
Abstract
Modified FOLFOX6 is an established therapy for patients with metastatic colorectal cancer (mCRC). We conducted a single-arm phase Ib study to address the hypothesis that addition of pembrolizumab to this regimen could safely and effectively improve patient outcomes (NCT02375672). The relationship between immune biomarkers and clinical response were assessed in an exploratory manner. Patients with mCRC received concurrent pembrolizumab and modified FOLFOX6. The study included safety run-in for the first six patients. The primary objective was median progression-free survival (mPFS), with secondary objectives including median overall survival, safety, and exploratory assessment of immune changes. To assess immunological impact, peripheral blood was collected at baseline and during treatment. The levels of soluble factors were measured via bioplex, while a panel of checkpoint molecules and phenotypically defined cell populations were assessed with flow cytometry and correlated with RECIST and mPFS. Due to incidences of grade 3 and grade 4 neutropenia in the safety lead-in, the dose of mFOLFOX6 was reduced in the expansion cohort. Median PFS was 8.8 months and median OS was not reached at data cutoff. Best responses of stable disease, partial response, and complete response were observed in 43.3%, 50.0%, and 6.7% of patients, respectively. Several soluble and cellular immune biomarkers were associated with improved RECIST and mPFS. Immunosuppressive myeloid and T cell subsets that were analyzed were not associated with response. Primary endpoint was not superior to historic control. Biomarkers that were associated with improved response may be informative for future regimens combining chemotherapy with immune checkpoint inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nearly two million cases of colorectal cancer (CRC) are diagnosed worldwide each year [1]. Of these, approximately 22% of patients present with metastasis at diagnosis and these patients have a dismal 5-year survival rate of 14% [2, 3]. The standard of care for metastatic CRC (mCRC) when this trial was initiated (2014) included 5-fluorouracil/leucovorin with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI), but biologics including bevacizumab are common in current clinical paradigms [4,5,6]. First line FOLFOX only extends median progression-free survival (mPFS) for mCRC to 9.0 months, a mark that we hypothesized may be improved by addition of immune checkpoint blockade [7]. As a monotherapy, the PD-1 neutralizing antibody, pembrolizumab is ineffective outside of the ~ 5% of CRC patients with DNA mismatch repair deficient/microsatellite instable (dMMR/MSI) tumors [8, 9]. Therefore, novel therapeutic strategies in advanced CRC are needed.
A prior phase Ib clinical trial demonstrated pembrolizumab can be safely combined with cytotoxic chemotherapy in solid tumors [10]. Further, other immunotherapies, including atezolizumab, bevacizumab, and nivolumab have been combined with FOLFOX and FOLFIRI in clinical trials for gastrointestinal malignancies [5, 6, 11]. Certainly, systemic chemotherapy can result in lymphopenia, which implies potential for adverse impacts on T cell-mediated immune responses. However, administration of chemotherapy may also have immunomodulatory potential by catalyzing release of tumor antigens [12,13,14]. Surprisingly few studies have carefully assessed dynamic immune changes following combined chemotherapy and immune checkpoint blockade.
Previous work identified immune checkpoints as aberrantly expressed and capable of suppressing anti-tumor T cell activity in mCRC [15,16,17]. We hypothesized that basal levels of immune checkpoint molecules on circulating T cells would inform response to our combination therapy. Additionally, we interrogated previously identified biomarkers of response to pembrolizumab including proliferating CD8+ T cells in the blood [18].
In addition to increasing tumor antigens, and immunogenic cell death, FOLFOX may also regulate prevalence of immunosuppressive T regulatory cells (Treg) and myeloid-derived suppressor cells (MDSC) [19, 20]. A component of FOLFOX, 5-fluorouracil, can kill MDSCs, and increase T cell-dependent antitumor immunity [21, 22]. Furthermore, when combined with bevacizumab, FOLFOX decreases Tregs, granulocytic MDSCs, and increases Th17 cells [23]. Notably, decreased MDSCs is associated with increased survival. Therefore, we also chose to assess changes in circulating levels of immunosuppressive cell populations during treatment and how they correlated with response.
We hypothesized pembrolizumab could be safely combined with mFOLFOX6 in mCRC patients and this combination therapy would provide an extension of mPFS and mOS compared to historical data on mFOLFOX6, or pembrolizumab, alone. To address this hypothesis, we initiated a multicenter single-arm phase Ib clinical trial (NCT02375672). In addition to safety and efficacy of combined mFOLFOX6 and pembrolizumab, this trial included exploratory correlative analysis of immune markers in peripheral blood.
Methods
Study design
This trial was a single-arm, multicenter, phase Ib study of mFOLFOX6 combined with pembrolizumab in mCRC patients who are treatment naïve (NCT02375672). All patients consented to the study following protocols established by the IRBs of each institution. The study enrolled 6 patients in the safety run-in cohort and 24 patients in the expansion cohort. The primary objective of the trial was to estimate median progression-free survival (mPFS) and compare it to a historical standard [7]. As such, the primary endpoint, mPFS, was measured from time of registration, to time of progression, per RECIST v1.1, or death. Secondary objectives included median overall survival (mOS), assessment of toxicity of combined pembrolizumab and mFOLFOX6 per CTCAE v4.0, and objective response rate (ORR) calculated using best RECIST response recorded in the observation period. Laboratory correlatives of cellular and soluble immune biomarkers were assessed as exploratory objectives.
Patients
Patients were eligible for the trial if they presented with histological or cytological evidence of colorectal adenocarcinoma with confirmation of metastatic disease measurable according to RECIST v1.1 [24]. Detailed inclusion criteria are in the supplemental methods.
Treatment
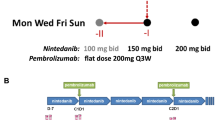
Modified FOLFOX6 was administered every two weeks and pembrolizumab administered every 3 weeks. Details on dosing and schedule are enumerated in the supplemental material. For correlative analyses, one “cycle” was defined as two weeks after administration of mFOLFOX6. Standard premedication was administered with recommendation to avoid steroids for antiemetic prophylaxis. For the safety run-in, patients were followed for four weeks to assess dose-limiting toxicities (DLT). DLT were defined as any grade 3–4 toxicity other than fatigue, nausea, vomiting, or diarrhea lasting less than 72 h. If DLT was observed in > 1 patient of the run-in cohort, doses of the mFOLFOX6 regimen were decreased.
Clinical assessment
Patients were evaluated every four weeks for study purposes and underwent imaging with CT or MRI every eight weeks to assess disease response.
Investigation of soluble and cellular biomarkers
Whole blood samples were collected for biomarker analysis at cycle 1 day 1 (C1D1) prior to initiation of therapy, cycle 1 day 15 (C1D15), and cycle 3 day 1 (C3D1) at time of first restaging. Blood samples were centrifuged at 575×g for 10 min to isolate plasma from cellular components. Plasma was collected and stored at − 80 °C until analysis. Peripheral blood mononuclear cells (PBMC) were isolated from cellular blood components using density-gradient centrifugation in Ficoll-Paque Plus (GE Healthcare, 17-1440-02) and cryopreserved.
Absolute levels of 40 soluble chemokines, cytokines, and growth factors were measured in plasma using Luminex Human XL Cytokine Discovery Premixed Kit (catalog # FCSTM18-45), with testing conducted by Biomarker Testing Service (R&D Systems, Racine WI). Flow cytometric analysis of PBMCs was performed on a FACS Canto II flow cytometer (BD Bioscience) and data analysis was performed in FlowJo (version 10.5.3; FlowJo, LLC, Ashland OR). A list of antibodies can be found in the supplemental information (Supplemental Table 1) with gating strategy (Supplemental Fig. 1).

Progression-free survival, overall survival, and RECIST response. The Kaplan–Meier method was used to determine median progression-free survival (PFS) (a) and median overall survival (OS) (b) in patients on the trial. Patients without events were censored and are indicated by tick marks on the survival curves. RECIST v1.1 was utilized to determine disease response (c) which is displayed with Agresti-Coull 95% confidence intervals. Tumor growth curves are displayed and color coded by RECIST response (c) or molecular alterations (d). The cutoffs for progressive disease and partial response are indicated by dotted lines. Overall response rate (ORR) and duration of response (DoR) are displayed (e). Blue bars indicate recorded period of response (CR and PR) in patients who responded (e). CI confidence interval, PFS progression-free survival, OS overall survival, CR complete response, PR partial response, SD stable disease, PD progressive disease, ORR overall response rate, DoR duration of response
Immunohistochemical analysis
Formalin-fixed paraffin-embedded (FFPE) samples from 13 patients with adequate archival tissue were analyzed by immunohistochemistry. Samples were stained for CD4 (Rabbit polyclonal; VWR, 102158-736), CD8 (Rabbit polyclonal; Abcam, ab4055), and PD-L1 (Rabbit polyclonal; Abcam, ab233482) and scanned on a Nanozoomer 2.0HT whole-slide scanner (Hamamatsu Photonic K.K). Percent-positive area was quantified in FIJI.
Statistical analysis
The 30 patients enrolled in this trial were analyzed as a single cohort for safety, efficacy, and predictive biomarkers. PFS and OS were estimated by Kaplan–Meier method with median values determined with 80% confidence intervals (CIs). Agresti-Coull 95% CIs were utilized to determine response rates. Descriptive statistics were employed to summarize duration of response and toxicities described with frequencies and proportions. The Common Terminology Criteria for Adverse Events (CTCAE) v4 was used to assess acute and late toxicities [25]. More information regarding statistical procedures is in the supplemental methods. Significant differences were considered those with P < 0.05. Analysis of immune-related correlates was performed in an exploratory manner and therefore false discovery rate adjusted P values were not employed.
Results
The combination of mFOLFOX6 and pembrolizumab is well-tolerated
From April 2015 to August 2016, 30 treatment naïve mCRC patients were enrolled and received combined pembrolizumab and mFOLFOX6 (Table 1). Most patients initially presented with stage IV disease and ECOG performance score of 0. Upon enrollment on the trial, all patients had progressed to stage IV disease. Many patients presented with liver metastases (73.3%) while others displayed metastases to lung (23.3%) and lymph nodes (16.7%). Dose-limiting toxicity for this study was defined as a grade 3 or 4 toxicity that delays treatment for over seven days. Overall, the combination was well-tolerated, with only two adverse events (AE) of grade 4 (Supplemental Table 2), both cases of neutropenia occurring in the safety lead-in. These AEs resulted in dose delay for mFOLFOX6 and dose reduction for the remainder of the trial. All patients in the safety lead-in received full dose mFOLFOX6 and all patients in the remainder of the trial received a reduced dose. Following dose reduction, no patients experienced dose-limiting toxicities.
At the data collection cutoff of 24 months after trial completion, in December 2018, 36.7% of patients had not progressed, and 66.7% were alive (Fig. 1a, b). Median follow-up was 19.9 months. At this time, 27 patients remained on trial. The mPFS was 8.8 months (80% CI, 7.7–11.3 months) and mOS was not reached (80% CI, NR-NR). Of 30 patients on trial, 2 (6.7%) experienced complete responses (CR), 15 (50.0%) had partial responses (PR), and 13 (43.3%) had stable disease (SD) (Fig. 1c). Four patients were confirmed as dMMR/MSI. One of these patients experienced SD and the remaining three experienced PR (Fig. 1d), while one had a sustained response. Of the two CR on trial, neither were assessed for dMMR/MSI status. We were unable to stratify patients by dMMR/MSI status due to incomplete characterization of all patients (Table 1). The overall response rate (ORR) when considering best responses was 56.7% (Fig. 1E). Of responding patients, the average duration of response (DoR) was 37.57 weeks.
Mean treatment exposure for all drugs administered on trial was greater than 30 weeks (Supplemental Table 3). Following treatment, some patients received surgical and medical interventions (Supplemental Table 4). Notably, some had resection of the parent colorectal tumor (10%) or site of liver metastasis (6.7%). Many patients received subsequent FOLFIRI (30%) and other chemotherapy. Further, biologics targeting VEGF and EGFR were administered to patients following this trial either alone, or combined with FOLFIRI.
Immune biomarkers are altered by treatment
We first sought to identify soluble and cellular biomarkers significantly altered early in treatment by comparing levels at cycle 1 day 15 (C1D15) to baseline (C1D1). This inquiry identified a slew of soluble biomarkers altered by mFOLFOX and pembrolizumab (Fig. 2a). Notably, we observed a significant increase in CXCL10, a chemokine tied to poor prognoses for CRC patients [26]. Additionally, we detected significant increases in circulating granzyme B, perhaps indicating increased T cell-mediated anti-tumor immunity.

Soluble and cellular biomarkers are significantly altered by mFOLFOX6 and pembrolizumab treatment. A heatmap displays soluble immune biomarkers that were assessed on this trial and how they were altered by treatment between baseline (C1D1) and cycle 1 day 15 (C1D15) (a). Significant changes in cellular biomarkers (b) are also displayed. Paired student’s t test used to compare C1D1 and C1D15 values. *P < 0.05. **P < 0.01, ***P < 0.001, ****P < 0.0001
Cellular biomarkers were also significantly altered by treatment, including increased proliferating CD8+ T cells, and decreased FasR expression on T cells (Fig. 2b), consistent with previous reports [18]. Levels of FoxP3+ CD4+ T cells with a regulatory phenotype were also increased by treatment. Finally, Tc1 and Tc17 cells were decreased by treatment, a noteworthy result given the pro-tumoral role for Tc17 cells in other malignancies [27]. We next sought to identify if these changes were associated with response to this regimen.
Soluble immune biomarkers are associated with RECIST response
First, we drew associations between baseline soluble immune biomarkers in plasma and RECIST response. Our results illustrated below detection limit levels of TNF-α were associated with improved RECIST response (Fig. 3a). Considering the dynamics of chemokine and cytokine levels during immune activation, we also assessed changes in soluble factors throughout the course of treatment, as they may indicate response to immunotherapy [18, 28, 29]. We compared circulating levels of soluble factors at C1D15 and C3D1 to levels at baseline (C1D1) to determine associations with changes in their levels and RECIST response. These data indicated that decreased CXCL10 from baseline to C1D15 was associated with improved RECIST response (Fig. 3b). No soluble biomarker changes between baseline and C3D1 were associated with response.

The baseline levels and change throughout treatment of soluble immune biomarkers are associated with RECIST response and mPFS. The circulating levels of numerous soluble immune biomarkers were measured in patient plasma. Soluble biomarkers whose baseline levels (a) or change over time (b) were significantly associated with RECIST response are displayed. Moreover, survival curves for soluble biomarkers whose baseline levels (c) or change over time (d) were significantly associated with mPFS are illustrated. Fisher’s exact test (a, b) or the log-rank method (c, d) utilized to assess statistically significant changes
We next determined if there was an association between soluble biomarker levels and mPFS. At baseline, above median circulating levels of two soluble factors, Flt3 ligand and TGF-α, were associated with improved mPFS (Fig. 3c). We also discovered an increase in CCL5 levels at C3D1 from baseline was associated with worse mPFS (Fig. 3d).
Alterations in immune checkpoint levels on T cells are associated with RECIST response and mPFS
Previous work has established that increased expression of immune checkpoints on T cells in mCRC is associated with suppressed T cell activity and worse prognosis [15,16,17]. Furthermore, T cell proliferation following pembrolizumab administration is associated with response in NSCLC [18]. Therefore, we next assessed if baseline expression of immune checkpoint molecules or proliferation markers on T cells, as well as changes in their expression over time were associated with RECIST response or mPFS.
Baseline frequency of immune checkpoint receptors on CD4+ and CD8+ T cell were compared to RECIST response. Patients with below median co-expression of PD-1 and the checkpoint molecules BTLA or LAG3 on CD4+ T cells at baseline had improved RECIST response (Fig. 4a). CD8+ T cells concurrently expressing PD-1 and LAG3 were also lower at baseline in patients with better RECIST response (Fig. 4a). Moreover, patients with below median levels of CD4+ T cells co-expressing CD27 and PD-1 or co-expressing CD28 and Ki67 had improved RECIST response (Fig. 4a).

Immune checkpoint expression is significantly associated with patient outcomes. Flow cytometry on peripheral blood mononuclear cells isolated from patient blood samples was used to assess the expression of various immune checkpoint molecules and associated proliferative markers on T cells. Those whose baseline levels (a) or change at later time points (b) were significantly associated with RECIST response are displayed. Survival curves illustrating immune checkpoint molecules and associated proliferative markers whose baseline levels (c) or change over time (d) were significantly associated with mPFS. C1D1 = cycle 1 day 1, C1D15 = cycle 1 day 15, C3D1 = cycle 3 day 1. Statistical significance determined with Fisher’s exact test (a, b), or the log-rank method (c, d)
For longitudinal changes, comparing differences in C1D15 values to C1D1 elucidated that patients who experienced a decrease in VISTA expression on CD8+ T cells had improved RECIST response (Fig. 4b). Here, differences were more frequently evident when comparing C3D1 values to C1D1. Increased frequency of BTLA expression on CD8+ T cells was associated with improved RECIST response. Additionally, patients with decreases in proliferating CD4+ T cells and VISTA+ CD4+ T cells experienced improved RECIST response (Fig. 4b).
To assess immune cell checkpoints in context of response, we also dichotomized patients based on RECIST criteria (CR/PR vs. SD/PD) and examined differences in the baseline levels of immune checkpoints on T cells. No differences in immune checkpoint expression were observed with this analysis.
To increase rigor and complement RECIST response data, we assessed if baseline levels (Fig. 4c) or changes (Fig. 4d) in checkpoint expression or cell proliferation throughout treatment were associated with mPFS. On CD8+ T cells, below median baseline co-expression of LAG3 and PD-1 on CD8+ T cells was associated with improved mPFS. Also, decreased CTLA-4 and PD-1 co-expression on CD8+ T cells between C1D15 and C1D1 was associated with improved mPFS.
Alterations in phenotypically defined immune cell subsets are associated with RECIST response and mPFS
In addition to immune checkpoint expression on T cells, we tested if there were other phenotypically defined immune cell subsets associated with patient outcomes (Fig. 5). Baseline frequency of two phenotypically defined CD8+ T cell subsets was significantly associated with improved RECIST response (Fig. 5a). Patients with below median baseline CD8+ T cells co-expressing PD-1 and FasR, or above median baseline levels of CD8+ T cells co-expressing PD-1 and RORγt had improved RECIST response. Below median levels of phenotypically defined CD4+ and CD8+ memory cells at baseline (Fig. 5d) were associated with longer mPFS. Moreover, below median baseline levels of Th17 cells (CD4+RORγt+) expressing PD-1 were associated with improved mPFS.

The levels of various phenotypically defined immune cell populations are associated with patient outcomes. Flow cytometry was used to assess the presence of various phenotypically defined immune cell populations in peripheral blood mononuclear cells isolated from patient blood samples. Those whose baseline levels (a) or change throughout treatment (b) were significantly associated with RECIST response are displayed. Phenotypically defined immune cell populations whose levels are significantly different at baseline (c) when comparing responders to non-responders are illustrated. Survival curves for populations whose baseline levels (d) or change over time (e) were significantly associated with mPFS are displayed. C1D1 = cycle 1 day 1, C1D15 = cycle 1 day 15, C3D1 = cycle 3 day 1. Statistical comparisons made with Fisher’s exact test (a, b), unpaired student’s t test (c), or the log-rank method (d, e)
Patients were alternatively dichotomized based on RECIST response (CR/PR vs. SD/PD), and differences in baseline levels of phenotypically defined immune cell populations were assessed (Fig. 5c). This analysis demonstrated patients with poor RECIST response displayed low levels of CD8+ T cells co-expressing RORγt or GATA3 and PD-1.
We also interrogated how changes in phenotypically defined cells throughout treatment (Fig. 5b, e) associated with RECIST response and mPFS. A decrease in both Tc17 cells expressing PD-1, and NK cells from baseline to C1D15 was associated with positive RECIST response (Fig. 5b). We also found two populations whose change over time associated with mPFS. When comparing C1D15 to C1D1, a decrease in CD4+ cells with a memory phenotype was associated with improved mPFS. Moreover, an increase in B cells between C3D1 and C1D1 was associated with improved mPFS (Fig. 5e).
Baseline levels and changes in circulating MDSC and Treg subsets are not associated with RECIST response or mPFS
Although previous work demonstrates chemotherapy can deplete MDSCs and Tregs to enhance immunotherapy efficacy [19, 20, 23, 30], a notable result was that baseline levels of suppressive myeloid or T regulatory cell subsets were not associated with RECIST response or mPFS. These results were consistent for phenotypically defined MDSCs, polymorphonuclear CD15-positive MDSCs, FoxP3-positive CD4+ T cells, and Tregs (Supplemental Fig. 2A, C). Changes between C1D15 or C3D1 versus C1D1 were also not associated with RECIST response or mPFS (Supplemental Fig. 2B, D).
CD4, CD8, and PD-L1 expression in FFPE tissue is not correlated with RECIST response
For a subset of patients (13/30), sufficient FFPE sections were available for immunohistochemical analysis. We stained sections with hematoxylin an eosin (H&E) to determine tumor areas and for CD4, CD8, and PD-L1 to assess associations with RECIST response (Supplemental Fig. 3). Notably, the percent-positive area for each marker was not correlated with RECIST outcomes.
Discussion
Considering the high prevalence and lethality of mCRC, development of new and optimized therapeutic regimens is vital [1,2,3]. Both the current standard of care, FOLFOX, and burgeoning immune therapies demonstrate limited efficacy in mCRC outside of a small subset of patients harboring highly immunogenic tumors that respond well to pembrolizumab [4, 8, 9]. In other cancers that are refractory to traditional chemotherapy and immunotherapy, notably NSCLC, TNBC, and GC/GEJC, combining the two approaches has yielded significantly improved responses compared to either approach alone [11, 31, 32]. Therefore, the question arises whether similar therapeutic approaches translate to mCRC. With this idea in mind, we initiated this phase Ib clinical trial to assess safety and efficacy of combined mFOLFOX6 and pembrolizumab in mCRC. This trial is one of the first to investigate combined cytotoxic chemotherapy and immunotherapy in the context of mCRC and the first to comprehensively analyze cellular and soluble immune biomarkers of response in this context.
In this single-arm trial, all patients received combined therapy with mFOLFOX6 and pembrolizumab. Therefore, we must resort to historic controls to determine efficacy relative to either mFOLFOX6 or pembrolizumab alone. The FOLFOX regimen as first-line therapy provides mPFS of 9.0 months, with mOS of 16.2 months in advanced CRC [7]. Moreover, in patients without mismatch repair deficiency or microsatellite instability, the ORR to pembrolizumab is 0%, with an immune-related PFS rate of 11% at 5 months [9]. In our study, at the data cutoff timepoint, mPFS was 8.8 months and mOS was not reached with median follow-up of 19.9 months. Considering mPFS in comparison to historical controls, combined pembrolizumab and mFOLFOX6 does not extend mPFS over FOLFOX alone. In this trial, we observed an ORR of 56.7%, aligned with FOLFOX alone or combined with other biologics like cetuximab or regorafenib [7, 33,34,35]. Our data support the hypothesis that cytotoxic chemotherapy and targeted biologics can be safely co-administered in mCRC patients.
Three patients with confirmed dMMR/MSI status experienced PR on this trial. Notably, one MSI-H patient had a long-term response. Neither of the CR observed were assessed for dMMR/MSI status. Following the trial, a subset of patients had their parent colorectal tumor (10%) or liver metastasis (6.7%) resected. This suggests that the combination of mFOLFOX6 and pembrolizumab may reduce tumor burden and allow patients with metastatic disease to pursue surgical options.
In addition to the primary objective of determining mPFS, we also included exploratory immune profiling of blood as a secondary objective. While we recognize that peripheral chemokine and cytokine levels do not necessarily reflect levels in tumors, circulating biomarkers may be prognostic [36, 37]. We first drew associations between circulating soluble immune biomarkers at baseline, or throughout treatment, with clinical outcomes. Below median baseline TNF-α levels associated with better RECIST response. This is notable considering the role for TNF-α in driving metastatic phenotype in CRC [38]. Above median baseline levels of two soluble factors, Flt3 ligand and TGF-α, associated with improved mPFS and increased CCL5 throughout treatment was associated with poor mPFS. Our findings that increased CCL5 is associated with poor mPFS support efforts targeting its receptor, CCR5, in mCRC [39, 40].
We next determined associations between immune checkpoint expression with RECIST response and mPFS. Importantly, we demonstrated that below median baseline expression of LAG3 on both CD4+ and CD8+ T cells was associated with improved RECIST response. Below median LAG3 at baseline on CD8+ cells was also significantly associated with improved mPFS. LAG3 is upregulated in MSI-H colorectal cancer and LAG3 blockade enhances infiltration and activity of cytotoxic T cells into CRC liver metastases [41, 42]. Clinical trials targeting LAG3 in CRC are underway and our data support the concept [43]. We found other immune checkpoints whose change in expression over time was associated with RECIST response or mPFS. For example, increased BTLA+CD8+ T cells and decreased VISTA+ CD4+ and CD8+ cells or proliferating CD4+ cells were associated with improved RECIST response. In total, this clinical data suggest combination therapy targeting multiple immune checkpoints may be necessary to invigorate T cell responses in advanced CRC.
Lower baseline levels of CD8+FasR+ T cells expressing PD-1 were associated with positive RECIST response. FasR induces apoptosis via the extrinsic pathway and mediates elimination of CD8 + T cells upon entry into the metastatic liver in pre-clinical models [44, 45]. These results possibly indicate patients with reduced cytotoxic T cells that were primed for apoptosis had improved response to this combination therapy. Further, we identified that high levels of Tc17 cells (CD8+RORγt+) expressing PD-1 at baseline was a positive prognostic factor, while a decrease in these cells throughout treatment was associated with improved response. This cell subset is functionally impaired in bile duct cancer [27]. Our data suggest that PD-1 inhibition may potentially restore functionality to these cells. These hypotheses will need to be examined with further studies.
Although informative, this trial does have limitations. First, this trial was single-arm, non-randomized, and did not stratify patients based on expression of PD-L1 or other biomarkers. Further, only a subset of patients had available data on dMMR/MSI status. These factors limited our ability to link clinical outcome with genomic subsets of mCRC. Additionally, this study was not designed to obtain tumor samples at baseline and post-treatment. Therefore, we are limited in making statements regarding changes in immune infiltrates and must rely on peripheral changes, which may not accurately reflect the tumor. We did utilize immunohistochemistry to analyze expression of CD4, CD8, and PD-L1 within the tumor and correlate it with response in a subset of patients where archival tissue samples were available, but found no association with RECIST response. Finally, due to the planned exploratory nature of our immune data, we did not see significant changes when correcting for multiple comparisons. This is a function of the large number of biomarkers analyzed in combination with a patient sample size required for clinical endpoints in an early phase trial. Nonetheless, these results are informative for hypothesis generation and lay the groundwork for future investigations.
Overall, this trial establishes that pembrolizumab can be safely combined with cytotoxic chemotherapy in mCRC. Moreover, our exploratory analysis uncovered a variety of potential cellular and soluble biomarkers of response to this therapy. These biomarkers should be interrogated more thoroughly in future studies. In addition, our data should inform future rational design of combination therapy for mCRC. In particular, addition of next-generation checkpoint blockades, namely inhibition of LAG3, to mFOLFOX6 and pembrolizumab may be an attractive avenue of investigation in mCRC.
Availability of data and material
All raw data and materials used to generate this manuscript and the included figures are available upon request from the corresponding author.
Code availability
Code utilized for the biostatistical analysis of this work is available from the corresponding author upon reasonable request.
References
Bray F et al (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424
Van Cutsem E, Oliveira J, EGW Group (2009) Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 20(Suppl 4):61–63
SEER Program (National Cancer Institute (U.S.)) et al (1993) SEER cancer statistics review, in NIH publication., U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute, Bethesda
Holch J, Stintzing S, Heinemann V (2016) Treatment of metastatic colorectal cancer: standard of care and future perspectives. Visc Med 32(3):178–183
Antoniotti C et al (2020) AtezoTRIBE: a randomised phase II study of FOLFOXIRI plus bevacizumab alone or in combination with atezolizumab as initial therapy for patients with unresectable metastatic colorectal cancer. BMC Cancer 20(1):683
Damato A et al (2020) Phase II study on first-line treatment of NIVolumab in combination with folfoxiri/bevacizumab in patients with Advanced COloRectal cancer RAS or BRAF mutated—NIVACOR trial (GOIRC-03-2018). BMC Cancer 20(1):822
de Gramont A et al (2000) Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol 18(16):2938–2947
Le DT et al (2020) Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol 38(1):11–19
Le DT et al (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372(26):2509–2520
Weiss GJ et al (2017) A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br J Cancer 117(1):33–40
Moehler M, Shitara K, Garrido M, Salman P, Shen L, Wyrwicz L, Yamaguchi K, Skoczylas T, Campos Bragagnoli A, Liu T, Schenker M, Yanez P, Tehfe M, Poulart V, Cullen D, Lei M, Kondo K, Li M, Ajani JA, Janjigian YY (2020) LBA6_PR-Nivolumab (nivo) plus chemotherapy (chemo) versus chemo as first-line (1L) treatment for advanced gastric cancer/gasgroesophageal junction cancer (GC/GEJC)/esophageal adenocarcinoma (EAC): first results of CheckMate649 study, in Ann Oncol
Galluzzi L et al (2017) Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17(2):97–111
Panaretakis T et al (2009) Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J 28(5):578–590
Tesniere A et al (2010) Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29(4):482–491
Zaravinos A et al (2019) Cytolytic activity correlates with the mutational burden and deregulated expression of immune checkpoints in colorectal cancer. J Exp Clin Cancer Res 38(1):364
Zhou E et al (2015) Up-regulation of Tim-3 is associated with poor prognosis of patients with colon cancer. Int J Clin Exp Pathol 8(7):8018–8027
Lines JL et al (2014) VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res 2(6):510–517
Kamphorst AO et al (2017) Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci USA 114(19):4993–4998
Meirow Y, Kanterman J, Baniyash M (2015) Paving the road to tumor development and spreading: myeloid-derived suppressor cells are ruling the fate. Front Immunol 6:523
Kanterman J et al (2014) Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res 74(21):6022–6035
Vincent J et al (2010) 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res 70(8):3052–3061
Wang Z, Till B, Gao Q (2017) Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology 6(7):e1331807
Limagne E et al (2016) Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res 76(18):5241–5252
Eisenhauer EA et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
National Cancer Institute (U.S.) (2009) Common terminology criteria for adverse events (CTCAE). Rev. ed. NIH publication, Bethesda, U.S. Dept. of Health and Human Services, National Institutes of Health, National Cancer Institute
Toiyama Y et al (2012) Evaluation of CXCL10 as a novel serum marker for predicting liver metastasis and prognosis in colorectal cancer. Int J Oncol 40(2):560–566
Chellappa S et al (2017) CD8+ T cells that coexpress RORgammat and T-bet are functionally impaired and expand in patients with distal bile duct cancer. J Immunol 198(4):1729–1739
Feun LG et al (2019) Phase 2 study of pembrolizumab and circulating biomarkers to predict anticancer response in advanced, unresectable hepatocellular carcinoma. Cancer 125(20):3603–3614
Keegan A et al (2020) Plasma IL-6 changes correlate to PD-1 inhibitor responses in NSCLC. J Immunother Cancer 8(2):e000678
Mougiakakos D (2011) Regulatory T cells in colorectal cancer: from biology to prognostic relevance. Cancers (Basel) 3(2):1708–1731
Paz-Ares L et al (2018) Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med 379(21):2040–2051
Gandhi L et al (2018) Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med 378(22):2078–2092
Ozaslan E et al (2017) Efficacy and safety of cetuximab plus FOLFOX in second-line and third-line therapy in metastatic colorectal cancer. J BUON 22(4):863–868
Schultheis B et al (2013) Regorafenib in combination with FOLFOX or FOLFIRI as first- or second-line treatment of colorectal cancer: results of a multicenter, phase Ib study. Ann Oncol 24(6):1560–1567
Lee JJ, Sun W (2016) Options for second-line treatment in metastatic colorectal cancer. Clin Adv Hematol Oncol 14(1):46–54
Hao Q, Vadgama JV, Wang P (2020) CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal 18(1):82
Ramsey ML et al (2019) Circulating interleukin-6 is associated with disease progression, but not cachexia in pancreatic cancer. Pancreatology 19(1):80–87
Kobelt D et al (2020) Pro-inflammatory TNF-alpha and IFN-gamma Promote Tumor Growth and Metastasis via Induction of MACC1. Front Immunol 11:980
Halama N et al (2016) Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell 29(4):587–601
Suenaga M et al (2019) Role of CCL5 and CCR5 gene polymorphisms in epidermal growth factor receptor signalling blockade in metastatic colorectal cancer: analysis of the FIRE-3 trial. Eur J Cancer 107:100–114
Llosa NJ et al (2015) The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 5(1):43–51
Zhou G et al (2018) Blockade of LAG3 enhances responses of tumor-infiltrating T cells in mismatch repair-proficient liver metastases of colorectal cancer. Oncoimmunology 7(7):e1448332
Ganesh K et al (2019) Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol 16(6):361–375
Zeestraten EC et al (2013) The prognostic value of the apoptosis pathway in colorectal cancer: a review of the literature on biomarkers identified by immunohistochemistry. Biomark Cancer 5:13–29
Yu J et al (2021) Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med 27(1):152–164
Acknowledgements
The authors are grateful for all the patients and families who participated in this clinical trial. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Funding
Research funding for the clinical trial and correlative research studies was provided to investigators through a sponsored research agreement between Merck and Co., Inc and Emory University or The Ohio State University. Research reported in this publication was supported in part by the Cancer Tissue and Pathology shared resource and the Pediatrics/Winship Flow Cytometry Core of Winship Cancer Institute of Emory University and NIH/NCI under Award Number P30CA138292.
Author information
Authors and Affiliations
Contributions
GBL, BFE, and SS conceptualized this project and obtained funding for the execution. MRF and TAM generated the raw data for each figure. BO, TB, AN, CM, WS, CW, BFE, and SS conducted the clinical work associated with this trial. CJH, YT, and ZL analyzed the data. CJH wrote the original draft. All authors participated in revisions of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
MRF, CJH, YT, ZL, AN, CM, and TAM have no conflicts of interest to declare. BO has consulted for Genentech, Amgen, and Merck. The terms of these arrangements have been reviewed and approved by Indiana University in accordance with its conflict-of-interest policies. TBS has consulted for Ipsen, Array Biopharma, Pfizer, Seattle Genetics, Bayer, Genentech, Incyte, and Merck through his institution. TBS has also consulted for Boehringer Ingelheim, Janssen, Eisai, Daichii Sankyo, Natera, TreosBio, Celularity, Exact Science, and Sobi and has been compensated for these services. TBS has received research funding from Agios, Arys, Boston Biomedical, Bayer, Amgen, Merck, Celgene, Lilly, Ipsen, Clovis, Seattle Genetics, Array Biopharma, Genentech, Novartis, Mirati, Merus, Abgenomics, Incyte, Pfizer, and BMS. The terms of these arrangements have been reviewed and approved by Mayo Clinic in accordance with their conflict-of-interest policies. WS has received honoraria from Merck, BMS, Lexicon, Mylan, Ipsen, Blueprint Medicine, Signatera, and Exilexis. Dr. Shaib has also received research support from Lexicon, Eli Lilly, and GSK. The terms of these arrangements have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. CW has consulted for Natera and Array Biopharma as well as received clinical trial funding from Vaccinex, Rapt Therapeutics, INBHRX, Boston Biomedical Inc., Seattle Genetics, Lycera, and Symphogen. The terms of these arrangements have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. BFE has consulted for Ipsen, Merck and Co., Bayer, AstraZeneca, Bristol-Myers Squibb, Inc. and been a speaker for Lexicon, Inc. He serves as a consultant to Merck and Co., and receives compensation for these services. He has also received research funding through a sponsored research agreement between Emory University and Bristol-Myers Squibb, Boston Biomedical, Novartis, Merck and Co, Bayer, Exelixis, Pfizer, AstraZeneca/Medimmune, Incyte, and EUSA. The terms of these arrangements have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. SS is currently employed by Eli Lilly. GBL has consulted for ProDa Biotech, LLC and received compensation. He has also received research funding through a sponsored research agreement between Emory University and Merck and Co., Bristol-Myers Squibb, Boerhinger-Ingelheim, and Vaccinex. The terms of these arrangements have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies.
Consent for publication
All authors and relevant funding sources have read and approved the final version of this manuscript and consent its publication.
Ethics approval and consent to participate
This study was conducted in accordance with the IRB procedures at Emory University (IRB00080442), Indiana University (1502701251), and The Ohio State University and was approved by their Ethics Boards. All patients were consented prior to participation in this trial (NCT02375672).This study was conducted in accordance with the IRB procedures at Emory University (IRB00080442), Indiana University (1502701251), and The Ohio State University and was approved by their Ethics Boards. All patients were consented prior to participation in this trial (NCT02375672).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Precis: We show that combining mFOLFOX and pembrolizumab is safe in patients with metastatic colorectal cancer. Correlative analyses identified immunotherapeutic targets, including LAG3, that warrant future investigation.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Herting, C.J., Farren, M.R., Tong, Y. et al. A multi-center, single-arm, phase Ib study of pembrolizumab (MK-3475) in combination with chemotherapy for patients with advanced colorectal cancer: HCRN GI14-186. Cancer Immunol Immunother 70, 3337–3348 (2021). https://doi.org/10.1007/s00262-021-02986-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-021-02986-5