Aul C (2000) Validation of the WHO proposal for a new classification of primary myelodysplastic syndromes: a retrospective analysis of 1600 patients. Leuk Res 24:983–992
Nösslinger T, Reisner R, Koller E, Grüner H, Tüchler H, Nowotny H et al (2001) Myelodysplastic syndromes, from the French-American-British to world health organization: comparison of classifications on 431 unselected patients from a single institution. Blood 98:2935–2941
Lee J-H, Lee J-H, Shin J-R, Kim W-K, Chi H-S, Park C-J et al (2003) Application of different prognostic scoring systems and comparison of the FAB and WHO classification in Korean patients with myelodysplastic syndromes. Leukemia 17:305–313
Howe B, Porwit-MacDonald A, Wanat R, Tehranchi R, Hellström-Lindberg E (2004) The WHO classification of MDS does make a difference. Blood 103:3265–3270
Malcovati L, Della Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E et al (2005) Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol 23:7594–7603
Strupp C, Gattermann N, Giagounidis A, Aul C, Hildebrandt B, Haas R, Germing U (2003) Refractory anemia with excess of blasts in transformation: analysis of reclassification according to the WHO proposals. Leuk Res 27:397–404
Germing U, Strupp C, Kuendgen A, Aivado M, Giagounidis A, Hildebrandt B et al (2006) Refractory anemia with excess of blasts (RAEB): analysis of reclassification according to the WHO proposals. B J Haematol 132:162–167
Germing U, Strupp C, Kuendgen A, Isa S, Knipp S, Hildebrandt B et al (2006) Prospective validation of the WHO proposals for the classification of myelodysplastic syndromes. Haematologica 91:1596–1604
Valent P, Horny HP, Bennet JM, Fonatsch C, Germing U, Greenberg P et al (2007) Definitions and standards in the diagnosis and treatments of the myelodysplastic syndromes: consensus statements and report from a working conference. Leuk Res 31:727–736
Mufti GJ, Bennet JM, Goasguen J, Bain BJ, Baumann I, Brunning R et al (2008) Diagnosis and classification of myelodysplastic syndromes: international working group on morphology of myelodysplastic syndrome. Haematologica 93:1712–1717
Breccia M, Latagliata R, Cannella L, Carmosino I, De Cuia R, Frustaci A et al (2009) Analysis of prognostic factors in patients with refractory anemia with excess of blasts (RAEB) reclassified according to WHO proposal. Leuk Res 33:391–394
Jädersten M, Hellström-Lindberg E (2008) Myelodysplastic syndromes: biology and treatment. J Int Med 265:307–328
Cazzola M, Malcovati L (2005) Myelodysplastic syndromes – coping with ineffective hematopoiesis. N Engl J Med 352:536–538
Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JCY, Schimmer AD (2007) Myelodysplastic syndromes: the complexity of stem-cell diseases. Nature Rev Cancer 7:118–129
Greenberg P, Cox C, Le Beau MM, Fenaux P, Morel P, Sanz G et al (1987) International system for evaluating prognosis in myelodysplastic syndromes. Blood 89:2079–2088
Solè F, Espinet B, Sanz G, Cervera J, Calasanz MJ, Luňo E et al (2000) Incidence, characterization and prognostic significance of chromosomal abnormalities in 640 patients with primary myelodysplastic syndromes. B J Haematol 108:346–356
Solè F, Luňo E, Sanzo C, Espinet B, Sanz GF, Cervera J et al (2005) Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica 90:1168–1178
Bernasconi P, Klersy C, Boni M, Cavigliano PM, Giardini I, Rocca B et al (2005) Incidence and prognostic significance of karyotype abnormalities in de novo primary myelodysplatic syndromes: a study on 331 patients from a single institution. Leukemia 19:1424–1431
Bernasconi P, Klersy C, Boni M, Cavigliano PM, Calatroni S, Giardini I et al (2007) World health organization classification in combination with cytogenetic markers improves the prognostic stratification of patients with de novo primary myelodysplastic syndromes. B J Haematol 137:193–205
Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B et al (2007) New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core data set of 2124 patients. Blood 110:4385–4395
Malcovati L, Germing U, Kuendgen A, Della Porta MG, Pascutto C, Invernizzi R et al (2007) Time-dependent prognostic scoring system for predicting survival and leukemic evolution in myelodysplastic syndromes. J Clin Oncol 25:3503–3510
Olney HJ, Le Beau MM (2001) The cytogenetics of myelodysplastic syndromes. Best Pract Res Clin Hematol 14:479–495
Bernasconi P, Boni M, Cavigliano PM, Calatroni S, Giardini I, Rocca B et al (2006) Clinical relevance of cytogenetics in myelodysplastic syndromes. Ann NY Acad Sci 1089:395–410
Tricot G, Boogaerts MA, de Wolf-Peeters C, Van den Berghe H, Verwilghen RL (1985) The myelodysplastic syndromes: different evolution patterns based on sequential morphological and cytogenetic investigations. B J Haematol 59:659–670
Benitez J, Carbonell F, Sanchez Fayos J, Heimpel H (1985) Karyotypic evolution in patients with myelodysplastic syndromes. Cancer Gen Cytogen 15:157–167
Horiike S, Taniwaki M, Misawa S, Abe T (1988) Chromosome abnormalities and karyotype evolution in 83 patients with myelodysplastic syndrome and predictive value for prognosis. Cancer 62:1129–1138
Suciu S, Kuse R, Weh HJ, Hossfeld DK (1990) Results of chromosome studies and their relation to morphology, course, and prognosis in 120 patients with de novo myelodysplastic syndrome. Cancer Gen Cytogen 44:15–26
Michalova K, Musilova J, Zemanova Z (1991) Consecutive chromosomal studies in patients with myelodysplastic syndrome (MDS). Czechoslovak MDS Cooperative Group. Ann Genet 34:212–218
White AD, Culligan DJ, Hoy TG, Jacobs A (1992) Extended cytogenetic follow-up of patients with myelodysplastic syndrome (MDS). B J Haematol 81:499–502
Ghaddar HM, Stass SA, Pierce S, Estey EH (1994) Cytogenetic evolution following the transformation of myelodysplastic syndrome to acute myelogenous leukaemia: implications on the overlap between the two diseases. Leukemia 8:1649–1653
White AD, Hoy TG, Jacobs A (1994) Extended cytogenetic follow-up and clinical progress in patients with myelodysplastic syndromes (MDS). Leuk Lymph 12:401–412
Tien HF, Wang CH, Chuang SM, Lee FY, Liu MC, Chen YC et al (1995) Acute leukaemic transformation of myelodysplastic syndrome-immunophenotypic, genotypic, and cytogenetic studies. Leuk Res 19:595–603
De Souza FT, Ornellas MH, de Carvalho LO, Tabak D, Abdelhay E (2000) Chromosomal alterations associated with evolution from myelodysplastic syndrome to acute myeloid leukaemia. Leuk Res 24:839–848
Mitelman F. (ed) (1995) ISCN: an international system for human cytogenetic nomenclature. Karger, Basel
Novotna B, Neuwirtova R, Siskova M, Bagryantseva Y (2008) DNA instability in low-risk myelodysplastic syndromes: refractory anemia with or without ring sideroblasts. Hum Mol Genet 17:2144–2149
Bernasconi P (2008) Molecular pathways in myelodysplastic syndromes and acute myeloid leukaemia: relationship and distinctions-a review. Brit J Haematol 142:695–708
Pomeroy C, Oken MM, Rydell RE, Filice GA (1991) Infection in the myelodysplastic syndromes. Am J Med 90:338–344
Kantarjian H, Giles F, List A, Lyons R, Sekeres MA, Pierce S et al (2007) The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 109:1705–1714
Germing U, Hildebrandt B, Pfeilstöcker M, Nösslinger T, Valent P, Fonatsch C et al (2005) Refinement of the International Prognostic Scoring System (IPSS) by including LDH as an additional scoring variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia 19:2223–2231
Kao JM, McMillan A, Greenberg P (2008) International MDs risk analysis workshop (IMRAW)/IPSS reanalyzed: impact of cytopenias on clinical outcomes in myelodysplastic syndromes. Am J Hematol 83:765–770
Starczynowski DT, Vercauteren S, Telenius A, Sung S, Tohyama K, Brooks-Wilson A et al (2008) High-resolution whole genome tiling path array CGH analysis of CD34+ cells from patients with low-risk myelodysplastic syndromes reveals cryptic copy number alterations and predicts overall and leukemia-free survival. Blood 112:3412–3424
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bernasconi, P., Klersy, C., Boni, M. et al. Does cytogenetic evolution have any prognostic relevance in myelodysplastic syndromes? A study on 153 patients from a single institution. Ann Hematol 89, 545–551 (2010). https://doi.org/10.1007/s00277-010-0927-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-010-0927-z