
Abstract
Bütschliite, K2Ca(CO3)2, occurring as inclusions in mantle minerals, is regarded as one of the key phases to understand phase relationships of dense potassium carbonates and thus to evaluate their potential role within the Earth’s deep carbon cycle. Accordingly, the high-pressure behavior of synthetic bütschliite has been investigated by in-situ single-crystal X-ray diffraction under isothermal compression up to 20 GPa at T = 298 K. The compression mechanism before and after the trigonal-to-monoclinic (R-3m to C2/m) phase transition at ∼6 GPa, found previously, is characterized in terms of the evolution of the cation polyhedra and carbonate groups. On this basis, the modulation of the axial compression is interpreted, and the contribution of the cation polyhedra into the bulk compression is estimated. The refined compressibility of the monoclinic phase (K0 = 44(2) GPa) fits to the trend of the carbonate bulk modulus versus average non-carbon cation radius. The analysis of the obtained and literature structural data suggests the distortion of a large cation polyhedron to be an effective tool to strengthen the carbonate structure at high pressure. On the other hand, the observed symmetrization of the cation polyhedra in trigonal bütschliite is apparently a crucial factor of its stabilization at high pressure upon the temperature rise observed previously. The structural crystallography provided in this study supports the enhanced stability of trigonal bütschliite at high P, T conditions and its significance of being considered as a constituent of the inclusions in deep minerals.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Bütschliite, K2Ca(CO3)2, is a mineral rarely found under conditions on the Earth’s surface. The main occurrence has been reported in wood-ash stones, representing the transformation product of its high-temperature modification known as the mineral fairchildite (e.g. Pabst 1974). The discovery of potassium carbonates entrapped in mineral inclusions from mantle rocks (Giuliani et al. 2012; Abersteiner et al. 2022) or found as melt inclusions hosted in diamonds (Jablon and Navon 2016; Logvinova et al. 2019) raised again the attention to the presence of potassium carbonates in the Earth’s mantle and their role in the deep carbon cycle of our planet. Such role is underlined by the presumed presence of potassium-rich alkali melts in the upper mantle. This is demonstrated not only by the findings of respective melt inclusions, but also through high-pressure experiments on partial melting of kimberlites, carbonated peridotites, eclogites and pelites under upper-mantle conditions (Yaxley and Brey 2004; Dasgupta and Hirschmann 2010; Litasov et al. 2010; Grassi and Schmidt 2011).
A rather complex phase diagram of the binary system K2CO3-СаCO3, obtained from thermal analysis (Arceo and Glasser 1995), experiments in autoclaves (Cooper et al. 1975) and multi-anvil press devices (Shatskiy et al. 2015; Arefiev et al. 2019), includes bütschliite as a sub-solidus phase at 0.1 and 6 GPa. At 3 GPa bütschliite even becomes a liquidus phase (Arefiev et al. 2019). At 0.1 GPa bütschliite, crystallizing in the trigonal space group R-3m (Pabst 1974), transforms to the hexagonal high-temperature (HT) polymorph fairchildite (P63/mmc) at about 550 oC (Cooper et al. 1975). At 3 and 6 GPa the upper temperature limit of bütschliite stability raises to about 980 oC, where it melts incongruently or transforms into double carbonates with different stoichiometry (Shatskiy et al. 2015; Arefiev et al. 2019).
It is remarkable that natural carbonates with the composition close to K2Ca(CO3)2 were found in the solidified melt inclusions hosted by ilmenite from a mantle xenolith (Giuliani et al. 2012) and also in primary inclusion trapped in host diamond (Logvinova et al. 2019). Moreover, the Raman spectrum of the last carbonate is the same as that of K2Ca(CO3)2 bütschliite synthesized by Arefiev et al. (2019). Bütschliite also forms, together with the other K-Ca carbonates, during quenching of carbonate melts in the experiments at 3 and 6 GPa (Arefiev et al. 2022a, b). In addition, it was shown that the gradual reduction of carbonates and their melts under mantle P-T conditions is accompanied by a change in their bulk composition towards enrichments in K-Na-Ca (Shatskiy et al. 2023). Therefore, bütschliite is a likely inclusion in diamonds crystallized from mantle carbonatite melts as a result of their partial reduction. Taken together, these data suggest potential significance of bütschliite as a solid phase or constituent of carbonate melts originating at upper-mantle conditions. This explains our motivation to investigate the crystallographic properties, in particular the structural behavior of bütschliite under high pressure (HP) conditions.
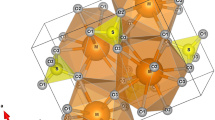
The trigonal crystal structure of K2Ca(CO3)2 bütschliite (Pabst 1974) can be presented as alternating layers of K and Ca cations separated by CO32− groups, which in turn are aligned parallel to each other with their triads determining the c-axis direction (Fig. 1a). The Ca coordination is octahedral (6-fold) with six equivalent Ca-O distances. The CaO6-octahedra are connected through the carbonate groups within the layer and share two faces with the K polyhedra of the adjacent layers (Fig. 1a). The 9-fold coordination around the K atoms includes six short (∼2.8 Å) and three long (∼3.0 Å) distances to the oxygens of the adjacent (CO32−-Ca-CO32−) sheets. Within the layer the KO9-polyhedra are interconnected through shared faces. The site symmetries of the cations and CO32− groups follow local trigonal symmetry.
The HP structure behavior of double carbonates is discussed in detail by Ross and Reeder (1992). In general, all trigonal carbonates are characterized by the typical compressional anisotropy, exhibiting a higher compressibility along the c axis and higher lattice stiffness in the directions perpendicular to it. This is due to the symmetry-constrained orientation of rigid planar CO32− groups parallel to the (001) plane direction, thereby making this direction less compressible compared to the c direction. The compression along c is entirely determined by the compression of the cation polyhedra. The polyhedral compression prevails over distortion in these rather symmetric structures.

Crystal structure of the K2Ca(CO3)2 bütschliite polymorphs as derived from the single-crystal data collected at (a) ambient pressure, (b) 7.52 GPa and (c) 20.65 GPa. The space group and pressure value are indicated on the top of each figure. The cation sites K (violet) and Ca (blue), and the carbonate groups (CO3) are labeled accordingly. In the monoclinic structure the coordination around the K atoms is displayed as KO9 instead of KO11 polyhedra for an easier comparison with the trigonal structure. The solid black line in (c) marks the angle O1-C1-C1, enlarged in (d), which accounts for the coupled polyhedral rotation (indicated by arrow) of CO3 and CaO6 building units. The structure is visualized using the program VESTA 3 (Momma and Izumi 2011)
In double alkali-bearing carbonates the large alkali cation coordination polyhedron is the most compressible and deformable structural unit, which defines the existence of transformations occurring at a relatively low pressure (Hou et al. 2022). This is demonstrated by several examples of carbonate phases belonging to the K2Mg(CO3)2−Na2Mg(CO3)2 join (Golubkova et al. 2015; Hou et al. 2022), as well as bütschliite K2Ca(CO3)2 (Zeff et al. 2024). In isostructural K2Mg(CO3)2 and K2Ca(CO3)2, a reversible transformation from trigonal (R-3m) to a monoclinic (C2/m) symmetry is predicted (Hou et al. 2022) and observed at about 8 and 6 GPa, respectively (Golubkova et al. 2015; Zeff et al. 2024). Tian et al. (2023) claimed the unconfirmed transition to a triclinic structure without providing actual data on the lattice metrics as derived from first-principle calculations. The confirmed monoclinic HP polymorphs of both compounds exhibit comparable structural topology with their LP phases, but a more distorted coordination around the alkali cation and CO32− groups being tilted off the former triad. In bütschliite, three more phase transitions were found in at ∼28, 34 and 37 GPa based on the Raman spectroscopy and X-ray powder diffraction data (Zeff et al. 2024).
As a result of the trigonal-to-monoclinic transition in bütschliite, the coordination of K structure polyhedron becomes enlarged to (9 + 3) or (9 + 2), if the K-O distances within 3.3 Å are included (Zeff et al. 2024). In contrast to the trigonal structure, compressed mainly through the contraction of the cation polyhedra, the compression of the monoclinic HP-K2Ca(CO3)2 proceeds through the inter-layer shifting and rotation of carbonate groups. The lateral shifting between layers in HP-K2Ca(CO3)2 is characterized by the increase of the monoclinic β-angle, which demonstrates a non-linear pressure evolution with the flattening at P > 11 GPa (Zeff et al. 2024). The origin for this non-linear behavior remains unclear.
Recent ab-initio calculations on four end-members of K-Na-Ca-Mg alkali-bearing double carbonates revealed clear correlation between their lower bulk modulus (K0) and a larger metallic ionic radius (Hou et al. 2022). In particular, for trigonal K2Ca(CO3)2 and K2Mg(CO3)2 the respective K0 are about 57 and 65 GPa. Notably, these values do not change appreciably upon the transition to the monoclinic phase; this agrees with the experimentally obtained data for K2Mg(CO3)2 (Golubkova et al. 2015). In this respect an extremely low K0 of ∼25 GPa, obtained for the HP-K2Ca(CO3)2 through the fit of the single crystal and powder diffraction data up to 20 GPa (Zeff et al. 2024), seems questionable.
The mentioned details show that, despite a general description of the pressure-induced structure evolution of bütschliite is given in most recent theoretical and experimental works (Hou et al. 2022; Zeff et al. 2024), some essential features of the structure behavior remained unresolved. Especially in the work of Zeff et al. (2024) only the interatomic distances and the angle of the carbonate-group tilting are stated among the structure parameters. To our opinion, it is important to consider also the polyhedral compressibilities, distortion and mutual interplay between the cation polyhedra. This could reveal more relations between the evolution of the structure units and bulk compression, and provide a deeper understanding of the compression and transition mechanism. Our interest arises also from our recent spectroscopic observations showing a pronounced high-pressure shift of the boundary between the trigonal and monoclinic bütschliite phase upon the temperature rise up to 300 oC (Likhacheva et al. 2024). A detailed analysis of the bütschliite structure evolution could provide explanation for the stabilization of the trigonal bütschliite under HP-HT conditions.
Here we report the compressibility and the structural behavior of synthetic bütschliite, with an emphasis on the evolution of the structure polyhedra, at high pressure up to 20 GPa and compare it with the previously obtained data for bütschliite and the other K, Ca-bearing carbonates.
Experimental
We used K2Ca(CO3)2 crystal specimen from a sample synthesized in a multi-anvil press at 900 oC and 6 GPa, as described and characterized by Arefiev et al. (2019). Its Raman spectrum collected in air (Online Resource 1) is identical to that of K2Ca(CO3)2 synthesized in the bütschliite stability field at 1 atm / 500 °C (Arefiev et al. 2019) and to that reported by Zeff et al. (2024).
The in situ HP single-crystal diffraction experiments were performed at the station ID15b at the European Synchrotron Radiation Facility. The crystals quality was checked prior to their loading in the diamond anvil cell (DAC). Membrane driven LeToullec type DAC, equipped with Boehler-Almax anvils with an opening half-angle of 32o, was used. The culet diameter was 600 μm. The sample chamber with diameter of 300 μm was drilled in the stainless steel gasket indented to about 80 μm. The K2Ca(CO3)2 single-crystal fragment of 0.05 × 0.05 × 0.03 mm was placed inside the sample chamber along with ruby sphere. Hydrostatic conditions were provided by the DAC loading with a helium pressure-transmitting medium. Pressures were determined using ruby fluorescence scale (Shen et al. 2020). The zero-point measurement at ambient pressure was made first, and then the DAC was loaded with He and sequentially pressurized from 0.3 to 20 GPa.
Monochromatic X-ray diffraction measurements were performed at a wavelength of 0.40986 Å with a beam size of 10 × 10 μm (Merlini and Hafland 2013). Diffraction patterns were collected using a EIGER2 XE CdTe 9 M hybrid pixel detector during ± 32° ω-rotation of the DAC in 0.5° steps. The patterns were then transferred into CrysAlisPro software using ESPERANTO protocol (Rothkirch et al. 2013) for indexing and integration. The structure refinement was performed at 27 pressure points using the computing system JANA2020 (Petříček et al. 2023). Due to the presence of two twin domains in the monoclinic phase on transformation from the trigonal LP-phase at P > 6 GPa, the structure was refined taking into account the twin axis along (110).
The structure at ambient pressure was refined using the atomic coordinates of synthetic bütschliite (Pabst 1974). For each subsequent pressure point, the structure refined at the previous point was used as a starting model. For the first pressure point (7.52 GPa) where the change to a monoclinic unit cell was detected, the structure was refined using the atomic coordinates of the high-pressure K2Mg(CO3)2 polymorph (space group C2/m, Golubkova et al. 2015) as a starting model. All structure refinements were performed using anisotropic displacement parameters for all atoms. The information on data collection and structure refinement (Online Resource 1) and the CIF-files for all pressure points (Online Resource 2) are attached to supplementary materials.
Additional in situ HP single-crystal diffraction measurements were performed at the Xpress beamline at the Elettra - Sincrotrone Trieste using ETH-type DAC (opening half-angle of 42o, an anvil culet diameter of 600 μm, 300 μm boreholes in steel gaskets). The DAC, loaded with the K2Ca(CO3)2 single-crystal fragment, ruby sphere as pressure sensor and Ne as pressure-transmitting medium, was sequentially pressurized from 5 to 10 GPa and backwards in several cycles, in order to evaluate more precisely the trigonal-to-monoclinic phase transition pressure and its possible hysteresis. Monochromatic X-ray diffraction measurements were performed at a wavelength of 0.4957 Å with a beam size of 50 × 50 μm. Diffraction patterns were collected using a Pilatus 6 M CdTe detector during ± 32° rotation of the DAC in 0.5° steps. The patterns were then transferred into CrysAlisPro software using ESPERANTO protocol (Rothkirch et al. 2013) for indexing and integration. The derived unit cell volume measured on the pressure rise and release is presented graphically in supplementary material (Online Resource 1).
Results
Compressibility of synthetic bütschliite up to 20 GPa
The HP evolution of the volume and lattice parameters of synthetic bütschliite, based on the data obtained at ESRF up to 20 GPa (Tables 1 and 2), is presented in Fig. 2. Up to 6.2 GPa bütschliite retains the initial R-3m symmetry. The data collected at P = 6.85 GPa can be indexed with both the trigonal and a monoclinic unit cell (space group C2/m). However, in the last case the agreement factors are much better (Rint(obs/all) = 1.29/1.39 versus 9.26/9.97 for the trigonal cell). More detailed measurements within the pressure range of 5–10 GPa (Online Resource 1) indicate the trigonal-to-monoclinic phase transition to proceed at 6.3(1) GPa with no appreciable hysteresis. The transition pressure is comparable to that of an analogous transition observed previously in K2Ca(CO3)2 between 5.5 and 6.0 GPa (Zeff et al. 2024). Our experimental findings on the lattice parameters of individual data points determined under hydrostatic conditions up to 21 GPa reveal no deviation from monoclinic lattice metrics and clearly excludes the occurrence of a triclinic HP-II phase as suggested by Tian et al. (2023).
The volume compression of synthetic bütschliite is rather monotonic within the whole pressure range (Fig. 2a). The fitting of the respective V-P data using the 2nd order Birch-Murnaghan equation of state (BM EoS) (Angel et al. 2014) gives: V0 = 455.3(4) Å3 (Z = 3) and 303.6(4) Å3 (Z = 2); K0 = 45.6(6) GPa (Online Resource 3). The fitting with the 3rd order BM EoS gives close parameters: V0 = 454.8(4) Å3 (Z = 3) and 303.2(4) Å3 (Z = 2); K0 = 48(1), K’ = 3.5(2) GPa (Online Resource 3). Since the K’ differs only slightly from the fixed K’ = 4, the 2nd order approximation can be acceptable to describe the volume compressibility of synthetic bütschliite within 20 GPa. A separate fitting of the V-P data for the LP- and HP-K2Ca(CO3)2 with the 2nd order BM EoS (Fig. 2a) provides a lesser K0 for the HP phase (44(2) GPa) compared to 48.8(8) GPa for the LP phase, whereas the V0 values are close to each other (LP V0 = 454.6(3) Å3, Z = 3; HP V0 = 304(3) Å3 for Z = 2 or 456(3) for Z = 3) and to the values obtained for the whole pressure range. The volume effect of the trigonal-to-monoclinic phase transition, estimated from the approximation of the EoS curves for the LP and HP phase, is 2(1) Å3, which comprises about − 0.5% relative to that of the LP phase.

Pressure dependencies of (a) the volume, (b) the unit-cell parameters for the trigonal LP-phase, (c) the unit-cell parameters for the monoclinic HP-phase, and (d) the monoclinic angle ß of the HP-phase of synthetic bütschliite K2Ca(CO3)2. Presented data correspond to the investigations on the sample compressed in helium. The volume data, normalized to Z = 3 and Z = 2 for the LP- and HP-phase in Fig. 2a, were fitted using a second-order Birch-Murnaghan EoS (Angel et al. 2014) (shown as solid black line). The error bars for the unit cell parameters of the LP-phase are within the size of the symbols
Despite the overall monotonic volume compressibility of synthetic bütschliite, the Eulerian finite strain plots as derived for the whole pressure range using the 2nd and 3rd BM EoS (Angel et al. 2014), reveal a more complex behavior for the trigonal and monoclinic phase (Online Resource 3). When fitted separately with the 3rd order BM EoS, both phases exhibit a distinct positive F-f trend.
The pressure dependencies of the lattice parameters for the LP-K2Ca(CO3)2 (Fig. 2b) show markedly anisotropic compression, the c direction being more compressible than the a direction, confirming the findings reported by Zeff et al. (2024). The compression anisotropy of the monoclinic phase (Fig. 2c) is less pronounced. Our data reveal minimal compressibility along the b axis and maximal along the a axis. Despite a rather large data scatter for the compression along c, one may note a slight bend on the c/P dependence at about 12 GPa. The pressure dependence of the monoclinic angle β (Fig. 2d) is rather regular but also slightly bends at 12–13 GPa, similarly to the data of Zeff et al. (2024).
Structure evolution at high pressure
The crystal structure of synthetic bütschliite is shown in Fig. 1, and the pressure dependences of its structural parameters (Tables 1 and 2) are presented in Figs. 3, 4, 5 and 6 and Online Resource 4.
The carbonate group
In agreement with the previous studies (e.g. Ross and Reeder 1992; Golubkova et al. 2015; Zeff et al. 2024), the carbonate group shows the most rigid behavior among all structure units, with the C-O bond distance getting reduced on average by just 0.1% up to 6 GPa. The lowering of the site symmetry of the CO32− group in the monoclinic HP phase at P > 7 GPa allows variations of the O-C-O angle (± 3o) and the individual C-O distances (± 0.03 Å), though the mean values remain constant on average (Online Resource 4). More importantly, the carbonate group is allowed to tilt relative to the a-b plane, off the direction corresponding to the former triad. The extent of tilting can be expressed by the angle between the bond C1-O1 and the a axis (corresponding to ∠ O1-C1-C1, Fig. 1c, d), in analogy to that proposed by Zeff et al. (2024). In the range of 7–20 GPa this angle increases remarkably from about 10o to 18o (Table 2), which also reflects the associated rotation of the CaO6-octahedron within the a-c plane, since it shares the joint atom O1 with the adjacent carbonate group (Fig. 1c, d).
The octahedral site
The pressure dependences of the bond distances (Fig. 3a) and volume of the Ca-octahedron in the bütschliite structure (Online Resource 4) are smooth in the whole pressure range studied, in agreement with the data of Zeff et al. (2024). The bulk compressibility of the Ca-octahedron, determined by the fitting of the volume data with the 2nd order Birch-Murnaghan EoS (Angel et al. 2014), is comparable in the structures of the HP and LP-phase (respective K0 are 59(5) and 65(3) GPa, Online Resource 4). In the HP phase the individual Ca-O bonds compress rather regularly at similar rates (Fig. 3a). However, the O-O distances reflecting the octahedron distortion exhibit more complex behavior.

Main interatomic distances and bond lengths of the octahedrally coordinated Ca in synthetic bütschliite K2Ca(CO3)2: pressure dependencies of (a) the cation-oxygen bond distances, (b) the O-O distances corresponding to the octahedral basal and lateral edges, and (c) and their normalized values. The trigonal-to-monoclinic phase transition pressure is marked by dash line
The evolution of the O-O distances within the Ca-octahedron is shown in Fig. 3b, c. In carbonates structures the shorter O-O octahedral edges lying within the cation layer are called basal O-O contacts, and those directed approximately perpendicular to the cation layer are referred to as lateral contacts (e.g. Ross and Reeder 1992). In the K2Ca(CO3)2 structure the octahedron face formed by the basal edges is shared with the K-polyhedron face (cf. Figure 1). The difference between the basal and lateral edge is remarkably large at ambient pressure, compared e.g. to that in dolomite structure (Online Resource 4). In the LP phase this difference is getting rapidly reduced with pressure through the predominant shortening of the lateral edge. Such behavior correlates somehow with the pressure dependence of the other parameters characterizing the octahedron distortion, quadratic elongation and bond angle variance (Robinson et al. 1971). Both these parameters, correlating with each other, decrease rapidly in the LP phase structure (Fig. 4a, b), so that the Ca-octahedron becomes almost regular (like in dolomite structure) on approaching the critical pressure of transition at 6 GPa.
The further deformation of the Ca-octahedron in the structure of monoclinic HP-K2Ca(CO3)2 proceeds smoothly, the distortion parameters remaining at the level achieved at 6 GPa. Notably, the basal O-O edge is almost incompressible in the range of 7–13 GPa (Fig. 3c), which makes the convergence between the basal and lateral O-O distances even more effective. Above 13 GPa the compression of the basal O-O edge is renewed with the rate similar to that of the lateral edge. In addition to smooth compression, the rotation of the Ca-octahedron proceeds within the a-c plane, so that it plunges into the K layer (Fig. 1c, d). As mentioned earlier, the octahedron rotation is characterized by the pressure dependence of the angle O1-C1-C1 (Table 2; Fig. 1c, d), which increases at a higher rate below 13 GPa and more slowly upon the further pressure rise (Fig. 4c).

The distortion of the octahedrally coordinated site in synthetic bütschliite K2Ca(CO3)2: pressure dependencies (a) of the quadratic elongation compared with those in dolomite (Ross and Reeder 1992) and (b) of the bond-angle variance. (c) The off-plane tilt angle O1-C1-C1 as a measure for the coupled rotation of carbonate groups and Ca-octahedra in the monoclinic HP-K2Ca(CO3)2. The trigonal-to-monoclinic phase transition pressure is marked by a vertical dash line
The K polyhedron
in the trigonal bütschliite structure can be regarded as a kind of truncated pyramid with sub-parallel base and top face formed by six and three O atoms, having short and long contacts with K, respectively (Online resource 4). The face formed by three oxygens is shared with the Ca-octahedron face, whereas the K-pyramid base contacts with the Ca-octahedra through shared edges. This determines different evolution of these six and three K-O bonds under pressure (Fig. 5).
In the LP-phase structure the set of three symmetry-equivalent K-O bonds is markedly more compressible compared to the six remaining K-O bonds (Fig. 5a). Nevertheless, the three K-O bonds remain appreciably larger than the six bond K-O below the transition (Fig. 5b). The transition to monoclinic phase is the origin of a significant diversification of the former three equivalent K-O bonds: two of them (K-O2) become the largest and one bond (K-O1) shortens significantly thus becoming comparable with one of the six K-O bonds (Fig. 5b, Online Resource 4). The former six equivalent K-O bonds retain similar values as before the transition (Fig. 5b). The compressibility of the mean K-O distance for the whole KO9-polyhedron is rather monotonic (Fig. 5a) and decreases in the HP-phase, which corresponds to the increase of the KO9-polyhedron bulk modulus from K0 = 37(1) GPa to 79(7) GPa. In addition to such a drastic stiffening, the K site exhibits a ∼2% volume expansion upon the transition (Fig. 6a); this is apparently due to the appreciable lengthening of the two K-O2 bonds out of the set of three symmetry-equivalent ones (Fig. 5b).

The bond lengths within the ninefold coordinated K site in synthetic bütschliite K2Ca(CO3)2: pressure evolution of (a) the normalized average K-O bond distances; (b) the individual K-O bond distances, and (c) and their normalized values. The trigonal-to-monoclinic phase transition pressure is marked by dash line
In addition to the nine O atoms forming the K coordination, in monoclinic structure the two of three O atoms of the adjacent CO32− group, as a result of its rotation, approach K at the distances of about 3.4 Å (Online Resource 4). This allows consider these two additional contacts within the K coordination (as 9 + 2), following the structure description given by Zeff et al. (2024). The pressure evolution of such a larger K polyhedron within the monoclinic structure differs from that of KO9, regarding the bulk compression (Fig. 6a) and the two parameters characterizing the geometry of cation site: the effective coordination number and the distortion index (Fig. 6b, c).
The effective coordination number (ECoN) is calculated as a weighted sum of the nearest atoms, the weight of an individual contact decreasing with its length (Hoppe 1979). For the KO9 polyhedron in the trigonal structure, this parameter slightly increases with the bond lengths shortening during the pressure rise up to 6 GPa (Fig. 6b). In monoclinic phase the change of the coordination number towards (9 + 2) exhibits a sudden increase of the ECoN of the K atoms, apparently due to a relatively quick shortening of the contacts with the new O atoms from the carbonate group (Fig. 6b; Online Resource 4). On the contrary, for the former KO9 polyhedron the effective coordination decreases, which can be attributed to the earlier described significant lengthening of the two K-O2 bonds (Fig. 5b). The further ECoN decrease in KO9 is apparently determined by a reduced compressibility of the K-O bonds, compared to those in the LP-phase.

Stereochemical and geometrical aspects on the KO9 (empty circles) and KO11 (filled circles) polyhedron and their evolution with pressure: (a) polyhedral volumes as a function of pressure; (b) the effective coordination number and (c) bond-length distortion index as a function of pressure. The polyhedral volumes were fitted using a second-order Birch-Murnaghan EoS (Angel et al. 2014) (shown as solid black line in Fig. 6a), the respective K0 values are indicated. The trigonal-to-monoclinic phase transition pressure is marked by dash line
The distortion index describes the variation of the bond lengths and angles with respect to an ideally regular polyhedron (Baur 1974). We used the program VESTA3 (Momma and Izumi 2011) where only the distortion of the bond lengths is calculated (Tables 1 and 2). Before the transition, the converging adjustment of the three and six K-O bonds towards more similar values (Fig. 5b) contributes to gradual decrease of the distortion index of KO9 on approaching the critical transition pressure. Due to the diversification into now symmetry-unconstrained individual K-O distances (Fig. 5b, c), the distortion index increases (Fig. 6c). After the transition the six bonds continue to diverge, which determines the further increase of this parameter. Compared to KO9, the KO11 polyhedron shows a much larger distortion at the moment of transition, but upon the further compression it becomes more regular rapidly, again, apparently due to a relatively quick shortening of the contacts with the new O atoms from the carbonate group (Online Resource 4). Notably, the KO11 bulk modulus K0 = 44(2) GPa is equal to the bulk compressibility of the whole structure (Fig. 2a).
Discussion
The role of cations in the compressibility of synthetic bütschliite
The obtained data on the axial compressibility of the LP-K2Ca(CO3)2 are consistent with the previously observed compression anisotropy of trigonal carbonates, related with the orientation of the rigid planar CO32− groups parallel to the (001) plane (e.g. Ross and Reeder 1992). The dependence of the compression along the c axis on the cation type is demonstrated by a higher axial compressibility of the LP-K2Ca(CO3)2 compared to the LP-K2Mg(CO3)2 (Golubkova et al. 2015; Online Resource 3), which is in perfect agreement with the data of Zeff et al. (2024). In the monoclinic structure the compression anisotropy is getting modified due to the remarkable inclination of the planar CO32− groups relative to the plane (001) (Fig. 1b). As a result of the tilt around the b axis, the carbonate groups remain parallel to only the b axis of the monoclinic cell, which is also the stiffest direction in both the HP-K2Ca(CO3)2 and K2Mg(CO3)2-II structures (Fig. 2c, Online Resource 3).
Regarding the bulk compressibility, the obtained results fit the general inverse relation between the cation size and the bulk modulus established for the alkali-bearing double carbonates (Golubkova et al. 2015; Hou et al. 2022; Zeff et al. 2024). Indeed, the average bulk modulus of K2Ca(CO3)2 of ∼46 GPa is smaller compared to that of K2Mg(CO3)2 of ∼58 GPa (Golubkova et al. 2015; Online Resource 3), in agreement with the presence of a larger and more compressible Ca cation instead of Mg. Notably, for both the LP trigonal and HP monoclinic polymorphs of K-bearing double carbonates a majority of the reported K0 values lies within a relatively narrow range of about 50–70 GPa (Golubkova et al. 2015; Hou et al. 2022). In this respect, the K0 = 24.8(44) GPa reported for the HP monoclinic K2Ca(CO3)2 by Zeff et al. (2024) seems to deviate from the main trend. The reason for such a discrepancy with, for example, our data (Online Resource 3) lies most probably in the pressure media with different hydrostatic properties (neon and silicon oil) used by Zeff et al. (2024). Notably, within the pressure range up to 6 GPa, where the deviation from hydrostatic conditions in silicon oil is expected to be not large (Klotz et al. 2009), both sets of the experimental points coincide, and the respective K0 values for trigonal K2Ca(CO3)2 are similar (Online Resource 3; Zeff et al. 2024).
It is worthy to compare the contribution of the cation polyhedra to the bulk compressibility of synthetic bütschliite. The bulk modulus of the LP trigonal phase (∼49 GPa) is roughly intermediate between the polyhedral moduli of the structure units CaO6 (65 GPa) and KO9 (37 GPa) (Figs. 2a and 6a; Online Resource 4); this reflects an almost equal contribution of the net contraction of these polyhedra into the compression of the whole structure. The bulk modulus of the HP monoclinic phase (44 GPa), as mentioned above, is equal to the polyhedral modulus for KO11. It is a natural consequence of the expansion of the K coordination towards a larger part of structure. In addition, in the HP structure the Ca-layer compresses rather through the rotation than pure contraction of the octahedral sites, which diminishes the contribution of the octahedral K0 into the bulk compression of the HP-K2Ca(CO3)2.
The volume contraction across the phase transition is very low, according to the previous data (∼2% per formula unit, Zeff et al. 2024) and our results (∼0.5%). Nevertheless, the transition is classified as a first order, based on a discontinuous character of the volume change and a pronounced hysteresis (Zeff et al. 2024). The revealed jump-like volume expansion of the K-polyhedron (Fig. 6a), constituting the main part of the structure, seems indeed compatible with the 1st order transition. The view of the HP phase structure as composed of the KO11 (instead of KO9) polyhedra is surely more reasonable, taking into account the above-mentioned identity of the bulk moduli of the K site and the whole structure.
The structure evolution up to the transition to monoclinic K2Ca(CO3)2
The obtained structural-chemical data allow to characterize the K2Ca(CO3)2 compression mechanism not only through the evolution of the interatomic distances, similarly to the work of Zeff et al. (2024), but also in terms of the relative deformations and interaction of the structure polyhedra. Within the pressure range up to 6 GPa, the compressibility of the trigonal LP-structure is dominated by the net polyhedral compression. This originates from the low degree of independent symmetry-unconstrained possibilities of atomic displacements. The bulk compressibility is anisotropic, with both the Ca-octahedron and K site compressing more strongly along the direction close to the c axis (Figs. 3b and 5b). This results in the less diversificated lengths of differently oriented bonds and more regular shapes of these polyhedra (Figs. 3, 4a and b, 5b and 6c).
The transition to the C2/m polymorph produces the enlargement and a marked distortion of the K-polyhedron (Fig. 6a, c), whereas the Ca-octahedron remains regular (Fig. 4a, b). The distortion of the K site is determined by the adjunction of the two distant contacts with the O atoms of carbonate group, as well as the differentiation of the three bonds with the O atoms forming the shared face with the Ca-octahedron (Online Resource 4; Fig. 5b). Such differentiation, in its turn, is related to the rotation and indentation of the Ca-octahedron face into the K-polyhedron, coupled with the rotation of planar (CO3)2− group (Fig. 1c, d). Such rotation can be regarded as a mean to allow further counter movement of the cation layers without shortening of the K-C distances.
The compression mechanism of monoclinic K2Ca(CO3)2
The further structure compression involves, apart from the polyhedral compression, gradual tilting/rotation and the Ca-octahedron getting increasingly pressed into the layer of interconnected K polyhedra. The deviation trends on the pressure dependences for the octahedral basal O-O distance and tilt angle at about 13 GPa (Figs. 3c and 4c) imply the presence of two slightly different compression regimes.
Within the pressure range of 7–13 GPa, the rotation of the Ca-octahedron proceeds to a higher extent; as a result, its axis is getting oriented closer to the c axis, thereby contributing to the increase of the octahedral layer thickness along the c direction (Fig. 1c, d). The shared face between the Ca- and K- polyhedra becomes a very rigid contact, taking into account the low compressibility of the affected three K-O bonds directed towards this face (Fig. 5a). Note that the area of this shared face remains nearly constant, because the basal O-O edge is almost incompressible within this pressure range (Fig. 3c). The change of the octahedron orientation through getting tilted, along with the rigidity of the K-O triad bonds oriented close to the c direction, may contribute to a relatively low directional compressibility along the crystallographic c axis (Fig. 2c).
Above 13 GPa, the octahedral tilting proceeds at a slower rate (Fig. 4c), and the structure compresses mainly through the polyhedral compaction associated with the increasing distortion of the K site (Fig. 6c). Hence, the lattice compressibility along the c axis slightly increases (Fig. 2c). The indentation of the Ca-octahedron face into the K-polyhedron induces re-orientation of the K-O bonds, mainly the set of three ones, and makes the K layer getting crimped (Fig. 1c).
These different compression regimes apparently manifest also in a subtle deviation of the pressure dependency of the monoclinic angle β at 12–13 GPa (Fig. 2d) from a linear fashion, very similar to that observed by Zeff et al. (2024). As noted by the authors, the increase of the β-angle characterizes the relative lateral shifting of the cation layers. It appears that the octahedral tilting contributes much to such shift, and the mentioned variation of the tilt rate (Fig. 4c) can indeed affect a slower growth of the β-angle above 13 GPa (Fig. 2d).
It should be noted that the described mechanism of lateral shifting through the octahedral tilt, coupled with the rotation of carbonate groups, mostly refers to the “softening” of the octahedral layer. At that, if the KO11coordination is considered, the octahedral layer is no more separated, but it turns fully merged into the K layer (Fig. 1b). This can explain the mentioned identity of the bulk modulii of the KO11 polyhedron and the whole structure in the HP phase.
The relative compressibility of structure units in alkali-earth and alkali-bearing carbonates
The bulk compressibility of alkali-earth and alkali-bearing carbonates demonstrates a roughly linear correlation with an average radius of a charge-balancing cation, as it is summarized in the works of Golubkova et al. (2015), Hou et al. (2022) and Zeff et al. (2024). At that, however, the individual cations in double carbonates can have drastically different compressibility even with the same coordination number. This depends on the structure type, which in turn is related to a counterpart cation. For example, octahedrally coordinated Ca and Mg in the LP-K2Ca(CO3)2 and K2Mg(CO3)2-I are much more compressible compared to those in dolomite (Online Resource 4); namely, the CaO6 bulk modulus in the LP-K2Ca(CO3)2 is 65 GPa compared to 91 GPa in dolomite (Ross and Reeder 1992). It appears that there is mutual influence of the both types of cations, and the presence of large and relatively “soft” alkali cation destabilizes more rigid octahedral Mg and Ca cations. The reverse example is given by the pair Ca-Mg. In double Ca-Mg carbonates the octahedral sites, occupied by Ca, tend to be more distorted under pressure than those containing Mg. The absence of displacive phase transitions in dolomite within 5 GPa, in contrast to calcite (Merrill and Bassett 1975), is attributed to the stabilization of the Ca-octahedra and the whole structure by the presence of Mg (Ross and Reeder 1992). These examples show that the cation radius itself may be not indicative in terms of the expected compressibility; notably, the Ca-O distance in more rigid Ca-octahedron in dolomite is about 5% larger compared to that for the CaO6 unit in the LP-K2Ca(CO3)2 (Online Resource 4).
Another factor which can influence the cation compressibility is the packing density, as it is shown for the middle-sized cations such as Ca and Mg (e.g. Ross and Reeder 1992). Regarding the large alkali cations, there is apparently no definite relation between their compressibility and this parameter. In Na-Ca carbonates the more tightly packed Na-polyhedra can be more compressible than those forming the less compact building units. For example, corner-sharing 8-fold Na polyhedra in shortite, Na2Ca2(CO3)3, have the bulk modulus comparable to that of the Ca-octahedron in K2Ca(CO3)2 (60–65 GPa), whereas the face-shared (Na, Ca)O8-units in burbankite Na3Ca2La(CO3)5 have the bulk modulus of 54(2) GPa (Vennari et al. 2018; Milani et al. 2022). Such “erratic” behavior is probably a consequence of a wide diversity of polyhedral distortions conditioned by a large size of the alkali cations.
Based on the available HP structural data, it seems probable that a more distorted coordination of alkali cation can be more stable under pressure. Notably, the shift of trigonal to a lower-symmetry phase transition in Na2Mg(CO3)2 to a higher pressure of about 14 GPa (compared to 8 GPa for K2Mg(CO3)2) is attributed to the presence of distorted NaO9-polyhedron in the initial structure of Na2Mg(CO3)2 (Golubkova et al. 2015). Another example is the structure of HP-K2Ca(CO3)2 with the KO11 polyhedron having a higher distortion index and the bulk modulus compared to those of the KO9 in the LP trigonal phase (Fig. 6a, c).
In contrast to the HP conditions, the temperature rise commonly leads to more symmetrical atomic arrangements and coordination. In this regard, the increase of the baric stability of the trigonal LP-K2Ca(CO3)2 upon the temperature rise (Likhacheva et al. 2024) can be interpreted based on the obtained structural data. On approaching the critical pressure of transition at 6 GPa, the shape of the Ca and K polyhedron becomes almost regular (Figs. 4a and b and 6c). Along with a higher crystal symmetry, compared to the monoclinic HP phase, this can favor the stabilization of the trigonal K2Ca(CO3)2 under temperature rise through the shift of the transition pressure from ∼6 to ∼8 GPa (Likhacheva et al. 2024).
Conclusions
The HP single-crystal investigations on synthetic bütschliite K2Ca(CO3)2 provide new details of the compression mechanism, which permit to relate the evolution of the structure units and the modulation of the axial compression, as well as to estimate the contribution of the cation polyhedra into the bulk compression. The obtained data show a similar bulk compressibility of the trigonal and monoclinic polymorphs of K2Ca(CO3)2, in agreement with the previous calculations (Hou et al. 2021) and experimental data on K-bearing double carbonates Golubkova et al. (2015), summarized in the work of Zeff et al. (2024). In particular, the bulk modulus K0 = 44 GPa of the HP-K2Ca(CO3)2 fits to the dependence of the carbonate bulk modulus versus average non-carbon cation radius (Zeff et al. 2024).
The comparison of the obtained and literature structural data on the double alkali-bearing carbonates suggests the distortion of a large cation polyhedron to be an effective tool to strengthen the carbonate structure at high pressure. On the other hand, the observed symmetrization of the structure units in the LP trigonal bütschliite is apparently a factor of its stabilization at high pressure upon the temperature rise, found previously (Likhacheva et al. 2024). Such conclusion contradicts the previous suggestion about the primary crystallization of bütschliite as the C2/m phase in the inclusions in deep-formed diamonds or during the equilibrium experiments in alkali carbonate systems (Zeff et al. 2024). The structural data of this study support the stability of trigonal bütschliite at high P, T conditions and, therefore, its significance as a constituent of the inclusions in deep minerals and respective carbonatite systems. This implies the use of the EoS parameters for trigonal K2Ca(CO3)2 to reconstruct the P-T history of the host minerals containing carbonate inclusions.
Data availability
Data are provided within the manuscript and supplementary information files.
References
Abersteiner A, Golovin A, Chayka I, Kamenetsky VS, Goemann K, Rodemann T, Ehrig K (2022) Carbon compounds in the West Kimberley lamproites (Australia): insights from melt and fluid inclusions. Gondwana Res 109:536–557. https://doi.org/10.1016/j.gr.2022.06
Angel RJ, Gonzalez-Platas J, Alvaro M (2014) EosFit-7c and a Fortran module (library) for equation of state calculations. Z Kristallogr 229:405–419. https://doi.org/10.1515/zkri-2013-1711
Arceo HB, Glasser FP (1995) Fluxing reactions of sulfates and carbonates in cement clinkering II. The system CaCO3–K2CO3. Cem Concrete Res 25(2):339–344. https://doi.org/10.1016/0008-8846(95)00019-4
Arefiev AV, Shatskiy A, Podborodnikov IV, Rashchenko SV, Chanyshev AD, Litasov KD (2019) The system K2CO3–CaCO3 at 3 GPa: link between phase relations and variety of K–Ca double carbonates at ≤ 0.1 and 6 GPa. Phys Chem Min 46:229–244. https://doi.org/10.1007/s00269-018-1000-z
Arefiev A, Shatskiy A, Bekhtenova A, Litasov K (2022a) Raman study of quench products of alkaline carbonate melt at 3 and 6 GPa: link to the pressure of origin. J Raman Spectr 53(12):2110–2122. https://doi.org/10.1002/jrs.6438
Arefiev AV, Shatskiy A, Bekhtenova A, Litasov KD (2022b) Quench products of K-Ca-Mg carbonate melt at 3 and 6 GPa: implications for Carbonatite inclusions in Mantle minerals. Minerals 12(9):1077. https://doi.org/10.3390/min12091077
Baur WH (1974) The geometry of Polyhedral distortions. Predictive relationships for the Phosphate Group. Acta Cryst B Struct Sci 30(5):1195–1215
Cooper AF, Gittins J, Tuttle OF (1975) The system Na2CO3–K2CO3–CaCO3 at 1 kilobar and its significance in carbonatite petrogenesis. Am J Sci 275(5):534–560
Dasgupta R, Hirschmann MM (2010) The deep carbon cycle and melting in Earth’s interior. Earth Planet Sci Lett 298(1–2):1–13. https://doi.org/10.1016/j.epsl.2010.06.039
Giuliani A, Kamenetsky VS, Phillips D, Kendrick MA, Wyatt BA, Goemann K (2012) Nature of Alkali-Carbonate fluids in the Sub-continental Lithospheric Mantle. Geology 40(11):967–970. https://doi.org/10.1130/G33221.1
Golubkova A, Merlini M, Schmidt MW (2015) Crystal structure, high-pressure, and high-temperature behavior of carbonates in the K2Mg(CO3)2–Na2Mg(CO3)2 join. Am Min 100(11–12):2458–2467. https://doi.org/10.2138/am-2015-5219
Grassi D, Schmidt MW (2011) The melting of Carbonated pelites from 70 to 700 km depth. J Petrol 52(4):765–789. https://doi.org/10.1093/petrology/egr002
Hoppe R (1979) Effective coordination numbers (ECoN) and mean fictive ionic radii (MEFIR). Zeit Krist - Cryst Mater 150:1–4
Hou B, Huang S, Qin S (2022) Phase transitions and compressibility of alkali-bearing double carbonates at high pressures: a first-principles calculations study. Phys Chem Min 49:34. https://doi.org/10.1007/s00269-022-01210-9
Jablon BM, Navon O (2016) Most diamonds were created equal. Earth Planet Sci Lett 443:41–47. https://doi.org/10.1016/j.epsl.2016.03.013
Klotz S, Chervin J-C, Munsch P, Le Marchand G (2009) Hydrostatic limits of 11 pressure transmitting media. J Phys D Appl Phys 42(7):075413. https://doi.org/10.1088/0022-3727/42/7/075413
Likhacheva AYu, Miloš S, Romanenko AV, Goryainov SV, Semerikova AI, Rashchenko SV, Miletich R, Shatsky A (2024) High-pressure behavior and stability of synthetic buetschliite K2Ca(CO3)2 up to 19 GPa and 300 oC. J Raman Spectr 55:517–524. https://doi.org/10.1002/jrs.6654
Litasov KD, Safonov OG, Ohtani E (2010) Origin of Cl-bearing silica-rich melt inclusions in diamonds: experimental evidence for an eclogite connection. Geology 38:1131–1134. https://doi.org/10.1130/G31325.1
Logvinova AM, Shatskiy A, Wirth R, Tomilenko AA, Ugap’eva SS, Sobolev NV (2019) Carbonatite melt in type Ia gem diamond. Lithos 342–343:463–467. https://doi.org/10.1016/j.lithos.2019.06.010
Merlini M, Hafland M (2013) Single-crystal diffraction at megabar conditions by synchrotron radiation. High Press Res 33:511–522. https://doi.org/10.1080/08957959.2013.831088
Merrill L, Bassett WA (1975) The crystal structure of CaCO3 (II), a high-pressure metastable phase of calcium carbonate. Acta Cryst B 3l:343–349
Milani S, Sparta D, Fumagalli P, Joseph B, Borghes R, Chend V, Maurice J, Bais G, Merlini M (2022) High-pressure and high-temperature structure and equation of state of Na3Ca2La(CO3)5 burbankite. Eur J Mineral 34351–358. https://doi.org/10.5194/ejm-34-351-2022
Momma K, Izumi F (2011) VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J Appl Crystallogr 44:1272–1276. https://doi.org/10.1107/S0021889811038970
Pabst A (1974) Synthesis, properties, and structure of K2Ca(CO3)2, bütschliite. Am Min 59(3–4):353–358
Petříček V, Palatinus L, Plášil J, Dušek M (2023) Jana2020 – a new version of the crystallographic computing system Jana. Zeit Krist - Cryst Mater 238(7–8):271–282. https://doi.org/10.1515/zkri-2023-0005
Robinson K, Gibbs GV, Ribbe PH (1971) Quadratic elongation: a quantitative measure of distortion in coordination polyhedra. Science 172:567–557
Ross NL, Reeder RJ (1992) High-pressure structural study of dolomite and ankerite. Am Min 77:412–421. https://api.semanticscholar.org/CorpusID:52241729
Rothkirch A, Gatta GD, Meyer M, Merkel S, Merlini M, Liermann H-P (2013) Single-crystal diffraction at the Extreme conditions beamline P02.2: procedure for collecting and analyzing high-pressure single-crystal data. J Synchrotron Rad 20:711–720. https://doi.org/10.1107/S0909049513018621
Shatskiy A, Borzdov YM, Litasov KD, Sharygin IS, Palyanov YN, Ohtani E (2015) Phase relationships in the system K2CO3–CaCO3 at 6 GPa and 900–1450°C. Am Min 100(1):223–232. https://doi.org/10.2138/am-2015-5001
Shatskiy A, Arefiev A, Litasov KD (2023) Change in carbonate budget and composition during subduction below metal saturation boundary. Geosci Front 14:101463. https://doi.org/10.1016/j.gsf.2022.101463
Shen G, Wang Y, Dewaele A, Wu C, Fratanduono DE, Eggert J, Klotz S, Dziubek KF, Loubeyre P, Fat’yanov OV, Asimow PD, Wentzcovitch RMM, other members of the IPPS task group (2020) Toward an international practical pressure scale: a proposal for an IPPS Ruby Gauge (IPPS-Ruby2020). High Press Res 40(3):299–314. https://doi.org/10.1080/08957959.2020.1791107
Tian H, Li Y, Zhang Q, Ningning S, Sun J, Xiao N, Liu Y, Zhang J, Liu H, Li Y (2023) Pressure-dependent compressibility of K2X(CO3)2 (X = ca, mg) under high pressure. Mater Lett 349:134895. https://doi.org/10.1016/j.matlet.2023.134895
Vennari CE, Beavers CM, Williams Q (2018) High-Pressure/Temperature behavior of the Alkali/Calcium Carbonate Shortite (Na2Ca2(CO3)3): implications for Carbon Sequestration in Earth’s Transition Zone. J Geophys Res: Solid Earth 123(8):6574–6591. https://doi.org/10.1029/2018JB015846
Yaxley GM, Brey GP (2004) Phase relations of carbonate-bearing eclogite assemblages from 2.5 to 5.5 GPa: implications for petrogenesis of carbonatites. Contrib Mineral Petrol 146(5):606–619. https://doi.org/10.1007/s00410-003-0517-3
Zeff G, Kalkan B, Armstrong K, Kunz M, Williams Q (2024) High pressure raman spectroscopy and X-ray diffraction of K2Ca(CO3)2 bütschliite: multiple pressure-induced phase transitions in a double carbonate. Phys Chem Min 51:2. https://doi.org/10.1007/s00269-023-01262-5
Acknowledgements
We appreciate profound and constructive criticisms of anonymous reviewers, which helped to improve the manuscript. This work is financially supported by the joint RFBR-FWF grant project (# 21-55-14001 and I 5046-N) and performed on state assignment of IGM SB RAS (No. 122041400176-0). We acknowledge the European Synchrotron Radiation Facility (ESRF) for the provision of synchrotron beamtime (proposal ES-1061) and we would like to thank Prof. Michael Hanfland and Dr. Davide Comboni for assistance and support in using beamline ID015b. The Elettra Sincrotrone Trieste is also acknowledged for provision of beamtime (exp. 20220403). We are grateful to Dr. Boby Joseph for assistance in using beamline Xpress.
Funding
AL, AR, SR and AS received financial support from RFBR (grant # 21-55-14001); AS was also supported by the state assignment of GEOKHI RAS; SM and RM received financial support from FWF (grant # I 5046-N).
Author information
Authors and Affiliations
Contributions
A.L., R.M.and A.S. wrote the main manuscript text; A.L. prepared all figures; A.L., S.R., A.R., S.M., R.M. and P.L. performed the experiment; A.R. refined the structure; A.S. synthesized the sample; All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Likhacheva, A.Y., Romanenko, A.V., Rashchenko, S.V. et al. Crystallographic mechanism of the elastic behaviour of synthetic bütschliite K2Ca(CO3)2 on compression to 20 GPa. Phys Chem Minerals 51, 29 (2024). https://doi.org/10.1007/s00269-024-01291-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-024-01291-8