
Abstract
The temperature dependences of the crystal structure and intensities of the (113) and (211) reflections in calcite, CaCO3, were studied using Rietveld structure refinements based on synchrotron powder X-ray diffraction data. Calcite transforms from \( R\overline{3} c \) to \( R\overline{3} m \) at about T c = 1,240 K. A CO3 group occupies, statistically, two positions with equal frequency in the disordered \( R\overline{3} m \) phase, but with unequal frequency in the partially ordered \( R\overline{3} c \) phase. One position for the CO3 group is rotated by 180° with respect to the other. The unequal occupancy of the two orientations in the partially ordered \( R\overline{3} c \) phase is obtained directly from the occupancy factor, x, for the O1 site and gives rise to the order parameter, S = 2x − 1. The a cell parameter shows a negative thermal expansion at low T, followed by a plateau region at higher T, then a steeper contraction towards T c, where the CO3 groups disorder in a rapid process. Using a modified Bragg–Williams model, fits were obtained for the order parameter S, and for the intensities of the (113) and (211) reflections.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There is considerable interest in the phase transitions in calcite, CaCO3 and nitratine, NaNO3, because they are chemically and structurally simple materials, but their transitions are not fully understood (Dove et al. 2005; Harris 1999). These materials are also of geological interest because calcite occurs in various geological environments and the orientational order–disorder transition influences the calcite–aragonite transition, which is used as a geobarometer and geothermometer (Redfern et al. 1989; Salje and Viswanathan 1976).
The transition in both isotypic calcite and nitratine involves orientational disorder of the anion groups. On heating, the symmetry changes from \( R\overline{3} c \) to \( R\overline{3} m \) and the (113) and (211) reflections disappear (indices based on hexagonal setting). Across the transition temperature, T c, the relation is c \(\left[ {R\overline{3} m} \right]\) = ½ c \(\left[{R\overline{3} c} \right].\) In \( R\overline{3} c \) calcite, successive layers of CO3 groups have apices that point in opposite directions (Fig. 1). In \( R\overline{3} m \)-disordered calcite, the apices, on average, have no such regular orientation. An individual CO3 group in disordered calcite has an equal probability of pointing in either direction (Carlson 1983; Megaw 1973; Strømme 1969a, b, 1972, 1975).

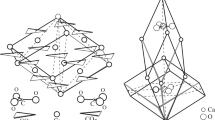
Structure of \( R\overline{3} c \) calcite: a unit cell showing ordering of the CO3 groups; in adjacent layers, the CO3 groups point in opposite directions. The Ca coordination is indicated
Detailed structure refinements are available for NaNO3 across T c (Antao et al. 2008), and for calcite to 1,073 K (Markgraf and Reeder 1985). Previous work on calcite and nitratine used various models to fit the intensities of the (113) and (211) reflections (Dove and Powell 1989; Dove et al. 2005; Harris 1999; Harris et al. 1990, 1998; Payne et al. 1997; Schmahl 1988; Schmahl and Salje 1989; Swainson et al. 1998). Theoretical studies on the transitions in calcite and nitratine were also carried out (e.g., Ferrario et al. 1994; Liu et al. 2001; Lynden-Bell et al. 1989).
Studies on the isomorphous phases (CaCO3, NaNO3, etc.) are important in understanding the transition sequences in CaCO3 and provide insights into the behavior of carbon in the mantle (e.g., Antao et al. 2004; Berg 1986). The purpose of this study is to examine the transition (at about T c = 1,240 K) in calcite on heating.
Experimental method
High purity Iceland spar calcite crystals were used in this study. An accurate room T structure was obtained using Rietveld structure refinement based on synchrotron high-resolution powder X-ray diffraction (HRPXRD) data from beam-line 11-BM, Advanced Photon Source, Argonne National Laboratory. The sample was loaded into a kapton capillary and rotated during the experiment at a rate of 90 rotations per second. The data were collected to a maximum 2θ of about 50° with a step size of 0.0005° and a step time of 0.1 s step−1. Beam-line optics consist of a platinum-coated collimating mirror, a dual Si (111) monochromator, and a platinum-coated vertically focussing mirror. The HRPXRD traces were collected with 12 silicon crystal analysers, which increase detector efficiency as well as reduce the angular range to be scanned, and therefore, allow rapid data acquisition. A silicon and alumina NIST standard (ratio of 1/3Si to 2/3Al2O3) was used to calibrate the detector response, zero offset, and to determine the wavelength used in the experiment [λ = 0.40231(2) Å]. Data were merged by interpolating measured counts onto a regularly spaced grid and by applying corrections for small differences in wavelength (~1 eV). Further details on the HRPXRD experimental set up are given in Lee et al. (2008) and Wang et al. (2008).
In situ high T synchrotron powder X-ray diffraction (XRD) experiments were performed at beam-line 1-BM, Advanced Photon Source. The sample was loaded into a quartz capillary (diameter = 0.7 mm) that was sealed and oscillated during the experiment over a θ range of 10°. The high T XRD data were collected using synchrotron radiation [λ = 0.61458(5) Å] at room pressure and from about 300 to 1,260 K. Elevated T was obtained using a heater and a thermocouple element placed close to the sample. Based on calibrations using various standards, the estimated experimental error in T was about ±0.5 K, but realistically maybe much larger at high T. Data were collected at a heating rate of about 13 K min−1 in regular intervals to a maximum of 2θ of about 30°. An image plate (IP) detector (Mar345), mounted perpendicular to the beam path was used to collect full-circle Debye–Scherrer rings with an exposure time of 5 s. An external LaB6 standard was used to determine the sample-to-detector distance, wavelength, and tilt of the IP. The two-dimensional diffraction rings recorded by the IP were integrated using the Fit2d program to produce conventional I-2θ XRD traces (Hammersley et al. 1996).
Rietveld structure refinement
The XRD traces were modeled using the Rietveld method (Rietveld 1969), as implemented in the GSAS program (Larson and Von Dreele 2000), and using the EXPGUI interface (Toby 2001). For the room T structure, the starting atomic coordinates, cell parameters, and space group, \( R\overline{3} c, \) were from Markgraf and Reeder (1985). The refined atomic coordinates were then used as input for the next higher T structure. Scattering curves for neutral atoms (Ca0, C0, O0) were used in all refinements. Two different models were used to refine the \( R\overline{3} c \) phase. In model-1, the O1 site at (x, 0, ¼) was fully occupied; this model was used from 298 to 653 K. In model-2, we introduced an O2 site at (−x, 0, ¼), while the O1 site was at (x, 0, ¼), and refined the occupancy parameter, x, for the O1 site; thus (1 − x) for the O2 site. The C–O1 and C–O2 distances were constrained to be equal. All the atoms of the CO3 groups were constrained to have the same U. Model-2 is a static disorder or Ising model and was used from 681 K to about T c (= 1,240 K). Similar to the NaNO3 structure refinements (Antao et al. 2008), we had no difficulty in the refinements of the calcite structure.
The background was modeled with a Chebyschev polynomial. The reflection-peak profiles were fitted using type-3 profile in the GSAS program. A full-matrix least-squares refinement varying a scale factor, cell parameters, zero shift, atomic coordinates, and isotropic displacement parameters converged rapidly. Towards the end of the refinement, all parameters were allowed to vary simultaneously, and the refinement proceeded to convergence. The number of observed reflections in a typical XRD trace was about 37 for the \( R\overline{3} c \) calcite (1-BM data). Synchrotron powder XRD patterns are shown in Figs. 2 and 3 as examples. The number of observed reflections in the HRPXRD trace was 509 for the \( R\overline{3} c \) phase (11-BM data, Fig. 3a). Table 1 contains structure refinement parameters for calcite, including occupancy factor, x, and normalized intensities (I 1/2) for reflections (113) and (211). Tables 2, and 3 contain the HRPXRD structural data for calcite.

Stacks of some XRD traces for calcite from 956 to 1,234 K. On heating, disappearance of (113) and (211) reflections at about T c = 1,240 K indicates the transition from \( R\overline{3} c \) to \( R\overline{3} m. \) The traces at 1,206 and 1,234 K show peaks for CaO formed by the breakdown of calcite

Comparison of the XRD traces for calcite: a part of the room T HRPXRD data, b 298 K, c 1,068 K, and d 1,149 K, together with the calculated (continuous line) and observed (crosses) profiles. The difference curve (I obs − I calc) is shown at the bottom. The short vertical lines indicate allowed reflection positions. Reflections (113) and (211) are strong in (a, b) and become weaker in c and d. In d, the (1, 0, 10) and (211) reflections overlap, whereas in a, b and c they are well separated
Results and discussion
Structure of \( R\overline{3} c \) calcite
From the stack of XRD traces at different T and Rietveld structure refinements, the \( R\overline{3} c \) to \( R\overline{3} m \) transition in calcite occurs at about T c = 1,240 K (Fig. 2). In the higher T traces, a few new peaks are seen from CaO, formed by the breakdown of calcite. The superstructure reflections, (hkl, l = odd) such as (113) and (211), are related to the ordering of the CO3 group, and they are indicated in Fig. 2. The (113) reflection is clearly visible up to 1,206 K, nearly absent at 1,234 K, and is undetectable at 1,262 K. The intensity for the (113) reflection in calcite decreases smoothly (Fig. 2). Examples of traces from the Rietveld refinement of the calcite structure at different T are given (Fig. 3).
The room T structural parameters obtained from the two different data sets are similar to each other, but that obtained by the superior HRPXRD set-up is more accurate than the IP XRD data (compare Fig. 3a and b). However, the close similarity between the two structures at room T gives us the confidence that detailed structural changes can be obtained at high T using the present IP XRD data.
Refinement of model-1 using the HRPXRD data for calcite gave a R F 2 of 0.0236 for 509 observed reflections, and the cell parameters are a = 4.9877648(1), c = 17.05242(2) Å, and V = 367.3911(4) Å3. The Ca–O = 2.3574(2) and C–O = 1.2836(3) Å compared well to 2.3595(5) and 1.280(1) Å obtained by Markgraf and Reeder (1985), or to 2.359(1) and 1.284(1) obtained by Maslen et al. (1993). Additional structural information is given in Tables 2, and 3. The \( R\overline{3} c \) structure of calcite using model-1 is similar to the \( R\overline{3} c \) structure of calcite obtained by Markgraf and Reeder (1985) and Maslen et al. (1993). The CO3 groups are planar and are along layers perpendicular to the c axis (Fig. 1). The CO3 groups in one layer have identical orientation, while those in adjacent layers are rotated by 180° with respect to the other. The coordination for the Ca atom is six (Fig. 1).
The only difference in model-2 is that two planar CO3 groups occur in the same plane that is parallel to (001) with z = ¼, and one CO3 group is rotated by 180° with respect to the other. The probability of finding the CO3 groups in any particular orientation is determined by an occupancy parameter, x, and gives rise to an order parameter, S = 2x − 1. Model-2 was confirmed by Fourier maps and subsequent successful structure refinements. When a difference Fourier (ΔF) map at 1,234 K was calculated, with the O1 and O2 sites omitted from the structural model, we observed their possible positions (Fig. 4a). The O1 and O2 sites at 1,234 K are shown in F obs map (Fig. 4b). With O1 site present (and O2 site omitted), a ΔF map at 1,177 K, shows the presence of the O2 site, indicating that model-2 is correct (Fig. 4c). However, F obs maps at 1,177 K (Fig. 4d) for model-1 (O1 site is present, O2 site omitted) did not show any O2 site, which was clearly observed in Fig. 4a, c. Dove et al. (2005) F obs map at 1,189 K showed no significant rotations of the anion group by 60° in calcite, and they indicated that calcite has considerable out-of-plane (001) disorder of the CO3 groups. The results from Dove et al. (2005) were not observed in the present study either on calcite or for NaNO3 (Antao et al. 2008). F obs maps may be biased by the structural model used to make the maps, whereas ΔF maps are usually less biased. Our ΔF map clearly shows that the CO3 group are rotated by 180° (Fig. 4a, c). The F obs maps for calcite showed that the O1 site is not smeared out (Fig. 4d), unlike that for Dove et al. (2005; their Fig. 5). Based on our results for NaNO3 and calcite, 60° rotational disorder appears to be present in both the materials.

Fourier maps for \( R\overline{3} c \) calcite at z = ¼. a ΔF at 1,234 K with the O1 and O2 sites omitted and showing their possible positions. b F obs at 1,234 K with both O1 and O2 sites present. c ΔF at 1,177 K with O2 site omitted (O1 site is present) and showing the presence of the O2 site, indicating that model 2 is correct. d F obs map at 1,177 K with only O1 site present and misleadingly does not show any O2 site. Dove et al. (2005), in their Fig. 5, observed a large spread of orientation of the CO3 groups in their F obs map
Molecular dynamics simulations indicate that the phase transition in NaNO3 and CaCO3 is initiated by a ±60° (equivalent to ±180°) reorientation of the NO3 and CO3 anions about the c axis, and the orientation of the anions are continuous with six preferred orientations above T c (Liu et al. 2001). The results from Liu et al. (2001) for the \( R\overline{3} c \) phase supports our model-2 as do the findings in earlier research (Ferrario et al. 1994; Lynden-Bell et al. 1989; Paul and Pryor 1971). Using a two-dimensional XY model, Harris (1999) indicated that the transition in NaNO3 and CaCO3 arise from orientational disorder that is driven by continuous planar rotations of the anion groups, which is not observed in the present study. The successful refinement of the \( R\overline{3} c \) calcite structure indicates that a good model for the transition is that in which the anion groups are rotated by 180°, and may be modeled using our structural model-2, as was done for NaNO3 (Antao et al. 2008).
Cell parameters
The variations of the cell parameters for the \( R\overline{3} c \) structure of calcite are shown (Fig. 5). In general, the variations of the cell parameters follow nearly the same non-linear trends observed by Markgraf and Reeder (1985), Dove and Powell (1989), and Dove et al. (2005). The a cell parameter shows a negative thermal expansion at low T, followed by a plateau region at higher T, then a steeper contraction near T c (Fig. 5a). A similar effect was observed by Dove et al. (2005). A contraction in the a parameter just before T c was also observed in NaNO3 (Antao et al. 2008). The other cell parameters (c, c/a, V; Fig. 5b–d) are similar to those reported, for example, by Dove et al. (2005).

Variations for the cell parameters: a a, b c, c c/a, d V. The a cell parameter shows a negative thermal expansion effect at low T, followed by a plateau region at higher T, then a steeper contraction from 1,149 K to 1,240 K = T c. In all cases, errors are smaller than symbols, if not seen
When the axial and volume expansions of the unit cell of CaCO3 are normalized and plotted with d/d o as a function of T, it can be seen that the expansion of the structure is strongly anisotropic with the axial expansion along the c axis (normal to the CO3 groups) being considerably larger than that along the a axis, and a negative thermal-expansion effect is seen for the a axis (Fig. 6).

Variation of the normalized a, c, and V parameters plotted as d/d 0, as a function of T. The a axis contracts slightly and large expansion occurs for the c axis
The volume thermal expansivity was obtained by fitting all the cell-volume data simultaneously to the expression:
where V(T) is the volume at any temperature T, V Tr is the volume at reference T and α V (T) is a polynomial expression for the volume thermal expansion coefficient: α V (T) = a 0 + a 1 T + a 2 T −2. The values a 0 = −5.68 (±0.83) × 10−5, a 1 = 1.02 (±0.08) × 10−7, and a 2 = 5.40 (±0.89) were obtained with V Tr = 367.45(2) Å3 at 298 K. This volume thermal expansion curve is shown in Fig. 5d.
Variation in structural parameters
The Ca–O distance increases, whereas the C–O distance gradually decreases (Fig. 7a). However, beyond 1,149 K, Ca–O distance decreases slightly, whereas the C–O distance increases steeply. These features are similar to that in NaNO3, however, near T c, both Na–O and N–O distances were almost constant in NaNO3 (Antao et al. 2008). The x coordinate for the O atom, xO, decreases to 1,149 K, beyond which it steeply increases (Fig. 7b). The displacement parameters (U) increase as T c is approached, particularly for the CO3 group (Fig. 7c).

a Variations of Ca–O and C–O distances in \( R\overline{3} c \) calcite with T. b Variation of the x coordinate of the O atom, xO, with T. c Variations of isotropic displacement parameter, U, with T. The U values for the CO3 groups increase steeply near T c. Bond distances from Markgraf and Reeder (1985) are shown as triangles and are fitted to a polynomial
It is interesting to note that at 1,075 K (=802°C), unusual effects were observed in previous studies. In high T Raman spectra of calcite, Salje and Viswanathan (1976) detected a discontinuity near 800°C in the variation of the phonon frequency with T for the 287 cm−1 mode. Using DTA and high T XRD, Mirwald (1979) observed a DTA transition at 800°C, accompanied by a reversal from negative to positive in the thermal-expansion coefficient in the a direction and a small increase in the c direction. However, we observed a further contraction in the a direction beyond 1,096 K (Fig. 5a).
The CO3 group is expected to behave as a rigid body, so no variation is expected for the C–O distances with T. The C–O distance obtained from the structure refinement is the distance between refined average positions, and they contain the effects from thermal and arc motions. The real instantaneous C–O bond lengths are unlikely to change much with T. The variations of the Ca–O and C–O distances are similar to that obtained by (Markgraf and Reeder 1985; Fig. 7a).
The order parameter, S, in model-2 indicates that partial anion disorder does not exist at ambient T where x = 1 (Table 1). From our high resolution powder X-ray diffraction (HRPXRD) data for calcite and nitratine at room T, the refined occupancy factor, x, is 0.932(1) for calcite, and 0.930(1) for nitratine, if ionized scattering curves were used for the ions (Ca2+, Na+, O2−), whereas x = 1 in both cases if neutral scattering curves were used for the atoms.
The variation of the order parameter, S, is shown in Fig. 8. A value of x = ½, corresponds to complete disorder where the O1 and O2 sites are equally populated. Once the CO3 groups begin to disorder considerably beyond about 1,122 K, the a axis contracts, and calcite reaches a fully disordered state at 1,240 K = T c. Thereafter, in the disordered \( R\overline{3} m \) structure, both the CO3 orientations occur in a 1:1 ratio. We observed the \( R\overline{3} c \) calcite structure close to T c without significant decomposition effects, but we did not observe the \( R\overline{3} m \) structure of calcite because of sample decomposition. However, after the phase transition in NaNO3, the two NO3 groups, one rotated by 180° with respect to the other, occur with equal probability, but they do not occur in the same plane (Antao et al. 2008).

Variations of the order parameter, S (=2x − 1), in \( R\overline{3} c \) calcite with T. The modified Bragg–Williams model (Eq. 3) fits the S data only near T c with a = − 0.3, b = 0 and T c = 1,240 K. Included for comparison are S parameter data for NaNO3, which is the same as that for calcite. Away from T c, the models do not match the experimental data
Thermodynamic model
A modified Bragg–Williams (BW) model was recently proposed for nitratine (Antao et al. 2008). As in the case of the NO3 − ions, we shall assume that the CO3 2− ions can only occur in two possible relative orientations, or states, designated by [1] and [2]. In a completely ordered lattice, all anions in a particular layer are present in state [1], which will be referred to as the “correct” orientation, whereas state [2] corresponds to a “wrong” orientation, inappropriate to that layer, where the anion groups are rotated by 60° with respect to the above position (our structural model-2).
The order parameter is defined as Q = 2p − 1, with p the fraction of carbonate groups in state [1]. The molar Gibbs energy of disordering will be taken to be of the form
with the molar entropy of disordering as
Equation 1 generalizes the expression given in our study of nitratine through addition of a sixth-order term in Q which, together with the fourth-order term, presumably accounts for the change in strain energy that accompanies the disordering process.
The condition for internal equilibrium (Gibbs energy of disordering minimized with respect to Q) then leads to the following relation among Q, T, and P:
T c is the critical temperature where the transition from partial (0 < Q < 1) to complete (Q = 0) disorder occurs. Parameters T c, a, and b are expected to depend exclusively on pressure, P.
Defining w as the work required to rotate a single anionic group through 60° in an otherwise perfectly ordered lattice, the BW formula for the critical temperature is (see Appendix 1 as an electronic supplementary material):
(k is the Boltzmann constant), which marks the \( R\bar{3}c \leftrightarrow R\bar{3}m \) transition between states with partial and complete orientational disorder of carbonate groups (Antao et al. 2008). The observed value for the critical temperature of CO3 group disordering at atmospheric pressure, T c = 1,240 K, can be used to estimate the energy to rotate one mole of CO3 groups in an ordered lattice as 20.6 kJ mol−1, according to Eq. 4.
The overall molar energy change of disordering, as predicted by the BW model (Eq. 1), is given as:
and, depending on the values of the fit parameters, would be equal to 5.8 kJ mol−1 (a = − 0.25, b = 0), 5.93 kJ mol−1 (a = − 0.3, b = 0), or 6.4 kJ mol−1 (a = − 0.114, b = − 0.560) for calcite. These should be compared with the calorimetric estimate of 10 ± 1.1 kJ mol−1 reported by Redfern et al. (1989).
It can be shown that for \(a \neq -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 3$}}\),
in the vicinity of T c, where \( t = 1 - T/T_{c} \) is the reduced temperature, so that the critical exponent β = ½ in this approximation, as compared to the experimental value of ¼ reported earlier for calcite (Dove et al. 2005). Only if b = 0, it is possible to unambiguously classify transitions as first-order if \(a < -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 3$}}\) and as second order if \(a > -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 3$}}\). In general, a transition is first-order (i.e., exhibits hysteresis if the temperature is scanned back and forth across T c) according to this model if, for given P, the equation T(Q, P) = T c(P) has a solution other than the trivial solution Q = 0. Otherwise, it is second order. No simple analytical expressions in terms of a and b can be given to characterize first- and second-order transitions.
In the case \(a = -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 3$}}\), tricritical behavior occurs if \(b = -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 5$}}\) according to the BW model, with
close to T c, so that the transition is second order if \(b > -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 5$}}\), and first order if \(b < -{\raise0.5ex\hbox{$\scriptstyle 1$} \kern-0.1em/\kern-0.15em \lower0.25ex\hbox{$\scriptstyle 5$}}.\)
Behavior of (113) and (211) reflections and order parameter, S
The intensities of the (113) and (211) reflections were used to fit a disorder model in calcite and NaNO3 because Q ∝ I 1/2 (e.g., Dove et al. 2005). The intensities for both the (113) and (211) reflections are affected by the same disordering process, so they are expected to behave in a similar manner, as was observed in NaNO3 (Antao et al. 2008). Table 1 contains the normalized intensities for the (113) and (211) reflections in \( R\overline{3} c \) calcite. Using our present data, and plotting I 1/2 versus T, the data points are shown and a curve is drawn with a = −0.25, b = 0 and T c = 1,240 K (Eq. 3), and the model fits both the (113) and (211) reflections only near T c (Fig. 9a). The value of a = − 0.25, b = 0 implies a second-order transition. Our data for both (113) and (211) reflections are close to each other and follow a similar curve. The overlap of the (1, 0, 10) reflection with (211) in a certain T range cannot account for the difference in behavior for the (113) and (211) reflections. In case of NaNO3, the fit to I 1/2 data for both (113) and (211) reflections were good (Antao et al. 2008; Fig. 9b). Instead of I 1/2 for NaNO3, I 2 was used for calcite by Dove et al. (2005) and they obtained a straight line for (113), but not for the (211) reflection (Fig. 9a). Our I 2 results for (113) and (211) in calcite show that they behave in a similar manner, but they deviate from the linearity near T c, especially for the (113) reflection, as is also the case for NaNO3 (Fig. 9a, b).

Variations of the normalized intensity (I 1/2 and I 2) for the (113) and (211) reflections in \( R\overline{3} c \) a calcite, and b nitratine. In calcite, a fit to the I 1/2 data points near T c is obtained with a = − 0.25, b = 0, T c = 1240 K (Eq. 3). Our I 2 data points for the (113) and (211) reflections are close to each other and do not vary linearly
Using our BW model (Eq. 3), a fit near T c was obtained for the order parameter, S (=2x − 1), with a = − 0.3, b = 0 and T c = 1,240 K for \( R\overline{3} c \) calcite; this fit is different from that using intensities (compare Figs. 8 and 9), and is indicative of a second order phase transition. Included in Fig. 8 are S data for NaNO3, which follows the same trend as that for calcite after scaling the temperature axis with respect to T c.
The fit of the thermodynamic models near T c and the continuous variation of the cell parameters across T c (in the case of NaNO3) indicate that the transition is second order in both the minerals (Figs. 8, 9). The thermodynamic model, Eq. 3 with b = 0 has some shortcomings in that it does not match the experimental data points throughout the T range.
The isomorphous calcite and nitratine should give similar trends for the order parameters S and the intensities for the (113) and (211) reflections that were obtained experimentally and a unique model should explain the order–disorder transitions in both isomorphic minerals. For both minerals, the S and intensity data points are compared (Fig. 10). Only the S data points are similar for both the minerals, while the intensities are different. Therefore, a model that explains the variations in the S data will uniquely describe the transition in both minerals. The model with b = 0 presented above does not work well throughout the T range. Allowing non-zero b values produces a common fit as shown for both calcite and nitratine in Fig. 10, and the match is excellent for a = −0.114, b = −0.560.

Variations of the order parameter S and normalized intensity (I 1/2) for the (113) and (211) reflections in \( R\overline{3} c \) calcite and nitratine. A common fit to only S data points for both calcite and nitratine is shown with a = − 0.114, b = − 0.560 (Eq. 3)
References
Antao SM, Mulder WH, Hassan I, Crichton W, Parise JB (2004) Cation disorder in dolomite, CaMg(CO3)2, and its influence on the aragonite + magnesite → dolomite reaction boundary. Am Mineral 89:1142–1147
Antao SM, Hassan I, Mulder WH, Lee PL (2008) The \( R\overline{3} c \to R\overline{3} m \) transition in nitratine, NaNO3 and implications for calcite, CaCO3. Phys Chem Miner. doi:10.1007/s00269-008-0232-8
Berg GW (1986) Evidence for carbonate in the mantle. Nature 324:50–51. doi:10.1038/324050a0
Carlson WD (1983) The polymorphs of CaCO3 and the aragonite-calcite transformations. Mineral Soc Am Rev Mineral 11:191–225
Dove MT, Powell BM (1989) Neutron-diffraction study of the tricritical orientational order–disorder phase-transition in calcite at 1260-K. Phys Chem Miner 16:503–507. doi:10.1007/BF00197019
Dove MT, Swainson IP, Powell BM, Tennant DC (2005) Neutron powder diffraction study of the orientational order–disorder phase transition in calcite, CaCO3. Phys Chem Miner 32:493–503. doi:10.1007/s00269-005-0026-1
Ferrario M, Lyndenbell RM, McDonald IR (1994) Structural fluctuations and the order–disorder phase-transition in calcite. J Phys Condens Matter 6:1345–1358. doi:10.1088/0953-8984/6/7/007
Hammersley AP, Svensson SO, Hanfland M, Fitch AN, Hausermann D (1996) Two-dimensional detector software: from real detector to idealised image to two-theta scan. High Press Res 14:235–248. doi:10.1080/08957959608201408
Harris MJ (1999) A new explanation for the unusual critical behavior of calcite and sodium nitrate, NaNO3. Am Mineral 84:1632–1640
Harris MJ, Salje EKH, Guttler BK (1990) An infrared spectroscopic study of the internal-modes of sodium-nitrate—implications for the structural phase-transition. J Phys Condens Matter 2:5517–5527. doi:10.1088/0953-8984/2/25/004
Harris MJ, Hagen ME, Dove MT, Swainson IP (1998) Inelastic neutron scattering, phonon softening, and the phase transition in sodium nitrate, NaNO3. J Phys Condens Matter 10:6851–6861. doi:10.1088/0953-8984/10/30/022
Larson AC, Von Dreele RB (2000) General structure analysis system (GSAS). Los Alamos National Laboratory Report, LAUR 86-748
Lee PL, Shu D, Ramanathan M, Preissner C, Wang J, Beno MA et al (2008) A twelve-analyzer detector system for high-resolution powder diffraction. J Synchrotron Radiat 15(Pt 5):427–432
Liu JJ, Duan CG, Ossowski MM, Mei WN, Smith RW, Hardy JR (2001) Simulation of structural phase transition in NaNO3 and CaCO3. Phys Chem Miner 28:586–590. doi:10.1007/s002690100191
Lynden-Bell RM, Ferrario M, Mcdonald IR, Salje E (1989) A molecular-dynamics study of orientational disordering in crystalline-sodium nitrate. J Phys Condens Matter 1:6523–6542. doi:10.1088/0953-8984/1/37/002
Markgraf SA, Reeder RJ (1985) High-temperature structure refinements of calcite and magnesite. Am Mineral 70:590–600
Maslen EN, Streltsov VA, Streltsova NR (1993) X-Ray study of the electron-density in calcite, CaCO3. Acta Crystallogr B 49:636–641. doi:10.1107/S0108768193002575
Megaw HD (1973) Crystal structures: a working approach. W. Saunders, Philadelphia
Mirwald PW (1979) Determination of a high-temperature transition of calcite at 800°C and one bar CO2 pressure. Neues Jahrb Miner Monatsh 7:309–315
Paul GL, Pryor AW (1971) The study of sodium nitrate by neutron diffraction. Acta Crystallogr B 27:2700–2702
Payne SJ, Harris MJ, Hagen ME, Dove MT (1997) A neutron diffraction study of the order–disorder phase transition in sodium nitrate. J Phys Condens Matter 9:2423–2432. doi:10.1088/0953-8984/9/11/010
Redfern SAT, Salje E, Navrotsky A (1989) High-temperature enthalpy at the orientational order–disorder transition in calcite—implications for the calcite–aragonite phase-equilibrium. Contrib Mineral Petrol 101:479–484. doi:10.1007/BF00372220
Rietveld HM (1969) A profile refinement method for nuclear and magnetic structures. J Appl Cryst 2:65–71. doi:10.1107/S0021889869006558
Salje E, Viswanathan K (1976) Phase-diagram calcite-aragonite as derived from crystallographic properties. Contrib Mineral Petrol 55:55–67. doi:10.1007/BF00372754
Schmahl WW (1988) Diffraction intensities as thermodynamic parameters—orientational ordering in NaNO3. Z Kristallogr 182:231–233
Schmahl WW, Salje E (1989) X-ray-diffraction study of the orientational order–disorder transition in NaNO3—evidence for order parameter coupling. Phys Chem Miner 16:790–798. doi:10.1007/BF00209703
Strømme KO (1969a) The crystal structure of sodium nitrate in the high-temperature phase. Acta Chem Scand 23:1616–1624. doi:10.3891/acta.chem.scand.23-1616
Strømme KO (1969b) On the crystal structure of potassium nitrate in the high temperature phases I and III. Acta Chem Scand 23:1625–1636
Strømme KO (1972) On the mechanism of the continuous transformation in sodium nitrate. Acta Chem Scand 26:477–482
Strømme KO (1975) Crystal-structures of high-temperature forms of strontium and barium carbonate and structurally related compounds. Acta Chem Scand A 29:105–110
Swainson IP, Dove MT, Harris MJ (1998) The phase transitions in calcite and sodium nitrate. Physica B 241:397–399
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Crystallogr 34:210–221
Wang J, Toby BH, Lee PL, Ribaud L, Antao SM, Kurtz C, Ramanathan M, Von Dreele RB, Beno MA (2008) A dedicated powder diffraction beamline at the advanced photon source: commissioning and early operational results. Rev Sci Instrum 79:085105. doi:10.1063/1.2969260
Acknowledgments
We thank the anonymous reviewers for useful comments. XRD data were collected at the X-ray Operations and Research beamlines 1-BM and 11-BM, Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Antao, S.M., Hassan, I., Mulder, W.H. et al. In situ study of the \( R\overline{3} c \to R\overline{3} m \) orientational disorder in calcite. Phys Chem Minerals 36, 159–169 (2009). https://doi.org/10.1007/s00269-008-0266-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00269-008-0266-y