
Abstract
The structural parameters and the thermal behavior of a complete series of Ca–Mg carbonates synthesized at high pressure and temperature (1–1.5 GPa, 1273–1373 K) in the range 0–50 mol% MgCO3 have been investigated by in situ powder synchrotron high-resolution X-ray diffraction at ambient and up to 1073 K under self-controlled CO2 partial pressure. The crystal structures are disordered Mg calcite in the range 1–41 mol% MgCO3, and Ca dolomite at 49 mol% MgCO3. New calibration curves of the cell parameters for the Mg content and thermal expansion from ambient to 1073 K are given. Short-range structural effects of cation substitution and ordering and their thermal behavior as a function of Mg content were identified from three sets of data: the peak broadening, the cell parameter strains and the Raman band enlargements. Both intra- and inter-crystalline levels of compositional heterogeneity are identified and allow splitting the Mg calcites into two groups: low- and high-Mg calcites. The low-Mg calcites (up to 22 mol% MgCO3) are homogeneous in Mg content with short-range ordering. High-Mg calcite (up to 41 mol% MgCO3) displays domains with different local ordering configurations and similar or slightly different Mg contents, and to which is added a compositional variation between crystals, as determined by EMP, of the order of ± 0.8 mol% MgCO3. The cation ordering in Ca-rich dolomites similarly occurs in high-Mg synthetic calcites. The role of (CO3)2− group ordering is shown to be an important factor in the formation of Ca–Mg carbonates.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among all carbonate systems, Ca–Mg carbonates on the calcite–dolomite join are the most studied due to their importance as geological and industrial materials. Low- to high-Mg calcites are main components of many biominerals, whereas Ca-rich dolomites with Ca excess up to 6 mol% occur in sedimentary rocks of diverse environments and ages. Most natural carbonates present morphology, structure, composition, stoichiometry, and/or cation ordering deviations from the ideal crystalline structures of calcite and dolomite (Deelman 2011; Rodriguez-Navarro et al. 2013). Such features may contain information about crystal growth. They may also reflect environmental factors that control Mg calcite and Ca dolomite formation and transformation in biominerals and sedimentary rocks.
Optically clear natural (OCN) calcite, dolomite and synthetic crystals on the calcite–dolomite join are commonly used as standards to study the physicochemical and structural properties of natural carbonates. Synthesized Mg calcites were produced either at high pressure and temperature from solid mixtures (Bischoff et al. 1983; Goldsmith et al. 1958, 1961; Jenkins et al. 2018) or by crystallization in aqueous solutions under ambient pressure and temperature (Glover and Sippel 1967) and references therein, (Zhang et al. 2010). As a reference work, Bischoff et al. (1983) synthesized up to 24 mol% MgCO3 Mg calcites at high pressure (2, 10 and 15 kbar) and temperature (973 K or 1273 K), and characterized the products by XRD and μRaman spectroscopy. The Mg calcites are reported as fully disordered calcite structures and display a nonlinear, but smooth, variation of unit cell parameters and volume with composition up to about 20 mol% MgCO3. Bischoff et al. (1983) reported negative excess volumes changing from negative to positive at 20 mol% MgCO3 (Fig. 1). Zhang et al. (2010) indicate that the cell parameters of Mg calcites synthesized in aqueous solutions deviate from a simple linear trend above 28 mol% MgCO3 (Fig. 1). Zhang et al. (2010) considered that this non-linear change of structural parameters is related to partially ordered arrangements of Mg and Ca and twinning which are well known for compositions close to dolomite (Reeder 1992; Rodriguez-Navarro et al. 2013; Wenk et al. 1983 and references therein). Thus, the relationship between Ca and Mg structural ordering observed by transmission electron microscopy (Fang and Xu 2019; Larsson and Christy 2008; Meike et al. 1988; Reksten 1990; Shen et al. 2014; Van Tendeloo et al. 1985; Wenk et al. 1991) and the cell parameter variations as a function of Mg content are still a matter of debate and require new structural studies of high-quality Mg calcites of known composition. Further evidence of structural ordering (or not) can be obtained by studying the evolution of the crystal structure with temperature through in situ heating experiments. An interesting relationship between structure and physical property is observed in carbonates in terms of thermal expansion and its relationship with anion group liberation and cation octahedral composition (Reeder and Markgraf 1986; Wang et al. 2018). Indeed, calcite displays negative and positive thermal expansions of a and c cell parameters, respectively, whereas dolomite and magnesite show positive, although quite anisotropic, thermal expansions of both a and c parameters. Noting this fact, Markgraf and Reeder (1985) wondered about the thermal expansion of the a cell parameter in magnesian calcites, and more specifically about the Ca:Mg ratio at which the thermal expansion of the a cell parameter changes from negative to positive.

a, c and V parameters of synthetic calcites according to previous studies. Note the discrepancies above 20 mol% MgCO3. Black triangles: Goldsmith et al (1961); Blue stars: Erenburg (1963); blue crosses: Zhang et al (2010); red squares: Bischoff et al (1983). The line in the V–X diagram corresponds to the calibration curve of Jenkins (2018): V (Å3) = 366.788–0.9326 × mol% MgCO3
The present study is a follow-up from a previous one on the same synthetic carbonate samples, already published by our group. The first study is a comprehensive characterization by μRaman spectroscopy giving accurate data (both in wavelength position and full width at half maximum (FWHM) peaks of carbonate group vibration bands) on the local ordering of the carbonate groups along the calcite–dolomite join (Perrin et al. 2016). The aims of the present study are (1) to analyze the powder diffractograms obtained by synchrotron high-resolution X-ray powder diffraction (HRXRPD), determine the cell parameters and identify the structural short- and (or) long-range ordering (if any) of Ca–Mg carbonates in the range 0–50 mol% MgCO3; (2) characterize in situ the evolution with temperature of the crystal structure of selected Mg calcites and determine their thermal expansion up to 1073 K under self-controlled CO2 partial pressure.
Materials and methods
Experimental syntheses
The samples were synthesized at high pressure and high temperature in a piston–cylinder apparatus (Perrin et al. 2016). The Mg calcites cover the range 2–50 mol% MgCO3 (2 and 5 mol% step intervals between 2 and 30 and 30 and 50 mol% MgCO3, respectively). The starting materials consist of pure synthetic calcite (Alfa Aesar) and natural magnesite from Brumado (Bahia, Brazil). After desiccation in an oven at 383 K, about 60 mg of the mixture were loaded in a 7-mm-long by 5-mm-outer diameter gold capsule and welded shut. The gold capsule was placed in a salt-glass assembly pressurized and heated for ~ 96 h in the piston–cylinder apparatus, at 1273 K and 1 GPa for compositions in the range 2–30 mol% MgCO3. For compositions in the range 35–50 mol% MgCO3, higher pressure and temperature were selected (1373 K and 1.5 GPa) to avoid exsolution in the calcite–dolomite solvus as determined by Goldsmith et al. (1961). Large thick-walled capsules yielded sufficient quantity of crystalline material in a single experiment to carry out different series of analyses while long run durations (~ 4 days) favored attainment of textural and thermodynamic equilibrium. All runs were quenched by the same procedure at high pressures, by cutting off the electrical power. The quenching time was about 20 s from 1273 to 573 K and 5 min to room temperature. After the experiment, the run product was extracted from the capsule, part of it was mounted in epoxy and polished for electron microprobe (EMP), scanning electron microscopy (SEM) and µRaman analyses (Perrin et al. 2016), while the remaining was ground and kept for X-ray diffraction (XRD). OCN calcite, dolomite, and magnesite crystals were also analyzed to serve as references. OCN calcite comes from a perfect rhombohedron of unknown origin. OCN dolomite and OCN magnesite come from Azcárate quarry, Eugui, Esteríbar (Spain), and Brumado, Bahia (Brazil), respectively. Note that the number associated with the sample name (e.g. MgCc2, MgCc50) refers to the nominal composition in mol% MgCO3 in the starting material. The actual concentrations measured by EMP are given in Table 1.
Analytical methods
Synchrotron high-resolution X-ray powder diffraction
HRXRPD of the samples was performed at the ID22 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). The instrument is equipped with a double-crystal monochromator Si(111) and crystal analyzer optical elements in the incident and diffracted beams, respectively. A description of the diffraction instrument is given by Fitch (2004). The setup allows high-quality powder diffraction patterns with high signal/noise ratio, combined with narrow peaks, accurate positions and intensities. The instrumental contribution to peak width (FWHM) does not exceed 0.003° 2θ. The typical resolution (Δd/d) is ∼ 10−4. Incident beam size on the sample is typically 1.5 mm × 1.5 mm. The selected wavelength λ = 0.41068(1) Å (30 keV) was calibrated with Si standard NIST 640c (certified cell parameter a = 5.4311946 (92) Å). One-millimeter-outer diameter quartz capillaries were filled with sample powder, then sealed, mounted horizontally, and spun at 1000 rpm during data collection to improve particle statistics. Data were collected in the 1° < 2θ < 45° range, in continuous motion 5°/min, and rebinned in 2θ step of 0.003°. Data acquisition time was 30 min for each sample at room temperature (RT) to ensure good counting statistics. For in situ heating measurements, the capillaries were heated with a hot air blower mounted vertically, perpendicular to the capillary. Temperature was controlled by a thermocouple located in the hot air stream. The measurements (three recording patterns of 10 min at each T) were performed at RT, then in steps of 60 K from 353 to 1073 K (heating rate of 10 K/min) and again at room temperature after cooling.
HRXRPD pattern analysis
Cell parameters
Both whole powder pattern and single peak analyses were performed with the Fullprof software suite (Rodriguez-Carvajal 1993). In the Rietveld refinements, the diffraction peak profiles were fitted with a pseudo-Voigt profile function and corrected for the asymmetry due to axial divergence (Rietveld 1969). All samples were refined in the rhombohedral system using the \( R\overline{3}c\) space group for calcite, Mg calcite and magnesite, and the \(R\overline{3}\) space group for dolomite and Ca-dolomite. Starting atomic coordinates, cell parameters, and isotropic temperature factors of calcite and magnesite are taken from Maslen et al. (1995) while those of dolomite and Mg calcite are from Althoff (1977). The atomic composition of each powder sample was determined by EMP with a mean value and a standard deviation. Consequently, (1) the random occupancy of Ca and Mg into octahedral sites for Mg calcite samples was not refined but constrained to the EMP composition, (2) the CO3 group was considered as a rigid structural unit. The C–O bond length was previously found to be very close to 1.284 Å for several carbonate phases, including magnesite and otavite (Reeder 1983; Zemann 1981). The C–O bond length was fixed as a function of Mg content (nMg), between the C–O bond lengths in calcite (nMg = 0, d(C–O)calcite = 1.2864 Å) and in dolomite (nMg = 0.5, d(C–O)dolomite = 1.2831 Å). It was constrained according to the linear relation d(C–O)MgCc = -0.000068 × nMg + 1.2864 (with nMg: mol% MgCO3 determined by EMP reported in Table 1). The background function, scale factors, unit-cell parameters, Gaussian and Lorentzian-broadening coefficients of the pseudo-Voigt profile function, and March-Dollase preferred orientation parameters were refined for each entire diffractogram. Atomic positions were refined only for OCN calcite, dolomite and magnesite. In the synthetic Mg calcites, where oxygen position and Ca and Mg occupations were constrained, typical agreement factors, χ2, Rp, and Rwp ranged from 3 to 13, 0.08 to 0.10, and 0.12 to 0.15, respectively.
Long-range and short-range atom ordering
The degree of order is of primary importance in the calcite–dolomite–magnesite system. As summarized by Putnis (1992), the degree of order involves two different concepts.
Long-range order involves the occupancies of the sites averaged over the complete crystal. It is defined in terms of relative occupancy of non-equivalent sites. In the case of dolomite, the disordered state occurs on sites of two distinct layers, Ca layers labelled α and Mg layers labelled β. The degree of cation order is described by the order parameter s defined as s = (nCa,α − nCa,β)/(nCa,α + nCa,β), where nCa,α and nCa,β are the occupancy of Ca-type cations in Ca-type layers and Mg-type layers, respectively. The long-range order parameter equals 0 in the fully disordered state and 1 in the fully ordered state. Other equivalent formulations for the s order parameter were found for stoichiometric dolomites: s = − 1 + 2 × nCa,α (Reeder and Wenk 1983) and for Ca-rich dolomite s = (2 × nCa,α − nCa,β)/(2 − nCa,α − nCa,β), where nCa,α and nCa,β are the occupancy of Ca-type cations in Ca-type layers and in Mg-type layers, respectively (Reeder 2000).
When the occupancy of Ca-type cations could not be refined, it is usual to calculate the long-range order parameter in dolomite structure from the intensity of b-type peaks. In the dolomite diffractograms, the periodic layer arrangement of the non-equivalent sites gives rise to b-type ordering peaks [(1 0 1), (0 1 5), (0 2 1)] characteristic of the \(R\overline{3}\) space group (Goldsmith and Heard 1961; Reeder and Nakajima 1982; Deelman 1999). In this case, the long-range order parameter is related to the intensities of normal (0 0 6) and b-type (0 1 5) peaks, and defined as \(\surd\)(I0 1 5/I0 0 6) ((Zucchini et al. 2012) and references therein).The order parameter \(\surd\)(I0 1 5/I0 0 6) was determined from diffraction patterns computed with Fullprof for stoichiometric dolomites (Supp. Mat. Fig. S1). From nCa,α = 1 to 0, \(\surd\)(I0 1 5/I0 0 6) = 0.0058 × (nCa)2 + 1.0621 × nCa + 0.0335, where nCa is the Ca occupancy at its normal site in the dolomite structure. The order parameter \(\surd\)(I0 1 5/I0 0 6) varies almost linearly between 1.1 and 0 from fully ordered (nCa = 1) to fully disordered (nCa = 0) dolomite. In the present study, the linear relation \(\surd\)(I0 1 5/I0 0 6) = 1.1 × nCa was used to quantify the disordering in dolomite structure.
Short-range order was first introduced and theorized by Warren et al. (1951), Cowley (1950, 1960). It involves the occupancies of the sites over a scale of a few Å. It is defined by the probability that an atom is in the first coordination sphere surrounding an arbitrary atom relatively to the random probability. In the case of Mg calcite, the disordered state occurs on one set of equivalent cation (Ca, Mg) sites. The degree of short-range order (α) is defined as α = 1 − PMg/nMg, = 1 − PCa/nCa where nMg or Ca is the Mg (or Ca) occupancy fraction, i.e. the random probability and PMg or Ca the probability of Mg (or Ca) atoms is within the first coordination sphere surrounding a central Ca (or Mg) atom. In the random structure α = 0, while in the fully ordered structure α = 1. In the Rietveld analysis of the Mg calcite powder diffractograms, the substitution of Mg for Ca could not be refined, it was considered as fully random and disordered. All kinds of short-range order being not considered, led to lower agreement factors values. Besides, short-range order such as preferential occupancy of Ca and Mg into octahedral sites within domains would generate variations of intensities and non-uniform broadening of select families of reflections (Radha et al. 2007). In particular, (h k l) stacking faults would selectively broaden the peaks due to the (h k l) family of reflections. The heat treatment of Mg calcites is expected to induce lattice parameter distortions and Ca and Mg re-ordering, but also change the average domain size and crystal lattice microstrain. For these specific microstructural characterizations, each single peak in the diffractogram was analyzed with the Winplotr program (Roisnel and Rodriguez-Carvajal 2001) and we used the Williamson–Hall plot method (Williamson and Hall 1953) in which the broadened peak is analyzed by a pseudo-Voigt function. The integrated intensity, coherent domain size ⟨L⟩ and average microstrain values ⟨ε⟩ are estimated from the refined profile width parameters. The coherent domain size was determined using the Scherrer (1918) formula: ⟨L⟩ = λ/βcos(θ), where λ, β, and θ are the wavelength, integral breath, and Bragg angle, respectively (Scherrer 1918). The microstrain effect is the variance of the lattice spacing (Δd/d) and the angular dependence of the form is defined as Δd/d = β/tan(θ) (Stokes and Wilson 1942).
Results and interpretations
HRXRPD characterization of magnesian calcite syntheses
Whole pattern analysis and cell parameters
HRXRPD patterns of OCN calcite, OCN dolomite, and synthetic magnesian calcite MgCc24 (~ 20.7 mol% MgCO3) are shown in Fig. 2. Note the narrowness of the peaks in the synthesized materials. No phase other than Mg calcite and/or dolomite was detected. No indication of extended basal cation stacking order such as b-type ordering reflections characteristic of the (\(R\overline{3}\)) ordered structure of dolomite was found in the synthetic carbonates in the range 2–45 mol% MgCO3. MgCc50 with 49.2 mol% MgCO3 is the only synthetic sample with this type of ordering reflections. (1 0 4) peaks of OCN calcite, OCN dolomite and synthetic samples in the range 2–50 mol% MgCO3 are shown in Fig. 3. The plots of the (3 0 0) and (0 0 12) peaks (distinctive a and c basal reflections, respectively) are provided as supplementary materials (Supp. Mat. Fig. S2). The three figures show the same general features and allow identifying two groups of syntheses: a first group comprising the MgCc2 to MgCc26 samples is characterized by narrow and symmetric peaks, a second group from MgCc28 to MgCc45 displays largely broadened and asymmetric peaks. The two groups will be referred to as low- and high-Mg synthetic calcites, respectively. Diffractograms of the high-Mg synthetic calcite group can be analyzed in different ways: the peak broadening corresponds to a distribution curve of single-phase Mg calcites with either (1) different Mg compositions, (2) different sizes and microdistortions (Ca/Mg disordering), or (3) different Mg compositions, sizes or microdistortions (Ca/Mg disordering). Table 1 reports the Rietveld refinement results of the simplest model involving three or four calcites with different compositions (and thus different cell parameters) and similar peak broadening. The origin of peak broadening will be discussed below. Refined unit cell parameters, microstructural parameters (FWHM broadening, size 〈L〉 and microstrain 〈ɛ〉) for, synthetic Mg calcites, synthetic dolomite and OCN crystals are given in Tables 1 and 2. Unit-cell parameters and volumes obtained for all synthetic samples are shown as a function of their Mg contents in Fig. 4. For the high-Mg synthetic calcites, values of the three phases used in the refinement are plotted in the diagram. The a and V parameters show a small negative deviation from the Cc-Dol straight line, whereas the c parameter displays almost no deviation. These parameters can be fit with a second-degree polynomial with R2 > 0.995:
(XMg: mole fraction of MgCO3 in the carbonate).

HRXRPD patterns of a OCN calcite (black), b synthetic magnesian calcite MgCc24 (blue) and c OCN dolomite (red) [wavelength of λ = 0.41068(1) Å (30 keV)]. Bragg peaks in the range 5.5 to 12° (2θ). \(R\overline{3}c\) space group reflections in black, and \(R\overline{3}\) space group b-type ordering reflections in red

Bragg (1 0 4) diffraction peaks of OCN calcite (black), synthetic magnesian calcites (MgCc2 to MgCc45), synthetic MgCc50 dolomite and OCN dolomite (red) [wavelength of λ = 0.41068(1) Å (30 keV)]

Unit cell parameters and volume as function of Mg composition for OCN calcite, OCN dolomite (black plus) and for synthetic carbonate (blue, red and green plus). Dashed line is the quadratic least-square adjustment as discussed in the text. Solid line is the straight line between OCN calcite and OCN dolomite
The fitting curves are shown in Fig. 4. As stated by Reeder (1983), d(1 0 4) has long been used to determine the Mg content of rhombohedral carbonates. In the present study, the following relationship is determined:
It should be noted that d(1 0 4) does not provide a unique description of the cell while lattice parameters do, and thus cell parameters should be preferred to determine Mg contents in Ca–Mg carbonates (Reeder 1983). For natural carbonates (including biominerals) that may contain significant amounts of other components such as Fe, Mn, Sr, S, organic matrix, etc., direct measurements of compositions (by EMP for instance) should be preferred.
Note again that the substitution of Mg for Ca in Mg calcite has been considered as fully random and disordered in this analysis of the entire diffractograms. A more precise structural model that would take into account preferential occupancy of Ca and Mg into octahedral sites within domains, would possibly lead to better agreement-factor values. However, in the present state of our knowledge, too many variables remain undetermined to allow this type of modelling with reasonable degree of confidence.
Single peak analysis and microstructural parameters
Short-range disorder in Mg calcites associated with the shift in position of atoms and distortion of octahedral sites must generate variations of (h k l) l = even intensities because of large differences in X-ray scattering factors between calcium and magnesium atoms. Misorientation of carbonate ions must generate variations of (h k l) l = odd intensities. As far as long-range order is concerned, the spatial extent and the structural nature of long-range order in Mg calcites domain must generate non-uniform widening of some families of reflections. Below, both intensities and FWHM of the diffraction peaks are analyzed to characterize both the structural short- and/or long-range order and the length of structural coherence.
Intensities of several first (h k l) peaks for even and odd (l)s are shown in Fig. 5a, b as a function of Mg contents, and they are compared to those calculated from randomly disordered Mg calcite structure. Intensities of all (h k l) peaks vary linearly as a function of Mg content. No particular (h k l) intensity variation as a function of Mg content is observed. Some of them increase while others decrease. The deviations from linearity (SD 0.10) are small and not correlated to Mg content and (h k l) families. A small deviation could be attributed to random distribution of structural distortions such as locally restricted cation ordering and anion positional disorder (off-planar tilting of the CO3 groups) (Althoff 1977; Paquette and Reeder 1990). In the present case, the deviations are erratic and prevent refining a better structural model with confidence.

Observed intensities of (h k l) peaks a l = even (distortion of octahedral sites); b l = odd (misorientation of CO3 groups) of synthetic carbonates as a function of Mg composition for synthetic carbonates (filled circles). The dashed line represents the intensities calculated for ideally disordered Mg calcites. All observed and calculated intensity are normalized to the same scale factor. Intensity axis is offset for each (h k l) for clarity
Concerning the FWHM, it has been mentioned earlier that two groups can be defined on the basis of peak FWHM differences, as observed in Fig. 3 (see also Supp. Mat. Fig. S2). In the low-Mg synthetic calcites (0–26 mol. % MgCO3), the FWHM variation of each (h k l) peak as a function of Mg content is small (usually less than 0.3%) (SD in Fig. 6). The largest values and variations are for (h k l) peaks with large l values such as (1 0 10), (2 0 8), (0 0 12), (0 2 10) and (1 1 12). The (h k l) families with large l values are sensitive to local basal stacking disorder, but here again we must remain cautious as differences with variations observed in OCN calcite are small (Fig. 6). The structural distortions determined with the Williamson–Hall plot method are given in Table 1. The mean coherent domain size ⟨L⟩ [1.0 µm (SD 0.2)] is similar to OCN calcite (Fig. 7a). However, the average microstrain ⟨ε⟩ = 2 × 10–4 (SD = 0.7 × 10–4), that is four times higher than in OCN calcite (Fig. 7b), could be related to the random distribution of structural distortions leading to small intensity deviations described above.

FWHM of OCN calcite (h k l) peaks (black crosses). Mean FWHM of (h k l) peaks of 13 samples of synthetic low-Mg carbonates (MgCc2 to MgCc26) (red dots, vertical bars are standard deviations)

a Mean coherent domain size ⟨L⟩; b average microstrain values ⟨ε⟩ calculated by the Williamson–Hall plot method for OCN calcite (black crosses) and for low-Mg synthetic carbonates (MgCc2–MgCc26) (red dots). Dashed lines are linear least-square fitting curves
In the case of high-Mg synthetic calcites, the FWHM of the diffraction peaks are three to four times larger than low-Mg calcites (Fig. 3, Supp. Mat. Fig. S2). In addition, the peaks are asymmetric whatever the Mg content, and in a similar way for each (h k l) peak family. Thus, the FWHM broadening cannot be interpreted in terms of domain size and microstructural defects but rather in terms of coexistence of tiny domains with different compositions in a single crystal (Table 1). This interpretation will be developed in the discussion below.
Special emphasis must be put on the MgCc50 sample that shows the characteristic superlattice peaks of dolomite. In this case and compared with OCN dolomite, no FWHM broadening of the (h k l) peaks indicative of disordered stacking sequences is observed (Figs. 3, 8). FWHM of the (1 0 4) peak (Fig. 3) and calculated size 〈L〉 and microstrain ⟨ɛ⟩ are similar to those of OCN dolomite (Tables 1, 2). For OCN dolomite, the long-range cation order parameter \(\surd\)(I0 1 5/I0 0 6) is equal to 1.07 whereas \(\surd\)(I0 1 5/I0 0 6) for MgCc50 dolomite equals 0.64 (as a reminder, \(\surd\)(I0 1 5/I0 0 6) of a fully ordered dolomite equals 1.1). Thus, OCN dolomite is almost fully ordered with only 3% cation disorder, while the synthetic MgCc50 dolomite is characterized by 36% cation disorder. Such highly disordered dolomite is expected at 1373 K since the critical temperature for the transition between long-range ordered and disordered dolomites has been located close to 1473 K (Goldsmith and Heard 1961). A similar degree of disorder (s = 0.75) is reported by Reeder and Wenk (1983) in a natural dolomite, thermally treated at T = 1373 K and P = 1.1 GPa.

HRXRPD diffractograms of the OCN and synthetic MgCc50 dolomites (in red and green, respectively) (wavelength of λ = 0.41068(1) Å (30 keV)). Bragg peaks in the 2θ range from 5.5 to 12 (°). Normal reflections and b-type ordering reflections for space group \(R\overline{3}\) are noted in black and red, respectively. The red (OCN dolomite) has been shifted in 2θ for clarity
Thermally processed magnesian calcite syntheses
OCN dolomite, and the synthetic MgCc24, MgCc40, and MgCc50 samples have been thermally processed following the step-mode heating process previously used for OCN calcite and MgCc10 (Floquet et al. 2015). In this previous work, no phase decomposition of OCN calcite or MgCc10 was observed until 1073 K. As expected, the calcite unit cell showed contrasting thermal contraction and expansion for the a and c parameters, respectively. For Mg calcite MgCc10, the a cell parameter decreased until 673 K, then increased, while the c parameter expanded monotonically all along the temperature range. Figures 9 and 10 display the XRD patterns collected at different temperatures for the MgCc24, MgCc40 and MgCc50 samples.

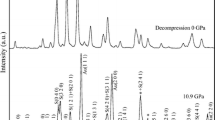
Selected HRXRPD diffractograms in the 10.9–11.7 2θ (°) range of the synthetic MgCc24 sample recorded from RT to T = 1073 K and then back to RT after heating (293 K A–H), showing (1) the (2 0 2) peaks of untransformed magnesian calcite (T in the range RT-653 K, indexes in italics) and partially decomposed product (at T > 653 K), and (2) the (2 0 0) peak of MgO resulting from partial decomposition (above 653 K). The black diffractogram collected at RT after annealing is shifted in intensity for clarity (black vertical arrow) [wavelength of λ = 0.41068(1) Å (30 keV)]

a HRXRPD patterns of synthetic MgCc40 recorded at RT (purple line, indexes in italics) and after annealing back to RT (black line). The original material (high-Mg calcite) is partially transformed into low-Mg calcite (~ 6.0 mol. % MgCO3) and MgO; b HRXRPD patterns of the synthetic dolomite MgCc50 recorded at RT before thermal processing (green line, and indexes in italics) and at RT after annealing at 1073 K (black line). The original dolomite has been totally transformed into low-Mg calcite with ~ 5.1 mol% MgCO3 and MgO [wavelength of λ = 0.41068(1) Å (30 keV)]
Thermal decomposition of Mg calcites and dolomite
Refined unit cell parameters and FWHM of (104) peaks of the samples as a function of temperature are given in Table 3 and as supplementary data (Supp. Mat. Tables S1 and S2). Partial decomposition (PD) [or ‘half decomposition’ according to many authors (e.g. (Valverde et al. 2015) and refs therein)] meaning that an original magnesian calcite progressively decomposes at high temperature into a less magnesian calcite plus MgO is observed for the three samples. The beginning temperature of partial decomposition (TPD), decreases as the Mg content increases. For instance, MgCc24, MgCc40, MgCc50 start forming low-Mg calcite (2.8–6 mol% MgCO3) and periclase at TPD of about 893 K, 833 K and 773 K, respectively (Figs. 9, 10, Table 3). After annealing at 1073 K for 20 min followed by cooling down to RT, MgCc24 and MgCc40 are only partially decomposed, whereas the dolomite sample (MgCc50) is completely transformed into low-Mg calcite (~ 5.1 mol. % MgCO3) and MgO. These observations are in great part in agreement with those of Reeder and Markgraf (1986) who observed the beginning of dolomite decomposition at ~ 973 K and total transformation into Mg calcite at ~ 1023 K in partial CO2 pressure. The average sizes of newly formed Mg calcite and periclase determined from the FWHM of the peaks using the Scherrer method, ⟨L⟩ ~ 80 and ~ 50 nm, respectively, are similar to those reported from XRD analysis of the thermal decomposition of dolomite (Hashimoto et al. 1980; Rodriguez-Navarro et al. 2012).
Variation of cell parameters with temperature
The variations of the unit cell parameters of MgCc24, MgCc40 and MgCc50 in the range RT-1073 K are plotted in Fig. 11. Note that in the experiments undergoing partial decomposition, cell parameters were determined on the remaining magnesian calcite that did not undergo partial decomposition. The unit cell shows a thermal expansion for both a and c parameters, even for MgCc24 with 20.7 mol% MgCO3. The temperature dependence of a, c and V is not linear over the entire temperature range, as best observed in the c/a vs T plot (Supp. Mat. Fig. S3). Note that for MgCc40, a model with two Mg compositions (MgCc40-1 and MgCc40-2) has been used to refine the diffractogram, hence two curves are plotted in Fig. 11, Supp. Mat. Fig. S3 for the MgCc40 experiment. The deviation from a linear expansion is positive for a, but negative for c, except for MgCc24 that shows a linear expansion for c. The c/a ratio of the unit cell deviates negatively from linearity at Tdif of about 653 K, 673 K and 773 K for MgCc24, MgCc40 and MgCc50, respectively (Supp. Mat. Fig. S3). A similar deviation occurs at ~ 673 K for MgCc10 due to the parabolic expansion of the a parameter (Floquet et al. 2015). The change of the thermal unit cell deformation starts at a temperature (Tdif) lower than the beginning of partial decomposition (TPD) for Mg calcite, and at the same temperature for the synthetic dolomite (Tdif ~ TPD).

Thermal expansion of the unit cell parameters a a parameter; b c parameter for synthetic MgCc24 (blue), MgCc40-1 and 2 (purple and pink) and MgCc50 (green), respectively
Above, two important temperatures are put forward: Tdif which is the temperature of the beginning of deviation from a linear expansion of the a, c (and V) parameter, and TPD the temperature of beginning of partial half decomposition of Mg calcites. The physical meaning of TPD is straightforward, as discussed above, while that of Tdif is not. On the basis of Raman characterizations, it has been argued that significant short-range Ca–Mg cation order is present in magnesian calcites that crystallized at 1273 K or 1373 K. Three reasons have been invoked: (1) the wavenumbers of magnesian calcites are close to the calcite–dolomite line (and not to calcite-magnesite), (2) the FWHM of the T, L, and ν4 modes reach a maximum around 30 ± 5 mol% MgCO3, and (3) the active mode (ν2, A2g) in dolomite at 880 cm–1 is observed in high-magnesian calcites above ~ 10 mol% MgCO3 (Perrin et al. 2016). It should be noted also that short-range order greatly depends on temperature, it increases as temperature decreases. Thus, Tdif is interpreted as the temperature at which re-ordering (by diffusion) of calcium and magnesium atoms is activated to reach a more stable short-range ordering configuration. For the studied Mg calcite samples, Tdif is lower than TPD and the two processes are expected to produce effects on Bragg peaks as a function of temperature, in addition to the thermal vibrational effect reflected in the Debye–Waller factors. Actually, the partial decomposition is mainly related to the decrease of all (h k l) peak intensities. In contrast, a short-range re-ordering should (1) produce thermal deviation excess of the cell parameters and (2) lead to different variations of the (h k l) peak intensities, and therefore could be detected by the variation of the intensity ratio as function of temperature.
Thermal expansion coefficients along the calcite–dolomite join
High-temperature single crystal (Markgraf and Reeder 1985; Reeder and Markgraf 1986) and powder (Antao et al. 2009; De Aza et al. 2002; Merlini et al. 2016) diffraction experiments have been conducted on calcite, dolomite and magnesite. The relative thermal expansion of the cell parameters a/a293 K, c/c293 K of OCN calcite, OCN magnesite and synthetic MgCc50 dolomite are compared to those of these previous data in Supp. Mat. Fig. S4. The comparison shows (1) a good agreement between OCN crystal data with powder diffraction experiments, but small discrepancies with single crystal experiments, (2) a marked deviation at about 773 K (Tdif) for synthetic MgCc50 dolomite compared to OCN dolomite. Similarly, the relative thermal expansion of the cell parameters for OCN calcite, synthetic MgCc10, MgCc24 MgCc40, and MgCc50 are presented in Supp. Mat. Fig. S5. The figure illustrates (1) the change of slope of the a parameter as a function of temperature, from negative to positive, at about 10 mol% MgCO3 (Supp. Mat. Fig. S5a), and (2) the deflection of slope of the c parameter as a function of temperature, at about 653 K (Tdif) (Supp. Mat. Fig. S5b). The thermal coefficients obtained by fitting the data to the usual second-order polynomial curve are reported in Table 4. The coefficient of thermal expansion of parameter x (x = a, c or V) \( \alpha_{x} = \left( {1/x} \right)({\text{d}}x/{\text{d}}T\)) is obtained by differentiating the polynomial expression of the thermal expansion x = f(T) and then dividing by the x parameter (Dubrovinsky 2002).
The mean coefficient of thermal expansion over the temperature range from T1 to T2 is expressed as \(\overline{{\alpha_{X} }} \) = \(\mathop \smallint \nolimits_{T1}^{T2} \alpha_{x} \times {\text{d}}T/\mathop \smallint \nolimits_{T1}^{T2} {\text{d}}T\). Because of the noticeable deviation of the thermal unit cell parameters at Tdif, the mean thermal expansion coefficients \( \overline{{\alpha_{a} }}\), \(\overline{{\alpha_{c} }}\) and \(\overline{{\alpha_{V} }} \) are calculated below and above Tdif, i.e. within two temperature ranges: 293–653 K (LT) and 653–1073 K (HT). The mean thermal expansion coefficients for the cell parameters are compared to those of OCN calcite, magnesite and dolomite (De Aza et al. 2002) in Fig. 12, no measurements on OCN dolomite was realized by ourselves.

Mean thermal expansion coefficients of the cell parameters αa, αc and αV as a function of Mg content for OCN calcite and OCN magnesite (double crosses), natural dolomite (De Aza 2002) (crosses), and for synthetic MgCc10, MgCc24, MgCc40 and MgCc50 (plus): a–c Low-temperature ranges (293–653 K) (blue) and d–f high-temperature ranges (653–923 K) (red)
The particular features of the thermal expansion coefficients of calcite, dolomite and magnesite, in both low- and high-T ranges, are (1) the negative \(\overline{{\alpha_{a} }}\) of calcite and (2) all coefficients of dolomite lie off of the calcite–magnesite line. These particularities were comprehensively studied and discussed by (Markgraf and Reeder 1985; Reeder and Markgraf 1986). In the Ca–Mg carbonate structure, the variation of the c parameter is related to the variation of lateral oxygen bonds of the (Ca, Mg)O6 octahedra, while the variation of the a parameter is related to the variation of basal oxygen bond which is limited by the resistance of rigid (CO2−)3 groups. Accordingly, the thermal c expansion and a contraction in calcite is related to the strong anisotropy of the thermal expansion of the CaO6 octahedra and the large magnitude of libration of the (CO2−)3 group (Markgraf and Reeder 1985; Reeder and Markgraf 1986). In magnesite, characterized by the a and c thermal expansion, the thermal expansion anisotropy of the MgO6 octahedra is moderate and the dominant motion of (CO2−)3 group is the translation. In dolomite, the corner sharing between CaO6 and MgO6 octahedra in adjacent layers induces a lack of anisotropy of the CaO6 octahedra and a low amplitude of both libration and translation motions of the (CO2−)3 group. Thus, the shift of the cell thermal expansion of dolomite from the calcite–magnesite line is explained by the perfect structural stacking alternating Ca and Mg basal planes on both sides of each CO3 plane. The systematic corner sharing between CaO6 and MgO6 octahedra in dolomite is ruled out in the disordered structures of MgCc calcites and dolomite. The thermal–structural relationships, as described by (Markgraf and Reeder 1985; Reeder and Markgraf 1986), imply that the thermal behavior of the synthetic MgCc calcites and MgCc50 dolomite is dependent of the structural arrangement of the CaO6 and MgO6 octahedra. The thermal coefficients are expected in the calcite–dolomite line for fully disordered MgCc calcites. Thus, a thermal coefficient excess from the calcite–dolomite line would be characteristic of the structural order of CaO6 and MgO6 octahedra.
In the low- and high-T range (Fig. 12), the mean thermal expansion coefficients of the synthetic MgCc calcites and MgCc50 dolomite lie off of both calcite–dolomite and calcite-magnesite lines. In the low-T range (Figs. 12a–c), a significant positive deviation from the calcite–dolomite line is observed for the \(\overline{{\alpha_{a} }}\) and \(\overline{{\alpha_{V} }}\) coefficients, with a maximum deviation close to 25 mol% MgCO3 for \(\overline{{\alpha_{a} }}\). Conversely, the \(\overline{{\alpha_{c} }}\) coefficients lie in between calcite–dolomite and calcite–magnesite line. In the high-T range, positive deviation from calcite–dolomite line occurs for the \(\overline{{\alpha_{a} }}\) coefficients (Fig. 12d). On the contrary, large negative deviation are observed for the \(\overline{{\alpha_{c} }}\) and \(\overline{{\alpha_{V} }}\) coefficients (Figs. 12e, f).
For the synthetic MgCc calcites and MgCc50 dolomite, in the LT range 273 K-653 K, the thermal expansion excess (with respect to the calcite–dolomite line) is positive for a, c and V (\(\Delta \overline{{\alpha_{a} }} = 1 \times 10^{ - 6}\), \(\Delta \overline{{\alpha_{c} }} = 1 \times 10^{ - 6}\), \(\Delta \overline{{\alpha_{V} }} = {3} \times {1}0^{{ - {6}}}\)). These results are consistent with the thermal–structural relationships proposed by Markgraf and Reeder (1985) and Reeder and Markgraf (1986), and could be related to the state of order–disorder as do the cell parameters that are similarly deviating from the calcite–dolomite line (Fig. 4). In the HT range, excess is unchanged for a (\(\Delta \overline{{\alpha_{a} }} = 1 \times 10^{ - 6}\)) but largely negative for c and V (\(\Delta \overline{{\alpha_{c} }} = - 5 \times 10^{ - 6}\), \(\Delta \overline{{\alpha_{V} }} = - {3} \times {1}0^{{ - {6}}}\)). The differences observed between the low- and high-T range comes from the positive and negative inflection of the a and c parameters, respectively, starting at Tdiff in all synthetic MgCc (Fig. 11 and Supp. Mat. Fig. S3) whereas not recorded in OCN dolomite powder (De Aza et al. 2002; Merlini et al. 2016) and in dolomite single crystal (Reeder and Markgraf 1986) (Supp. Mat. Fig. S4).
Intensities and FWHMs of the Bragg peaks as a function of T: interpretation of the data
In the case of MgCc24, both intensity effects are detected. The variations of the (1 0 4) peak intensities as a function of T for MgCc24 are shown in Fig. 13a. The regular shift of the (1 0 4) peak towards lower 2θ values is due to the progressive change of cell parameters with temperature. Concerning intensities, different zones can be defined: (1) intensities increase between 293 and 413 K, (2) intensities decrease regularly between 413 and 713 K (= Tdif), (3) a plateau is observed between Tdif (713 K) and TPD (953 K), (4) intensities decrease past TPD. Interestingly, the two patterns determined at RT before (initial sample) and after annealing (remaining undecomposed sample) are almost identical (Supp. Mat. Table S1). Besides, the intensity ratio I(1 0 4)/I(1 1 0) and I(1 0 4)/I(1 1 3) shown in Fig. 13b displays two marked slope disruptions at 713 K and 953 K relevant to both diffusion and decomposition processes. However, no notable FWHM effect on the Bragg peaks was detected. The variations of (h k l) FWHMs as a function of T are unrelated to the Mg content of the samples, the variations among different samples are not significant (Supp. Mat. Table S1), and no irregularity is observed even for (h k l) peaks with large l values such as (0 1 8) (0 0 12) (0 2 10) and (1 1 12).

a Bragg (1 0 4) diffraction peaks for MgCc24; b Intensity ratio I(1 0 4)/I(1 1 3) and I(1 0 4)/I(1 1 0) for MgCc24 (wavelength of λ = 0.41068(1) Å (30 keV)) recorded from RT to T = 1073 K. Note the slope breaks at 713 K and 953 K
Concerning the synthetic dolomite MgCc50, Tdif equals TPD and normal and b-type peak intensities display different behaviors. Normal peak intensities (e.g. 0 0 6) plotted in Fig. 14 decrease progressively up to Tdif = TPD = 773 K, then faster above Tdif. This observation is valid for all normal peaks. On the other hand, b-type peak intensities [e.g. (0 1 5)] decrease up to Tdif, then increase up to 1013 K (Fig. 14).

a HRXRPD patterns in the 2θ range corresponding to the (0 0 6) and (0 1 5) peaks; b intensity ratio I(0 1 5)/I(0 0 6) (green plus) and \(\surd\)(I0 1 5/I0 0 6) order parameter (black crosses) for MgCc50 (Ca-dolomite) from RT to T = 1073 K (wavelength of λ = 0.41068(1) Å (30 keV))
These data indicate that cation long-range reordering within the dolomite structure starts at 773 K. The order parameter \(\surd\)(I0 1 5/I0 0 6) shows that the MgCc50 dolomite is almost fully ordered at T = 1073 K, with less than 10% cation disordering (Fig. 14b) which is not surprising since ordered dolomite is expected at 1073 K. (Hammouda et al. 2011; Zucchini et al. 2012). In addition, (Goldsmith et al. 1961; Reeder and Wenk 1983) concluded that cation ordering in dolomite results in a decrease of c and a slight increase of a, thus a marked decrease of the c/a ratio. Data reported above with c/a of MgCc50 deviating from linearity at Tdif (Supp. Mat. Fig. S3) is interpreted as an effect of cation ordering activated by temperature, in a disordered dolomite initially synthesized at 1373 K. Similarly to MgCc50, the changes of cell parameters arising at Tdif = 653 K, 673 K for MgCc10, MgCc24 and MgCc40 are interpreted in terms of ordering process in partially disordered Mg calcite samples synthesized at 1273 K or 1373 K. Note that in the case of MgCc50, both short- and long-range ordering is implicated while in more calcitic syntheses only short-range ordering would be implied. Summarizing, in the range RT–Tdif, the variation of cell parameters is simply due to a thermal effect. Above Tdif, both thermal effect and cation ordering affect the cell parameters. More interactive and interlinked effects are expected once the partial decomposition starts.
Discussion
Comparison with previous data and interpretation of high-Mg calcite XRD patterns
Our data are consistent with previous experimental studies (Byrnes and Wyllie 1981; Goldsmith et al. 1955; Goldsmith and Heard 1961; Harker and Tuttle 1955; Irving and Wyllie 1975; Newton et al. 1969) that predict a continuous (or almost continuous) solid solution between CaCO3 and (Ca0.5Mg0.5)CO3, in the pressure and temperature conditions of our experiments. Indeed, Goldsmith and Heard (1961) locate the top of the calcite–dolomite solvus at a temperature of 1348 K and a composition of 43 mol% MgCO3. The variation of cell parameters as a function of composition are in agreement with most previous studies (Bischoff et al. 1983; Goldsmith et al. 1958, 1961) but disagree with the results of Zhang et al. (2010) that strongly deviate from previous data, particularly above 30 mol% MgCO3 (Fig. 1).
The identification of two groups characterized either by narrow and symmetric peaks (low-Mg synthetic calcites: MgCc2–MgCc26) or broadened and asymmetric peaks (high-Mg synthetic calcites: MgCc28–MgCc45) is of major importance and deserves further interpretation. First of all, the features on which the separation into two groups are robust since (1) all MgCc above 26 mol% MgCO3 display similar patterns, (2) higher temperatures (1373 K instead of 1273 K) and pressure (1.5 GPa instead of 1 GPa) are not responsible for the peak widening because two of the high-Mg calcites (MgCc28 and 30) synthesized at 1273 K, 1 GPa display similar broad peaks. Actually, the broadening of the peaks corresponds to (1) a compositional variability in the high-Mg calcites of the order of ± 1 mol% MgCO3, (2) a mean apparent strain ⟨ε⟩ of about 4 × 10–3 against 2 × 10–4 in low-Mg calcites, (3) both effects of compositional heterogeneity. One of them could be related to compositional variation between crystals as determined by EMP of the order of ± 0.8 mol% MgCO3. But the second, finer level of compositional heterogeneity could be due to the coexistence in a given crystal of coherent scattering domains, the components of which have different cation ordering and slightly different compositions and are producing microstructural strain. The actual distribution of Ca and Mg in each microcrystal cannot be determined because the broadening of the diffraction peaks depends also on the strain, size and distribution of the coherent scattering domains. Nanoscale heterogeneous microstructures are very common in Ca-rich dolomite natural samples. They were reported in numerous HRTEM studies by interpreting HAADF image modulations and satellite reflections in the diffraction patterns (Fang and Xu 2019; Larsson and Christy 2008; Meike et al. 1988; Reksten 1990; Shen et al. 2014; Van Tendeloo et al. 1985; Wenk et al. 1991). But actually, if many microstructures were proposed at the nanometric scale, no further structural details of the domain model would be provided at the micrometric scale by single crystal diffraction method (Reeder 2000—natural dolomites with 44 and 45.5 mol% MgCO3−sorder = 0.84 and 0.71) and by comprehensive Rietveld techniques (Drits et al. 2005—natural dolomites with 45.1, 47.8 and 48.4 mol% MgCO3). Considering all this, it is very likely that the constraints on cation order in Ca-rich dolomite similarly occur in the high-Mg synthetic calcite. Clearly, further studies are required to validate the nanoscale compositional heterogeneity, but additional valuable insights at the short-range-order scale could be given from our previous Raman data (Perrin et al. 2016), as discussed below.
Length scales of the cationic disorder state
Unit-cell parameters and volume obtained for all the MgCc samples show small deviations from the almost straight calcite–dolomite–magnesite line. Assuming that a total disordered MgCc corresponds to an ideal solid solution and plots along the straight line, the strain associated with the partial order (p-ord)–total disorder (t-dis) transformation is defined as εX = (Xp-ord − Xt-dis)/Xt-dis = ∆X/Xt-dis where Xp-ord is the value of a, c or V obtained for a MgCc sample and Xt-dis is the value of a, c or V calculated for the same composition from the straight line through the end-members. The strains εa, εc, εV calculated for the a, c and V parameters, for all the MgCc samples are plotted as a function of the Mg content in Fig. 15.

Strain in magnesian calcites at RT as a function of Mg content a εa; b εc; c εV (black: OCN calcite and OCN dolomite; blue: low-Mg synthetic samples; red: high-Mg and MgCc50 synthetic samples)
The strain values are negative for εa and εV and generally positive for εc; they display different behavior for low- and high-Mg synthetic calcites. In the range 0–22 mol% MgCO3 (low-Mg: MgCc2–26), all strain values increase as the Mg content increases, while in the high-Mg synthetic calcites, εa and εV decrease, and εc increases. The transition between the two groups is marked by a discontinuity (negative jumps for εa and εv, negative for εc). In the carbonate structure, strongly bonded coplanar (CO3)2− groups are arranged parallel to the (0 0 1) plane and connected through much weaker bonds to 6-coordinated divalent cations. Thus, the negative strain along the a-axis could be related to the distance O1–O2 of the MO6 octahedra (M = Ca or Mg), connecting two rigid (CO3)2− carbonate groups in the (001) plane. On the other hand, the positive strain along the c-axis could be related to the variation of O1–O6 bond in the MO6 octahedra. Cation ordering affects the orientation of the (CO3)2− groups (1) by their rotation (δ) around the c axis and (2) tilting out (γ) of the carbonate groups in the (001) plane. The rotation generates a negative a strain, while tilting generates a positive c strain. On this basis, both in-plane rotation (δ) and tilting out (γ) of the (CO3)2− groups increase as the Mg content increases in the low-Mg calcites, whereas in-plane rotation (δ) decreases and tilting out (γ) increases in the high-Mg calcites, as the Mg content increases. The basal rotation (δ) and tilting out (γ) of the (CO3)2− groups are maximal at 25 and 50 Mg mol. %, respectively. The above correlation indicates that the deviation with respect to an ideal solid solution in the calcite–dolomite range is related to the local ordering or clustering of Ca and Mg in cation layers involving reorientation of (CO3)2− groups, associated with an inverse strain of the unit cell parameters, negative for a and positive for c.
Neither loss of rotational symmetry nor long-range cation ordering is observed by X-ray diffraction in the synthetic magnesian calcites, only structural distortion. Thus, partial ordering can only be short-range. This conclusion is in agreement with the Raman study of the same synthetic magnesian calcites reported in a previous study (Perrin et al. 2016). As shown in Supp. Mat. Fig. S6, the FWHMs of the Raman bands change as a function of Mg content and type of vibrations within the (CO3)2− groups [bending deformation modes (doubly degenerate Eg) that involve both carbon and oxygen motion, and stretching mode (A1g) which is like the respiration mode of oxygen]. A comparison of Figs. 15 and S6 shows that the unit cell strain determined by XRD and the Raman band enlargement are correlated. The strain εa for the a parameter (Fig. 15a) and the FWHM of the bending modes [particularly the internal translation mode (T) (Supp. Mat. Fig. S6a) are similar]. On the other hand, the strain εc for the c parameter (Fig. 15b) is more consistent with the enlargement observed for the single stretching mode ν1 (Supp. Mat. Fig. S6e). Below 25% Mg content, the heterogeneity between local cationic configurations increases inside the crystals. Above 25% Mg content, the single phase–multi-phase transition from homogeneous to heterogeneous crystals linked to the strain relaxation is marked for both modes with a decrease of the local cationic configuration heterogeneity towards dolomite like ordering. The heterogeneous MgCc crystals could be made of different domains with different local ordering configurations having similar or slightly different magnesium contents.
Thus, both the bending deformation and stretching modes observed by Raman spectroscopy can be related to the two types of site-symmetry of the (CO3)2− group: the off planar tilting (γ) and the in plane rotation (δ), respectively. The genetic connection between the off plane tilting (γ) specific to the dolomite structure and the stretching mode (ν1, A1g) is also supported by the out of plane bending mode Raman peak (ν2, A2g). This peak, which is active in dolomite structure, is weak in synthetic magnesian calcites but can be detected from about 10 mol% MgCO3 and its intensity progressively increases thereafter until 50 mol% MgCO3 (Perrin et al. 2016). As seen above, the synthetic MgCc50 dolomite is characterized by 36% cation disordering in both types of cationic layers. No anomalous intensity (except for the b-type reflection) nor FWHM broadening of the (h k l) peaks (potentially indicative of ordered stacking sequence) were detected. The cation disordering corresponds to the maximal positive strain of the c parameter. A medium negative strain of the a parameter is observed (Fig. 15). Short-range disordering can be characterized from previous Raman data. As observed in Supp. Mat. Fig. S6, both enlargement of the stretching mode (ν1, A1g) and intensity of the dolomite bending mode (2ν2, A2g) are maximal (Fig. 7 in (Perrin et al. 2016)), whereas enlargements of the other bending modes start decreasing above 25 mol% MgCO3. Thus, the short-range Ca, Mg disorder set to 36% from √(I0 1 5/I0 0 6) order parameter, is characterized by a decrease of the a parameter, an increase of the c parameter, and an increase of both in plane rotation and off-plane tilting variability.
Conclusion
These data confirm the presence of an almost continuous solid solution between calcite and dolomite above 1273 K, and the presence of a second-order transition between long-range disordered Mg calcite and dolomite in the interval 41–49 mol% MgCO3 at 1373 K. In the Mg calcites, the deviation from the Cc-Dol straight line in the a/Mg, c/Mg, or V/Mg diagrams is interpreted in terms of cationic short-range ordering in the Mg calcites. This short-range order is accompanied by in plane rotation and the off planar tilting of the (CO3)2− groups in the anion layer that are maximal at 25 and 50 Mg mol. %, respectively. As a result, the role of (CO3)2− group ordering is an important factor in the crystal structure of Ca–Mg carbonate. The thermal expansion of MgCc10, 24, 40 and 50 have been determined up to 650-750 K. The thermal expansivity deviates from the Cc-Dol straight line. Compared with calcite, dolomite and magnesite previous results, this trend is interpreted in terms of interactive contributions of CaO6 and MgO6 octahedra and CO3 groups to the thermal expansion and thus explained by the structural short-range ordering observed at RT. At higher temperatures, complications arise from cation reordering or partial decomposition of the samples. The excess of thermal expansion coefficients from the Cc-Dol straight line are consistent with the thermal–structural relationships proposed by Markgraf and Reeder (1985) and Reeder and Markgraf (1986), and supports the cationic short-range ordering in the Mg calcites. The peak width of the diffractograms allow separating the synthetic Mg calcites into two groups: low- and high-Mg synthetic calcites. Low-Mg synthetic calcites are homogeneous in Mg content while high-Mg synthetic calcites display domains with slight differences in Mg content and degree of short-range order. The short-range Ca, Mg disorder could be at random or set into a mosaic of the different planar ordering configurations reported in TEM studies for dolomite and Ca-rich dolomite (Fang and Xu 2019; Larsson and Christy 2008; Meike et al. 1988; Reksten 1990; Shen et al. 2014; Van Tendeloo et al. 1985; Wenk et al. 1983) and by computer simulation approach (Burton and Van de Walle 2003; Lacivita et al. 2017; Reeder 2000; Wang et al. 2011). Further studies are required to better characterize the size and spatial distribution of these domains. One of the ways to quantify the local ordering state at the length scale of a few unit cells could be to combine Z-contrast imaging from scanning transmission electron microscopy of appropriate samples and image simulation, a novel approach developed by Fang and Xu (2019). This new complete series of structural data on Mg calcite including thermal properties will serve as references in many fields of research involving synthetic crystals obtained in solution-based crystallization processes (nanocrystals, mesocrystals, calcite/polymers crystals), biogenic crystals and geological minerals. It will also provide a robust basis for studies to come on the physical chemistry of precipitation, dissolution and ionic diffusion of Mg carbonates, and for the thermodynamic modeling of solid solutions which is an important research topic in many areas of materials science and geoscience.
References
Althoff PL (1977) Structural refinements of dolomite and a magnesian calcite and implications for dolomite formation in marine environment. Am Mineral 62(7–8):772–783
Antao SM, Hassan I, Mulder WH, Lee PL, Toby BH (2009) In situ study of the R-3c toR-3m orientational disorder in calcite. Phys Chem Miner 36(3):159–169. https://doi.org/10.1007/s00269-008-0266-y
Bischoff WD, Bishop FC, Mackenzie FT (1983) Biogenically produced magnesian calcite; inhomogeneities in chemical and physical properties; comparison with synthetic phases. Am Mineral 68(11–12):1183–1188
Burton BP, Van de Walle A (2003) First-principles-based calculations of the CaCO3–MgCO3 and CdCO3–MgCO3 subsolidus phase diagrams. Phys Chem Miner 30(2):88–97. https://doi.org/10.1007/s00269-002-0294-y
Byrnes AP, Wyllie PJ (1981) Subsolidus and melting relations for the join CaCO3-MgCO3 at 10 kbar. Geochim Cosmochim Acta 45(3):321–328. https://doi.org/10.1016/0016-7037(81)90242-8
Cowley JM (1950) An Approximate Theory of Order in Alloys. Phys Rev 77(5):669–675. https://doi.org/10.1103/PhysRev.77.669
Cowley JM (1960) Short- and long-range order parameters in disordered solid solutions. Phys Rev 120(5):1648–1657. https://doi.org/10.1103/PhysRev.120.1648
De Aza AH, Rodriguez MA, Rodriguez JL, De Aza S, Pena P, Convert P, Hansen T, Turrillas X (2002) Decomposition of dolomite monitored by neutron thermodiffractometry. J Am Ceram Soc 85(4):881–888
Deelman JC (1999) Low-temperature nucleation of magnesite and dolomite. Neues Jahrbuch Fur Mineralogie-Monatshefte 7:289–302
Deelman JC (2011) Low–temperature formation of Dolomite and Magnesite. Compact Disc Publications Geology Series. Compact Disc Publications, Eindhoven, The Netherlands. http://www.jcdeelman.demon.nl/dolomite/bookprospectus.html
Drits VA, McCarty DK, Sakharov B, Milliken KL (2005) New insight into structural and compositional variability in some ancient excess-Ca dolomite. Can Mineral 43:1255–1290. https://doi.org/10.2113/gscanmin.43.4.1255
Dubrovinsky L (2002) Thermal expansion and equation of state. In: Buschow KHJ, Cahn RW, Flemings MC et al (eds) Encyclopedia of materials: science and technology. Elsevier, Oxford, pp 1–4. https://doi.org/10.1016/B0-08-043152-6/01817-9
Fang YH, Xu HF (2019) A new approach to quantify the ordering state of protodolomite using xrd, tem, and z-contrast imaging. J Sediment Res 89(6):537–551. https://doi.org/10.2110/jsr.2019.29
Fitch AN (2004) The high resolution powder diffraction beam line at ESRF. J Res Nat Inst Stand Technol 109(1):133–142. https://doi.org/10.6028/Jres.109.010
Floquet N, Vielzeuf D, Ferry D, Ricolleau A, Heresanu V, Perrin J, Laporte D, Fitch AN (2015) Thermally induced modifications and phase transformations of red coral Mg-calcite skeletons from infrared spectroscopy and high resolution synchrotron powder diffraction analyses. Cryst Growth Des 15(8):3690–3706. https://doi.org/10.1021/acs.cgd.5b00291
Glover ED, Sippel RF (1967) Synthesis of magnesium calcites. Geochim Cosmochim Acta 31(4):10. https://doi.org/10.1016/0016-7037(67)90037-3
Goldsmith JR, Heard HC (1961) Subsolidus phase relations in the system CaCO3-MgCO3. J Geol 69(1):45–74. https://doi.org/10.1086/626715
Goldsmith JR, Graf DL, Joensuu OI (1955) The occurrence of magnesian calcites in nature. Geochim Cosmochim Acta 7(5):212–230. https://doi.org/10.1016/0016-7037(55)90033-8
Goldsmith JR, Graf DL, Chodos AA, Joensuu OI, Mcvicker LD (1958) Relation between lattice constants and composition of Ca–Mg carbonates. Am Mineral 43(1–2):84–101
Goldsmith JR, Graf DL, Heard HC (1961) Lattice constants of the calcium magnesium carbonates. Am Mineral 46(3–4_Part_1):453–459
Hammouda T, Andrault D, Koga K, Katsura T, Martin A (2011) Ordering in double carbonates and implications for processes at subduction zones. Contrib Mineral Petrol 161:439–450. https://doi.org/10.1007/s00410-010-0541-z
Harker RI, Tuttle OF (1955) Studies in the system CaO-MgO-CO 2; Part 2, Limits of solid solution along the binary join CaCO 3-MgCO 3. Am J Sci 253(5):274–282. https://doi.org/10.2475/ajs.253.5.274
Hashimoto H, Komaki E, Hayashi F, Uematsu T (1980) Partial decomposition of dolomite in CO2. J Solid State Chem 33(2):181–188. https://doi.org/10.1016/0022-4596(80)90118-8
Irving AJ, Wyllie PJ (1975) Subsolidus and melting relationships for calcite, magnesite and the join CaCO3-MgCO3 36 kb. Geochim Cosmochim Acta 39(1):35–53. https://doi.org/10.1016/0016-7037(75)90183-0
Jenkins DM, Holmes ZF, Ishida K, Manuel PD (2018) Autocorrelation analysis of the infrared spectra of synthetic and biogenic carbonates along the calcite–dolomite join. Phys Chem Miner 45(6):563–574. https://doi.org/10.1007/s00269-018-0942-5
Lacivita V, D’Arco P, Mustapha S, Faria Bernardes D (2017) On the use of the symmetry-adapted Monte Carlo for an effective sampling of large configuration spaces. The test cases of calcite structured carbonates and melilites. Comput Mater Sci 126:217–227. https://doi.org/10.1016/j.commatsci.2016.09.037
Larsson AK, Christy AG (2008) On twinning and microstructures in calcite and dolomite. Am Mineral 93(1):103–113. https://doi.org/10.2138/am.2008.2520
Markgraf SA, Reeder RJ (1985) High temperature structure refinements of calcite and magnesite. Am Mineral 70(5–6):590–600
Maslen EN, Streltsov VA, Streltsova NR, Ishizawa N (1995) Electron density and optical anisotropy in rhombohedral carbonates. III. Synchrotron X-ray studies of CaCO3, MgCO3 and MnCO3. Acta Crystallogr Section B 51(6):929–939. https://doi.org/10.1107/S0108768195006434
Meike A, Wenk HR, Okeefe MA, Gronsky R (1988) Atomic resolution microscopy of carbonates—interpretation of contrast. Phys Chem Miner 15(5):427–437. https://doi.org/10.1007/bf00311121
Merlini M, Sapelli F, Fumagalli P, Gatta GD, Lotti P, Tumiati S, Abdellatief M, Lausi A, Plaisier J, Hanfland M, Crichton W, Chantel J, Guignard J, Meneghini C, Pavese A, Poli S (2016) High-temperature and high-pressure behavior of carbonates in the ternary diagram CaCO3-MgCO3-FeCO3. Am Mineral 101(6):1423–1430. https://doi.org/10.2138/am-2016-5458
Newton RC, Goldsmith JR, Smith JV (1969) Aragonite crystallization from strained calcite at reduced pressures and its bearing on aragonite in low-grade metamorphism. Contrib Mineral Petrol 22(4):335–348. https://doi.org/10.1007/BF00400128
Paquette J, Reeder RJ (1990) Single crystal X-ray structure refinements of 2 biogenic magnesian calcite crystals. Am Mineral 75(9–10):1151–1158
Perrin J, Vielzeuf D, Laporte D, Ricolleau A, Rossman GR, Floquet N (2016) Raman characterization of synthetic magnesian calcites. Am Mineral 101(11):2525–2538. https://doi.org/10.2138/am-2016-5714
Putnis A (1992) An Introduction to Mineral Sciences. Cambridge University Press, Cambridge
Radha AV, Kamath PV, Shivakumara C (2007) Order and disorder among the layered double hydroxides: combined Rietveld and DIFFaX approach. Acta Crystallogr Sect B 63(2):243–250. https://doi.org/10.1107/S010876810700122X
Reeder RJ (1983) Crystal chemistry of the rhombohedral carbonates. Rev Mineral Geochem 11(1):1–47
Reeder RJ (1992) Carbonates; growth and alteration microstructures. Rev Mineral Geochem 27(1):380–424
Reeder RJ (2000) Constraints on cation order in calcium-rich sedimentary dolomite. Aquat Geochem 6(2):213–226. https://doi.org/10.1023/a:1009659122772
Reeder RJ, Markgraf SA (1986) High-temperature crystal chemistry of dolomite. Am Mineral 71(5–6):795–804
Reeder RJ, Nakajima Y (1982) The nature of ordering and ordering defects in dolomite. Phys Chem Miner 8(1):29–35. https://doi.org/10.1007/BF00311160
Reeder RJ, Wenk H-R (1983) Structure refinements of some thermally disordered dolomites. Am Mineral 68(7–8):769–776
Reksten K (1990) Superstructures in calcite. Am Mineral 75(7–8):807–812
Rietveld HM (1969) A profile refinement method for nuclear and magnetic structures. J Appl Crystallogr 2:65
Rodriguez-Carvajal J (1993) FullProf: a program for rietveld refinement and profile matching analysis of complex powder diffraction patterns (ILL). Phys B 192:55
Rodriguez-Navarro C, Kudlacz K, Ruiz-Agudo E (2012) The mechanism of thermal decomposition of dolomite: new insights from 2D-XRD and TEM analyses. Am Mineral 97(1):38–51. https://doi.org/10.2138/am.2011.3813
Rodriguez-Navarro C, Ruiz-Agudo E, Nieto F, Livi KJT, Oberti R (2013) Carbonates: an overview of recent TEM research. Minerals at the nanoscale. Mineralogical Society of Great Britain and Ireland, London. https://doi.org/10.1180/EMU-notes.14.10
Roisnel T, Rodriguez-Carvajal J (2001) WinPLOTR: a Windows tool for powder diffraction pattern analysis. In: Delhez R, Mittemeijer EJ (eds) Epdic 7: European powder diffraction, Pts 1 and 2, vol 378–3. Materials Science Forum. Trans Tech Publications Ltd, Zurich, pp 118–123. https://doi.org/10.4028/www.scientific.net/MSF.378-381.118
Scherrer P (1918) Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr Ges WissGöttingen 26:98–100
Shen ZZ, Konishi H, Szlufarska I, Brown PE, Xu HF (2014) Z-contrast imaging and ab initio study on “d” superstructure in sedimentary dolomite. Am Mineral 99(7):1413–1419. https://doi.org/10.2138/am.2014.4647
Stokes AR, Wilson AJC (1942) A method of calculating the integral breadths of Debye-Scherrer lines. Math Proc Cambridge Philos Soc 38(3):313–322. https://doi.org/10.1017/S0305004100021988
Valverde JM, Perejon A, Medina S, Perez-Maqueda LA (2015) Thermal decomposition of dolomite under CO2: insights from TGA and in situ XRD analysis. PCCP 17(44):30162–30176. https://doi.org/10.1039/C5CP05596B
Van Tendeloo G, Wenk HR, Gronsky R (1985) Modulated structures in calcian dolomite—a study by electron-microscopy. Phys Chem Miner 12(6):333–341. https://doi.org/10.1007/bf00654343
Wang Q, Grau-Crespo R, de Leeuw NH (2011) Mixing thermodynamics of the calcite-structured (Mn, Ca)CO3 solid solution: a computer simulation study. J Phys Chem B 115(47):13854–13861. https://doi.org/10.1021/jp200378q
Wang ML, Shi GH, Qin JQ, Bai Q (2018) Thermal behaviour of calcite-structure carbonates: a powder X-ray diffraction study between 83 and 618K. Eur J Mineral 30(5):939–949. https://doi.org/10.1127/ejm/2018/0030-2768
Warren BE, Averbach BL, Roberts BW (1951) Atomic size effect in the X-ray scattering by alloys. J Appl Phys 22(12):1493–1496. https://doi.org/10.1063/1.1699898
Wenk H-R, Barber DJ, Reeder RJ (1983) Microstructures in carbonates. Rev Mineral Geochem 11(1):301–367
Wenk HR, Hu MS, Lindsey T, Morris JW (1991) Superstructures in ankerite and calcite. Phys Chem Miner 17(6):527–539
Williamson GK, Hall WH (1953) X-ray line broadening from filed aluminium and wolfram. Acta Metall 1(1):22–31. https://doi.org/10.1016/0001-6160(53)90006-6
Zhang F, Xu H, Konishi H, Roden EE (2010) A relationship between d104 value and composition in the calcite-disordered dolomite solid-solution series. Am Mineral 95(11–12):1650–1656. https://doi.org/10.2138/am.2010.3414
Zucchini A, Comodi P, Katerinopoulou A, Balic-Zunic T, McCammon C, Frondini F (2012) Order–disorder–reorder process in thermally treated dolomite samples: a combined powder and single-crystal X-ray diffraction study. Phys Chem Miner 39(4):319–328. https://doi.org/10.1007/s00269-012-0489-9
Acknowledgements
This work has been supported by the Centre National de la Recherche Scientifique (CNRS)—Institut National des Sciences de l'Univers (INSU) through grant INTERRVIE 2017 to DV, by the Agence Nationale pour la Recherche through ANR MOBi 2018, and by the Centre Interdisciplinaire de Nanoscience de Marseille (CINaM) through internal grants. We would like to thank A. Saùl and A. Ricolleau for constructive discussions. This is contribution ANR MOBi 2. We are grateful to the European Synchrotron Radiation Facility for providing access to the instruments and we would like to thank Andy Fitch for assistance in using beamline ID22.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Floquet, N., Vielzeuf, D., Heresanu, V. et al. Synchrotron high-resolution XRD and thermal expansion of synthetic Mg calcites. Phys Chem Minerals 47, 48 (2020). https://doi.org/10.1007/s00269-020-01115-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00269-020-01115-5