
Abstract
Cryopreservation of peripheral blood stem cells (PBSC) mobilized by chemotherapy combined with or without granulocyte colony-stimulating factor (G-CSF) is an essential part of procedure for anti-cancer strategies. We evaluated whether a higher cell concentration (2×108/ml) without the use of a programmed freezer was acceptable for the storage of mobilized PBSC in an autologous setting. Mobilized PBSC were enriched to mononuclear cells (MNC) by Percoll separation and then frozen at cell concentrations of 2–5×107/ml (group I, n=20) or 2×108/ml (group II, n=44) without the use of a programmed freezer using 5% DMSO, 6% hydroxy ethyl starch, and 4% autologous serum or human albumin. CD34+ cells purified by ISOLEX300 were frozen at 2×107/ml (group III, n=22) using the same method. The median recovery rates of CD34+ cells and CFU-GM were, respectively, n.d. (not determined) and 88% in group I, 103 and 64% in group II, and 98 and 53% in group III. There was a statistical significance between the recovery rate of CFU-GM in group III and that in group I (p=0.02). The median percentage of cell viability after thawing in each group was 89, 87, and 75%, respectively. The median numbers of days after PBSCT to achieve a WBC of >1.0×109/l, an absolute neutrophil count of >0.5×109/l, and a platelet count of >50×109/l were, respectively, 11, 11 and 15 in group I; 12, 12 and 16 in group II; and 12, 12 and 27 in group III. These results suggest that enriched MNC from mobilized PBSC could be frozen at a higher cell concentration (2×108/ml) without the use of a programmed freezer, leading to reduction of the toxicities associated with infusion of thawed cells and of costly space required for cell storage.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peripheral blood stem cell transplantation (PBSCT) has replaced autologous marrow transplantation in the treatment of various types of cancers [1].Improvements in mobilization methods with recombinant cytokines enable us to collect larger numbers of PBSC. Harvested peripheral blood stem cells (PBSC) are also expected to be a source of targeting cells for various types of future cell therapies.
Cryopreservation of PBSC is essential in an autologous setting. It has been previously reported that cell concentration (2×107/ml) and rate control (−1°C/min) are critical in cell freezing procedure [2]. In case of PBSCT, the traditionally recommended concentration of 2×107/ml would result in large product volumes with correspondingly increased amounts of cryoprotectant, which could be toxic for the recipients [3, 4, 5]. On the other hand, rate-controlled freezing with a programmed freezer is expensive as a routine clinical procedure. To make this procedure simpler and more economical, a non-rate-controlled freezing method has been reported by several investigators [6, 7, 8].It has been shown that rate-controlled cryopreservation with a programmed freezer at a higher cell concentration does not impair the post-thaw recovery of hematopoietic stem/progenitor cells [9].
However, the effect of cryopreservation at a higher cell concentration without the use of a programmed freezer on autologous PBSCT has been tested very little [10]. In this study, we compared the recovery of CFU-GM, CD34+ cells and engraftment kinetics of cryopreserved products, which were frozen without the use of a programmed freezer at low and higher cell concentrations.
Methods and materials
Subjects
Patients who underwent PBSC collection and autologous PBSCT from January 1992 to December 1999 in the Department of Pediatrics, University of Tokushima, were enrolled into this study after obtaining consent. The study subjects consisted of patients with acute lymphoblastic leukemia (ALL, n=26), acute nonlymphocytic leukemia (ANLL, n=11), non-Hodgkin’s lymphoma (NHL, n=3), and various solid tumors (n=46). The solid tumors included neuroblastoma (n=13), brain tumor (10), Wilms tumor (6), ovarian cancer (5), breast cancer (4), retinoblastoma (2), rhabdomyosarcoma (2), testicular tumor (2), peripheral neuroectodermal tumor (1), and juvenile rheumatoid arthritis (1). Of 86 patients, 42 were male and 44 were female. Their ages ranged from 1 to 56 years, with a median age of 10 years.
Mobilization and collection of PBSC
PBSC were mobilized by intensive chemotherapy with or without recombinant human granulocyte colony-stimulating factor (G-CSF) as previously reported [11].They were collected in the recovery phase of chemotherapy with a Baxter CS3000 plus continuous-flow blood cell separator (Baxter Healthcare, Deerfield, IL, USA) [12].Collection was performed on the day the patient achieved a WBC of >3×109/l and a platelet count of >100×109/l in the recovery phase after chemotherapy. In some patients, PBSC were mobilized by G-CSF alone [13].In these cases, patients received 10 μg/kg of G-CSF once a day by subcutaneous injection for 5 days.Apheresis was initiated from days 4 to 6 after G-CSF injection, and 200–300 ml/kg (max. 10 liters) were processed per session.
Cryopreservation and thawing procedures
Apheresis-collected cells were separated using discontinuous gradients of 40 and 60% Percoll and centrifugation [14].Cells were resuspended in Dulbecco’s modified Eagle’s minimum essential medium (DMEM) supplemented with 10% autologous serum, as previously reported. The freezing method reported by Makino et al. was introduced with minor modifications from the beginning of this study [7].Briefly, Percoll-separated cells were resuspended in DMEM with 10% autologous serum or 8% human albumin (Albumin Yoshitomi, Yoshitomi Pharmaceutical, Osaka) and mixed slowly with an equal volume of freezing solution containing 12% HES and 10% DMSO to give final concentrations of 5% DMSO and 6% HES. Prior to May 1995, PBSC were cryopreserved at concentrations of 2–5×107/ml (Group I, n=20). Purified CD34+ cells were frozen at 2×107/ml using the same method (Group III, n=22). Subsequently, a concentration of 2×108/ml was adopted (Group II, n=44). In all methods, cells were transferred to 5-ml polypropylene cryo-tubes (MS4605 W, Sumitomo Bakelite, Osaka), placed directly into a −80°C electric freezer, and then transferred to −135°C on the following day. The cells were stored in the same freezer until use.
Cells were thawed rapidly in a water bath maintained at 37°C. We divided stored cells into two portions that were infused over 2 days, when the volume of cells suspension was >300 ml [15]. For recovery analysis, an aliquot of cell suspension was quickly transferred to a 50-ml tube and diluted with thawing medium consisting of 10% fetal bovine serum (FBS, Filtron, Brooklyn, Australia) and 50 IU/ml of deoxyribonuclease (Sigma DN-25, Aldrich Japan, Tokyo) in DMEM by the stepwise addition of this medium at room temperature with gentle agitation. The cells were then collected by centrifugation, washed three times with the thawing medium, and resuspended in DMEM supplemented with 10% FBS for further experiments. Trypan blue staining method was used to measure cell viability.
CD34+ cell purification
G-CSF-mobilized PBSC collected by apheresis were enriched for CD34+ cells using an ISOLEX-300 (Baxter Healthcare, Deerfield, IL, USA) according to the manufacturer’s suggestions. Briefly, excess platelets were removed by centrifugation for 20 min at 200×G at room temperature. Cells were incubated in phosphate-buffered saline (PBS, Nissui, Tokyo) containing 0.5% human-globulin (Gammagard, Baxter Japan, Tokyo) for 15 min to block Fc-receptors. One vial of anti-CD34 monoclonal antibody (9C5, 2 mg) was added to the cell suspension that contained <5×1010 cells. After 30 min of incubation at room temperature with gentle rotation (4/min), cells were washed three times with PBS containing 1% human serum albumin (Albumin-Yoshitomi, Yoshitomi Pharmaceutical, Osaka). Sensitized cells were incubated with sheep anti-mouse IgG1-coated paramagnetic microspheres (Dynabeads, 10 ml; Dynal, Oslo). Cells rosetted with beads were captured on permanent magnets, and released by chymopapain or peptide capture included in the kit. These cells were frozen by the same method as described above.
Flow cytometry
CD34+ cells were assayed by Otsuka Assay Institute (Tokyo). Sample cells were shipped by air-cargo and assayed within 24 h. Cells that expressed the surface CD34 antigen were identified by flow cytometry analysis. Briefly, 100 μl of cell suspension were added to a test tube (Falcon 2052, Becton Dickinson, Lincoln Park, NJ, USA) containing isotype control (phycoerythrin-mouse IgG1) and phycoerythrin-conjugated CD34 monoclonal antibody (Anti-HPCA2 antibody, Becton Dickinson) at a concentration of 1 μg antibody/106cells. Samples were analyzed with a FACScan flow cytometer (Becton Dickinson). After function was verified, samples were drawn into the flow cytometer using FSC and SSC, as gating parameters, along with debris subtraction techniques to determine the characteristics of the cells. A total of 20,000 events were counted to identify the mononuclear cell fraction. The flow cytometric data were analyzed using a gated analysis via a set of SSC-FL parameters for CD34+ cells to calculate the percentage of positive cells. When a sample was substantially contaminated with RBC, it was lysed with a solution consisting of 0.826% (w/v) NH4CL, 0.1% KHCO3, and 0.004% EDTA-4Na.
Hematopoietic progenitor assay
Colony-forming cells were incubated in methylcellulose cultures supplemented with 20% FBS, 450 µg/ml of human transferrin (Sigma T-1147), 2 U/ml of recombinant human erythropoietin (Kirin Brewery, Tokyo), 1% deionized delipidated BSA (Calbiochem 12657, Hoechst Japan, Tokyo), and a combination of recombinant human G-CSF (filgrastim, Kirin), interleukin-3 (Kirin), and stem cell factor (Kirin). These stimulating factors were used at a final concentration of 20 ng/ml, which was the previously determined optimal concentration in our laboratory. Triplicate or quadruplicate cultures were plated in volumes of 0.4 ml in 24-well tissue culture plates (Corning 258201, New York, NY) that were then placed in an ESPEC N2-O2-CO2 BNP-110 incubator (Tabai ESPEC, Osaka, Japan), which maintained a humid atmosphere of 5% carbon dioxide, 5% oxygen, and 90% nitrogen at 37°C. Plates were incubated for 13–15 days and three types of colonies, including colony-forming unit for granulocyte-macrophage (CFU-GM), were counted using an inverted microscope. The mean number of colonies in four wells was calculated.
Transplant procedures
The transplant procedures in our institute have been previously described in detail [16]. Briefly, frozen cells were thawed rapidly in a water bath maintained at 37°C. The patients were given 5 mg/kg of hydrocortisone and/or antihistamines to prevent allergic reactions before infusion. The recovery speed after autografting was evaluated in terms of the number of days to achieve a WBC of >1×109/l, an absolute neutrophil count (ANC) of >0.5×109/l, and a platelet count of >50×109/l. G-CSF was given to the patients only in group III after autografting.
Statistics
The Mann Whitney U-test was used to analyze the significance of differences. Data were analyzed using StatView (Version 4.5; Abacus Concepts, Berkeley, CA, USA) for a Macintosh computer.
Results
Frozen cells
After Percoll separation (groups II and I) or the purification procedure (group III), the median (range) numbers of frozen cells per kilogram of recipient body weight were determined, as shown in Table 1. MNC in group III (median, 2.6×106/kg) was significantly lower than those in groups II (5.9×108/kg) and I (13×108/kg) (p<0.001 each). The number of CD34+ cells in group II (5.7×106/kg) was also significantly higher than that in group III (2.2×106/kg) (p=0.002). The number of CFU-GM in group II (16×105/kg) was significantly higher than those in groups III (4.2×105/kg) and I (6.1×105/kg) (p<0.001 each). However, there was no difference in the number of CFU-GM between groups III and I.
Recovery rate and cell viability
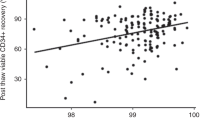
Cell recovery rates after cryopreservation/thawing are shown in Table 2. These numbers ranged widely. Recovery rates of MNC were statistically identical among the three groups, but with different p values (II and I; 0.770, III and I; 0.100, III and I; 0.144). The viability of MNC after thawing in group III [75% (66–89)] was significantly lower than those in groups II and I (p<0.001, each). There was no difference in cell viability between groups II and I (p=0.332). The recovery rate of CFU-GM in group III [53% (13–202%)] was lower than that in group I (p=0.023). However, there were no significant differences between groups II and I (p=0.091), or groups III and II (p=0.271).
Engraftment kinetics
The median volume of cell suspension infused was 180 ml (range, 85 –700 ml) in group I, 60 ml (23–145 ml) in group II, and 8 ml (3.5–15 ml) in group III. The number of infused CFU-GM in group I [median, 5.6 (range, 1.5–20) ×105/kg] was statistically identical to that in group II [9.1 (0.3–90) ×105/kg] (p=0.086). The number of infused CD34+ cells in group III [2.1 (0.43 – 7.0) ×106/kg] was significantly lower than that in group II [5.3 (0.12–75) ×106/kg] (p=0.001), and the number of CFU-GM in group III [2.2 (0.3–12) ×105/kg] was significantly lower than those in groups II [9.1 (0.3–90) ×105/kg] and I [5.6 (1.5–20) ×105/kg] (p<0.001 each).
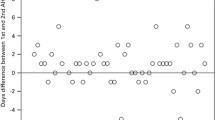
The engraftment rates determined by the number of days to achieve a WBC of >1×109/l and ANC of 0.5×109/l after autografting were identical among the three groups. However, the platelet engraftment rate determined by the number of days to achieve a platelet count of >50×109/l in group III was significantly delayed (p=0.022 and 0.002 versus groups II and I, respectively; Table 3).
Discussion
The rapid hematopoietic recovery after myeloablative therapy has prompted the use of PBSC in preference to BM cells [17]. Apheresis after mobilizing chemotherapy with or without G-CSF enables the collection of a large numbers of PBSC in comparison with marrow aspiration under general anesthesia. However, the collection and cryopreservation of PBSC is associated with intense labor and requires ample space for storage. Optimal conditions for PBSC cryopreservation have not yet been defined. In particular, an increase in the volume of cell suspension which will be frozen results in a concomitant increase in the volume of cryoprotectant, such as DMSO, which may become toxic at cell infusion [3, 5]. Under these circumstances, we applied a gradient centrifugation method with double-layered Percoll to deplete red cells, granulocytes, and platelets for clinical use in pediatric patients [18].
Rowley et al. reported that stem cell survival, as reflected in the post-thaw recovery of MNC, CFU-GM and CD34+ cells, was unaffected even when nucleated cells were frozen at a concentration of 3.7±1.9×108/ml [9].In addition, cryopreservation at different cell concentrations did not predict the time to engraftment or duration of aplasia [9]. Another study showed a reduced recovery of CFU-GM when a higher cell concentration of PBSC was compared with a lower cell concentration of bone marrow at the time of freezing. However, this did not translate into delayed hematopoietic recovery in clinical transplantation [19].Based upon these studies, we have initiated to cryopreserve PBSC at higher cell concentrations without the use of a programmed freezer. Benefit and efficacy of the procedure was evaluated by comparing hematopoietic recovery after autologous PBSCT.
In this study, we did not observe a significant difference in the freeze/thaw recovery rates of CD34+ cells or CFU-GM between groups II and I. However, recovery rate of CFU-GM and the number of reinfused cells in group III were significantly smaller than in the other two groups. The speeds of engraftment were not significantly different between groups II and I, although patients in group III showed a slower recovery of platelets. The neutrophil recovery might be enhanced by the administration of G-CSF in group III. On the other hand, there is another possibility that purification procedures with ISOLEX300 selectively affect on platelet-lineage progenitor cells or make them vulnerable to freeze/thaw procedure. Further investigations will be required to solve this problem.
Balint et al. reported that presence of 10% DMSO is an essential part of the cryopreservation procedure of very primitive murine stem cells [20]. On the other hand, DMSO is the primary factor related to toxicities at graft infusion and reduction of amount of DMSO by reducing the total volume of grafts should merit patients, particularly pediatric population. Thus, there is no suspicion for the superiority of using 5% DMSO when recovery rates of frozen cells and engraftment kinetics are identical.
In conclusion, the present results indicate that MNC in apheresis products be cryopreserved at 2×108/ml without the use of a programmed freezer, without jeopardizing their engraftment potential. Definition of the upper limit for the cryopreservation cell concentration will require further studies.
References
Goldman JM, Schmitz N, Niethammer D, Gratwohl A (1998) Allogeneic and autologous transplantation for hematological disease, solid tumors and immune disorders: current practice in Europe in 1998. Bone Marrow Transplant 21:1–7
Goldman JM, Th’ng KH, Park DS, Spiers AS, Lowenthal RM, Ruutu T (1979) Collection, cryopreservation and subsequent viability of haemopoietic stem cells intended for treatment of chronic granulocytic leukemia in blast-cell transformation. Br J Haematol 40:185–195
Stroncek DF, Fautsh SK, Lasky LC, Hurd DD, Ramsay NK, McCullough J (1991) Adverse reactions in patients transfused with cryopreserved marrow. Transfusion 31:521–526
Okamoto Y, Takaue Y, Saito S, Shimizu T, Suzue T, Abe T, Sato J, Hirao A, Watanabe T, Kawano Y, Kuroda Y (1993) Toxicities associated with cryopreserved and thawed peripheral blood stem cell autografts in children with cancer. Transfusion 33:578–581
Al Alessandrino P, Bernasconi P, Caldera D, Colombo A, Bonfichi M, Malcovati L, Klersy C, Martinelli G, MaiocchiM, Pagnucco G, Varettoni M, Perotti C, Bernasconi C (1999) Adverse events occurring during bone marrow or peripheral blood progenitor cell infusion: analysis of 126 cases. Bone Marrow Transplant 23:533–537
Rowley SD (1992) Hematopoietic stem cell cryopreservation: a review of current techniques. J Hematother 1:233–250
Makino S, Harada M, Akashi K, Taniguchi S, Shibuya T, Inaba S, Niho Y (1991) A simplified method for cryopreservation of peripheral blood stem cells at −80°C without rate-controlled freezing. Bone Marrow Transplant 8:239–244
Takaue Y, Abe T, Kawano Y, Suzue T, Saito S, Hirao A, Sato J, Makimoto A, Kawahito M, Watanabe T, Shimokawa T, Kuroda Y (1994) Comparative analysis of engraftment after peripheral blood stem cell autografts cryopreserved by controlled vs uncontrolled-rate method. Bone Marrow Transplant 13:801–804
Rowley SD, Bensinger WI, Gooley TA, Buckner CD (1994) Effect of cell concentration on bone marrow and peripheral blood stem cell cryopreservation. Blood 83:2731–2736
Cabezudo E, Dalmases C, Ruz M, Sanchez JA, Torrico C, Sola C, Querol S, Garcia J (2000) Leukapheresis components may be cryopreserved at high cell concentrations without additional loss of HPC function. Transfusion 40:1223–1227
Takaue Y, Kawano Y, Kuroda Y (1995) Application of recombinant granulocyte colony-stimulating factor in peripheral blood stem-cell transplantation: a pediatric experience. In: Levitt D, Mertelsmann R (eds) Hematopoietic stem cells. Marcel Dekker, New York, pp 611–630
Makimoto A, Kawano Y, Abe T, Okamoto Y, Sato J, Nakagawa R, Watanabe H, Watanabe T, Kuroda Y, Sweet L, Takaue Y (1999) Comparative evaluation of procedures with a Baxter CS-3000 cell separator for collecting peripheral blood cells from children. J Hematother 8:305–310
Kawano Y, Takaue Y, Watanabe T, Abe T, Okamoto Y, Iwai A, Watanabe A, Ito E, Makimoto A, Nakagawa R, Watanabe H, Sato J, Suenaga K, Suzuya H, Ohnishi T, Kanamaru S, Kaneko S, Kuroda Y (1999) Efficacy of the mobilization of peripheral blood stem cells by granulocyte colony-stimulating factor in pediatric donors. Cancer Res 59:3321–3324
Takaue Y, Kawano Y, Watanabe A, Eguchi H, Abe T, Makimoto A, Okamoto Y, Kuroda Y (1996) Transplantation with purified or unmanipulated mobilized blood stem cells in children. In: Ikehara S, Takaku F, Good RA (eds) Bone marrow transplantation: basic and clinical studies. Springer, Berlin, Heidelberg, New York, pp 246–249
Kawano Y, Takaue Y, Watanabe T, Saito S, Abe T, Hirao A, Sato J, Ninomiya T, Suzue T, Koyama T, Shimokawa T, Yokobayashi A, Asano S, Masaoka T, Takaku F, Kuroda Y (1993) Effects of progenitor cell dose and preleukapheresis use of human recombinant granulocyte-macrophage colony-stimulating factor on the recovery of hematopoiesis after blood stem cell autografting in children. Exp Hematol 21:103–108
Takaue Y, Kawano Y, Abe T, Okamoto Y, Suzue T, Shimizu T, Saito S, Sato J, Makimoto A, Nakagawa R, Watanabe T, Ito M, Kuroda Y (1995) Collection and transplantation of peripheral blood stem cells in very small children weighing 20 kg or less. Blood 86:372–380
To LB, Roberts MM, Haylock DN, Dyson PG, Branford AL, Thorp D, Ho JQ, Dart GW, Horvath N, Davy ML, Olweny LM, Juttner CA (1992) Comparison of haematological recovery times and supportive care requirements of autologous recovery phase peripheral blood stem cell transplants, autologous bone marrow transplants and allogeneic bone marrow transplants. Bone Marrow Transplant 9:277–284
Takaue Y, Watanabe T, Kawano Y, Koyama T, Huq M, Suzue T, Abe T, Sato J, Shimokawa T, Kosaka M, Shimizu M, Ogura T, Ninomiya T, Kuroda Y (1989) Isolation and storage of peripheral blood hematopoietic stem cells for autotransplantation into children with cancer. Blood 74:1245–1251
Keung YK, Cobos E, Morgan D, Park M, Dixon S, Wu K, Park CH (1996) High cellular concentration of peripheral blood progenitor cells during cryopreservation adversely affects CFU-GM but not hematopoietic recovery. J Hematother 5:73–77
Balint B, Ivanovic Z, Petakov M, Taseski J, Jovcic G, Stojanovic N, Milenkovic P (1999) The cryopreservation protocol optimal for progenitor recovery is not optimal for preservation of marrow repopulating ability. Bone Marrow Transplant 23:613–619
Acknowledgements
The authors are grateful to Ms. Yasuda for her excellent technical assistance and to the nursing staff of the pediatric ward at the University Hospital of Tokushima.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kawano, Y., Lee, C.L., Watanabe, T. et al. Cryopreservation of mobilized blood stem cells at a higher cell concentration without the use of a programmed freezer. Ann Hematol 83, 50–54 (2004). https://doi.org/10.1007/s00277-003-0817-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-003-0817-8