
Abstract
Porcine parvovirus (PPV) virus-like particles (VLPs) are a potential vaccine candidate for the prevention of parvovirus-induced reproductive failure in pregnant sows. Currently, the Escherichia coli (E. coli) expression system is the most cost-efficient to express recombinant proteins. To overcome the limitations of protein misfolding and to prepare soluble highly bioactive antigen and high yields of protein, we optimized the PPV-VP2 gene, subcloned it into pET24a, pET26b, pET28a, and pET30a, and transformed it into E. coli BL21(DE3)-Tf16 competent cells. The pET28a plasmid was selected for further manipulations because it expressed high levels of the bioactive PPV-VP2 protein. Under optimal high-density fermenting conditions in a 70-L fermenter, the total yield of wet weight E. coli cells was 124.86 g/L, and PPV-VP2 protein was 2.5 g/L. After large-scale purification with Triton X-114 two-phase extraction as well as activated carbon powder adsorption, hemagglutination (HA) titers in the purified PPV-VP2 protein reached 219 and endotoxin was reduced to 2500 EU/mL. Dynamic light scattering (DLS) and transmission electron microscopy (TEM) results indicated that the purified PPV-VP2 protein self-assembled into VLPs. Immunogenicity assays in guinea pigs and pigs indicated that the ISA-201 VG adjuvanted PPV-VP2 VLP vaccine elicited hemagglutination inhibition (HI) and virus neutralization (VN) antibody titers comparable with PPV commercial inactivated vaccines, whereas viral loads in the spleen and liver of challenged guinea pigs were significantly reduced. In conclusion, our study provides a method for producing the PPV-VLP vaccine against PPV infection in E. coli and may offer a novel strategy for the soluble expression of other vaccine antigens.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine parvovirus (PPV) is a major cause of reproductive failure in swine. Fetal death, mummification, stillbirths, and delayed return to estrus are some of the predominant clinical signs commonly associated with PPV infections (Meszaros et al. 2017). Since it was first discovered in Germany in 1965, PPV has resulted in considerable losses to the swine industry throughout the world (Oh et al. 2017). PPV belongs to the genus Parvovirus, subfamily Parvovirinae, family Parvoviridae (Song et al. 2010). It is a non-enveloped, single-stranded DNA genome in T = 1 icosahedral protein capsids that are 18–26 nm in diameter (Cotmore and Tattersall 2013). Two structural proteins, VP1 and VP2, in a ratio of roughly 1:20, form the porcine parvovirus capsids (Sumana et al. 2013). The structural VP2 protein can self-assemble into VLPs alone in vitro, which is responsible for eliciting neutralizing antibodies in immunized animals (Ji et al. 2017).
Several types of vaccines have been developed to counteract PPV disease, including live attenuated, inactivated, and new-generation VLP vaccines (Antonis et al. 2006; Wrathall et al. 1984). However, due to several drawbacks in traditional vaccines, such as difficulties in production and safety concerns, alternative ways for the development of safer vaccines are needed. VLPs offer several advantages since they present conformational epitopes similar to native particles, resulting in identical immune responses and the production of relevant antibodies in a safer way than live vaccines (Zhou et al. 2010). Protein expression systems, such as insect, yeast, mammalian cells, or bacteria, have been used to produce PPV-VP2 VLPs, and all these VLP vaccines exhibited improved immune responses and protection compared with traditional vaccines (Guo et al. 2014; Rymerson et al. 2003). Escherichia coli (E. coli) is one of the organisms of choice for the expression of recombinant proteins at a low cost, which is especially crucial for the production of veterinary vaccines. E. coli cells are widely used as a cell factory for the high-level production of heterologous proteins (Zhou et al. 2018; Zhou et al. 2012).
In the present study, following up on our previous work (Ji et al. 2017), we developed high-density fermentation and downstream processing of PPV-VP2 VLPs in E. coli. Safety tests in pigs indicated that the MONTANIDE™ ISA-201 VG adjuvanted PPV-VP2 VLP vaccine had no side effects. Potency tests showed that the VLP vaccine could induce stronger immune responses in both guinea pigs and pigs as compared with a commercial inactivated vaccine. Following the challenge in guinea pigs, lower viral loads in the spleen and liver were detected. All results indicated that the ISA-201 VG adjuvanted PPV-VP2 VLP vaccine is a realistic alternative that could compete with the currently available classical PPV inactivated vaccines.
Materials and methods
Cell and virus
Porcine kidney (PK-15) cells were obtained from the China Center for Type Culture Collection (Wuhan, China) and kept by the Henan Provincial Key Laboratory of Animal Immunology (Zhengzhou, China). PK-15 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640, Solarbio) medium supplemented with 10% fetal bovine serum (FBS, Hyclone) at 37 °C in a 5% CO2 incubator. PPV reference strain 7909 was purchased from the China Institute of Veterinary Drug Control (Beijing, China). When the monolayer of PK-15 cells was about 60% confluent, PPV 7909 was added and incubated at 37 °C for 2 h. Then the cell supernatant was discarded, and new media, containing 3% FBS, was added. The virus was cultured at 37 °C for 72 h. When the cytopathic effect was observed in 80% of the cells, the supernatant was collected; and the virus was stored at − 80 °C. PPV was titrated using the Reed-Muench formula (Opriessnig et al. 2011).
Optimization and screening of PPV-VP2 strains
The gene encoding PPV-VP2 protein (GenBank accession number MN708487) with a His-tag in N-terminal was optimized according to the codon preference of E. coli and subcloned into different vectors: pET24a, pET26b, pET28a, and pET30a. The recombinant plasmids were confirmed and transformed into BL21(DE3)-Tf16 competent cells, prepared according to the manufacturer’s protocol (Takara, China). The PPV-VP2 protein expression was induced in 100 mL LB medium. Protein yields were compared with SDS-PAGE and hemagglutination (HA) test. The colony with the highest protein expression level was used as the seed strain in the subsequent fermentation.
Fed-batch fermentation of PPV-VP2 protein
For fed-batch fermentation, 50 μL of frozen glycerol seeds were added to 100 mL LB medium containing chloramphenicol 25 mg/L and kanamycin 50 mg/L, and then cultured in a 500-mL flask at 37 °C for 16 h. These were the primary seeds for future VLPs production. This procedure was repeated in 1000-mL flasks for the generation of secondary seeds. For high-density fermentation, 1000 mL secondary seeds were inoculated into a 70-L fermenter (Tofflon, China) containing 35 L sterilized medium (tryptone 15 g/L, yeast extract 30 g/L, glycerol 15 g/L, MgSO4·7H2O 0.3 g/L, K2HPO4 9.31 g/L, KH2PO4 2.5 g/L, L-Arabinose 1 g/L, pH 7.0) supplemented with chloramphenicol and kanamycin. Seeds were cultured at 37 °C for 16 h until glycerol was exhausted, as detected by a sudden increase in both dissolved oxygen (DO) and pH. Then, a total of 5 L fed-batch fermentation medium (tryptone 50 g/L, yeast extract 21 g/L, glycerol 26.5 g/L, MgSO4·7H2O 0.63 g/L) containing L-Arabinose (10 g/L) was sterilized separately and added into the fermentation medium continuously with 400 mL each hour. The isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the fermentation medium at a final concentration of 0.15 mM, and the temperature was adjusted to 25 °C gradually. During the entire fermentation process, pH was kept at 7.0 by adding 25% (v/v) ammonia and 1 mol/L HCl when required. The DO level was maintained at least at 30% by controlling the cascading impeller speed (between 150 and 250 rpm) and the airflow rate. One percent antifoam (Sigma-Aldrich, USA) was added manually to reduce bubbles when necessary. Cell growth was monitored by measuring the optical density (OD) of the E. coli culture at 600 nm using a UV-visible spectrophotometer (Eppendorf, Germany) during the entire fermentation process. Culture supernatants were collected by centrifugation, resuspended in buffer A (150 mM NaCl, 50 mM Tris, pH 8.0), and lysed by a high-pressure homogenizer. Fermentation yields of the VP2 protein were detected by SDS-PAGE and HA tests.
Large-scale processing and purification of PPV-VP2 protein
For industrial-scale purification, the PPV-VP2 protein was first purified with Triton X-114 two-phase extraction. Triton X-114 was added to the PPV-VP2 protein at a final concentration of 1%. After stirring continuously with a magnetic stirrer for 30 min, the mixture was incubated at 4 °C for 1 h. Subsequently, the sample was raised to 25 °C by incubation in a 37 °C water bath with continuous stirring. Then, centrifugation was performed at 25 °C (12,000×g, 20 min) to separate the aqueous phase sample and bottom Triton X-114 phase. After removing the Triton X-114 phase, another 2 cycles of treatment were performed with the top aqueous phase. Finally, 2% activated carbon powder was added to the aqueous phase sample and stirred for 10 min. The purified PPV-VP2 protein was recovered by centrifugation (12,000×g, 30 min) at 25 °C, which was then filtered with 0.45-μm and 0.22-μm polyethersulfone ultrafiltration membranes successively. The content of bacterial endotoxin in the purified PPV-VP2 protein was measured by a Chromogenic End-point Tachypleus Amebocyte Lysate (CE TAL) assay kit (Houshiji, China) following the kit’s instructions. A standard curve was established based on the relationship between the absorbance value of the standard and the endotoxin content. The purified PPV-VP2 protein was inactivated with 0.1% formaldehyde at 37 °C for 24 h and stored at − 20 °C.
Characterization of PPV-VP2 VLPs
The purified PPV-VP2 protein was detected by 12% SDS-PAGE, western-blot, and HA tests. HA test was carried out using 0.6% (v/v) guinea pig erythrocytes as previously described (Feng et al. 2014). PPV-VP2 VLPs were detected by transmission electron microscopy (TEM) using the negative staining method (van Beurden et al. 2011) and dynamic light scattering (DLS) (Malvern, China).
PPV-VP2 VLP vaccine preparation
MONTANIDE™ ISA 201 VG (Seppic, France), a water-in-oil-in-water (W/O/W) adjuvant, was autoclaved at 121 °C for 20 min and kept at 31 °C. The temperature of the purified PPV-VP2 antigen was raised to 31 °C, and added into equal ISA201 VG adjuvant to prepare the PPV-VP2 VLP vaccine as described in the manufacturer’s manual. PPV-VP2 VLP vaccine production containing 215 HA titers of VP2 antigen per mL, and all other procedures were performed under aseptic conditions.
Vaccination and challenge of guinea pigs
Fifty female guinea pigs weighing between 300 and 400 g were randomly divided into five groups to evaluate the immunogenicity and protective efficacy of the PPV-VP2 VLP vaccine. Guinea pigs in group 1 were immunized with 2.8 μg of the PPV-VP2 VLP vaccine, whereas groups 2 and 3 were inoculated with 8.3 μg and 25 μg of the VLPs, respectively. Group 4 was inoculated with 200 μL of the commercial inactivated PPV vaccine (Keqian, China), and group 5 was inoculated with 200 μL of PBS as a negative control. All the immunogens were injected into the tibialis cranialis muscle of both rear legs. At 35 days post immunization (dpi), three guinea pigs from each group were sacrificed for T lymphocyte proliferation assay. Four guinea pigs from each group were challenged with 100 μL of 106.8 50% tissue culture infection dose (TCID50)/mL PPV 7909. The challenged guinea pigs were monitored for 10 days and sacrificed at 45 dpi in a humane way. Spleens and livers were separated from the challenged guinea pigs for PPV virus detection by real-time PCR (RT-PCR). Blood samples were collected from the remaining three guinea pigs in each group once every 2 weeks until 84 dpi.
PPV-VP2 VLP vaccine safety test
Sixteen 6-to-8-week-old piglets weighing between 10 and 20 kg were equally divided into four groups to analyze the safety of the PPV-VP2 VLP vaccine. Pigs in groups 1, 2, and 3 were inoculated intramuscularly with 2 mL, 5 mL, and 10 mL of the PPV-VP2 VLP vaccine containing 28 μg, 70 μg, and 280 μg of the VP2 protein, respectively. Pigs in group 4 were used as negative controls. Symptoms, such as temperature, loss of appetite, and diarrhea, were observed in all immunized and unimmunized pigs every day for 28 days after injection.
Vaccination and antibody monitorization in pigs
Twenty 150-day-old pigs were equally divided into five groups. Group 1 was inoculated intramuscularly with 28 μg of the PPV-VP2 VLP vaccine, group 2 was given 83 μg, and group 3 was given 250 μg. Group 4 was inoculated with 2 mL of the commercial inactivated PPV vaccine and group 5 was inoculated with PBS as a negative control. Blood samples were collected from pigs once every 2 weeks in the first 3 months and then once a month until 180 dpi. Hemagglutination inhibition (HI) assay and virus neutralization (VN) assay were carried out.
Hemagglutination inhibition and virus neutralization assay
For HI assay, serum samples were heat-inactivated at 56 °C for 30 min and then incubated with 25% kaolin and 20% guinea pig erythrocytes in PBS for 30 min. Then, 25 μL of the treated serum samples were twofold serially diluted into 96-well V-shaped microtiter plates and then mixed with equal four hemagglutination units of PPV 7909. After incubation at 37 °C for 30 min, 25 μL of 0.6% guinea pig erythrocytes were added to the plates. Results were read after 2 h at room temperature. HI titers were defined as the highest serum dilution that completely inhibited hemagglutination.
VN assay was performed as described below. Briefly, 100 μL heat-inactivated serum samples were twofold diluted and incubated with PPV 7909 (200 TCID50) at 37 °C for 1 h. Then the mixture was added to 96-well plates containing 6 × 105 PK15 cells/well at 37 °C for 72 h in a 5% CO2 incubator. VN titers of the serum were defined as the highest dilution that completely neutralized PPV 7909 by the immunoperoxidase monolayer assay (IPMA).
Lymphocyte proliferation assay
Spleen lymphocytes were isolated from immunized guinea pigs using hydroxypropylmethyl cellulose (Solarbio, China) according to the product instructions. Cells were suspended at 2 × 107 cells/mL in RPMI-1640 medium supplemented with 10% FBS and seeded in triplicate in 96-well plates. Cells were stimulated with 100 TCID50 of PPV 7909, and non-stimulated cells were set as the control. After incubating the cells at 37 °C for 48 h, CCK 8 was added and incubated at 37 °C for 2 h. The stimulation index (SI) was calculated as:
SI = mean OD450 nm of PPV-stimulated cells/mean OD450 nm of non-stimulated cells.
Quantitative RT-PCR for PPV in tissues
Genomic DNA was extracted from the spleen and liver of the challenged guinea pigs and PPV viral load was detected by RT-PCR as previously described (Chen et al. 2009; Ji et al. 2017). Homogenized tissues of 0.1 g spleen or liver in 2 mL PBS were digested with proteinase K (Solarbio, China). Fifty microliters genomic DNA was extracted by using the TIANamp Genomic DNA Kit (Tiangen, China) from 200 μL of homogenized tissues according to the product instructions. The conserved region of the VP2 gene (194 bp) was amplified by a pair of primers (F: CCAAAAATGCAAACCCCAATA; R: TCTGGCGGTGTTGGAGTTAAG), and subcloned into the pEASY-T1 vector as a standard plasmid. The amplification reactions were carried out in a total volume of 20 μL, containing 2 μL of template DNA, 10 μL of 2 × FastStart Universal SYBR® Green Master (Rox) (Roche, Germany), 0.4 μL of each forward and reverse primer (2.5 μM), and 7.2 μL of RNAse-free water. RNAse-free water was used as the template to act as the negative control. All samples were performed in triplicate. Viral load was calculated according to the standard curve plotting Ct values against different dilutions of the standard plasmid.
Statistical analyses
All values were tested for significance by one-way analysis of variance (ANOVA) using GraphPad Prism version 7.00 (GraphPad Software, USA). Data were shown as the mean ± SEM. No significance (NS) between groups was set at P > 0.05. Statistical significance was determined at p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****) between the experimental groups.
Results
Screening of high-level expression strains of PPV-VP2
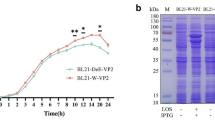
To improve the yields and bioactivity of the PPV-VP2 protein, the gene encoding VP2 protein was subcloned into pET24a, pET26b, pET28a, and pET30a, and then transformed into E. coli BL21(DE3)-Tf16 competent cells. Chaperonin Tf16 was induced with L-Arabinose before induction, in order to ensure the VP2 protein folded correctly. Results from SDS-PAGE (Fig. 1a) and HA tests (Fig. 1b) indicated that recombinant E. coli BL21(DE3)-pET28a offered the highest yield.

Screening and optimization of high-level expression strains of the PPV-VP2 protein. The PPV-VP2 protein was expressed in different vectors. After induction with 0.15 mM IPTG and 2 mg/ml L-Arabinose at 25 °C for 12 h, culture supernatants were collected by centrifugation, resuspended in buffer A, and lysed by ultrasonic treatment on the ice. The supernatant and the precipitate were analyzed with SDS-PAGE and HA tests. a SDS-PAGE of the PPV-VP2 protein expressed in different vectors. M, Marker; Lanes 1–4, supernatants of the PPV-VP2 protein in pET24a, pET26b, pET28a, and pET30a with IPTG induction; Lanes 5–8, precipitates of the PPV-VP2 protein in pET24a, pET26b, pET28a, and pET30a with IPTG induction. b HA titers of the PPV-VP2 protein expressed in different vectors. HA test in triplicate analyzed the PPV-VP2 protein expressed in pET24a, pET26b, pET28a, and pET30a vector
High-density fermentation and industrial-scale purification of PPV-VP2 VLPs in E. coli
To obtain the PPV-VP2 protein at the highest possible yield, high cell density culture was performed in a 70-L fermenter, and cell growth and HA titers were monitored during the entire fermentation process as described above. IPTG and L-Arabinose induced the PPV-VP2 protein and chaperonin Tf16 expression, and the fed-batch medium was fed to supply enough nutrition in fermentation anaphase. HA test showed that the yields of the PPV-VP2 protein exhibited sustained growth and reached a plateau by 20 h after adding 0.15 mM IPTG (Fig. 2); about 4.37 kg wet weight E. coli cells were obtained from 35 L media. HA titers in supernatants of the PPV-VP2 protein reached 222, implying that the PPV-VP2 protein obtained from high-density fermentation had natural PPV and VLP activity.

Fermentation results of the PPV-VP2 protein in a 70-L fermenter. The culture was grown in fermentation medium and induced with 0.15 mM IPTG at OD600 of 20.2 for expression of the VP2 protein. Cell growth was monitored and analyzed with OD600 every 4 h. The DO concentration was controlled at least at 30% of air saturation, and pH was monitored at 7.0 during the fermentation. After induction, culture supernatants were taken every 4 h to detect the expression of the PPV-VP2 protein by HA assay in triplicate. The starting point of induction was indicated with the arrow
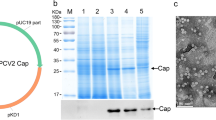
An industrial-scale purification method, using the physical characteristics of Triton X-114 to realize phase separation, was established to remove the endotoxin and purify PPV-VLPs from the lysed bacterial supernatant. Results from SDS-PAGE showed two bands at 64 kDa and 55 kDa corresponding to the PPV VP2 protein and chaperonin Tf16, respectively (Fig. 3a). Western blot analysis showed that the VP2 protein reacted specifically with the anti-His-tag antibody (Fig. 3b). After 3 cycles of Triton X-114 two-phase extraction, as well as activated carbon powder adsorption, the endotoxin level in the purified PPV-VP2 protein was reduced to 2500 EU/mL. Importantly, HA titers of the PPV-VP2 protein were not obviously decreased during the purification process (Table 1). The purified VP2 protein showed high HA titers reaching 219, like the natural PPV (Fig. 3c). The PPV-VP2 VLP vaccine was prepared by mixing ISA 201 VG adjuvant with the purified VP2 protein, which contained 215 HA titers antigen per mL.

Expression and purification of the PPV-VP2 protein. a SDS-PAGE analysis of the PPV-VP2 protein. b Western blot analysis of the PPV-VP2 protein with an anti-His monoclonal antibody. M, Marker; Lane 1, supernatants of the PPV-VP2 protein; Lane 2, purification of the PPV-VP2 protein with Triton X-114 extraction for 1 cycle; Lane 3, purification of the PPV-VP2 protein with 2% activated carbon powder adsorption. c Hemagglutination assay of the PPV-VP2 protein. 1, supernatants of the PPV-VP2 protein; 2, the purified PPV-VP2 protein; 3, positive control of PPV 7909
Characterization of PPV-VP2 VLPs
TEM, DLS, and HA tests were carried out to analyze the self-assembly of PPV-VP2 protein with His-tag and its biological activity. As shown in Fig. 4a, TEM result revealed that the purified VP2 protein was assembled into VLPs with a consistent diameter of 25 nm. Result from DLS indicated that more than 95% of the purified VP2 protein self-assembled into particles with a diameter of about 25 nm (Fig. 4b). HA test showed that the purified VP2 protein had high HA titers reaching 219, implying efficient VLPs assembly (Fig. 3c).

Characterization of VLPs from the purified PPV-VP2 protein. a Negative staining electron microscopy result of PPV-VLPs; bar size, 200 nm. VLPs were indicated with arrows. b Dynamic light scattering result of PPV-VLPs
Antibody response in guinea pigs
For the evaluation of the effectiveness of the PPV-VP2 VLP vaccine, HI and VN assays were performed to analyze the serum collected from five groups of immunized guinea pigs. As shown in Fig. 5a, HI titers of PPV-VP2 VLP vaccine groups were significantly higher than those in the commercial inactivated vaccine group. The same trend was found for PPV VN test (Fig. 5b), and there was no specific PPV antibody in the PBS group. Interestingly, guinea pigs in the low-dose group 1 (2.8 μg) showed higher HI and VN antibody titers, and no significant difference was observed between group 1 (2.8 μg), group 2 (8.3 μg), and group 3 (25 μg), i.e., the titers did not correlate with different immunizing doses.

Production of anti-PPV antibodies in guinea pigs. Female guinea pigs in each group were immunized with 2.8 μg, 8.3 μg, and 25 μg of the VLP vaccine, inactivated PPV vaccine, and PBS. Sera from three guinea pigs in each group were sampled at 42 dpi, and assayed for anti-PPV antibody titers by HI and VN assays. a HI antibody titers of immunized guinea pigs at 42 dpi. b VN antibody titers of immunized guinea pigs at 42 dpi. Error bars indicated the SEM (n = 3). Statistical significance was determined at p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), and p < 0.0001 (****)
T lymphocyte proliferation
At 35 dpi with a single-dose immunization, spleen lymphocytes of guinea pigs from each group were isolated, and T lymphocyte proliferation was measured. As shown in Table 2, SI indexes of the immunized groups were higher than the PBS group, and the highest level of proliferation was detected in group 3 (25 μg), indicating that the PPV-VP2 VLP vaccine induced T cell response in guinea pigs.
PPV-VP2 VLP vaccine protected guinea pigs against PPV infection
Four guinea pigs from each group were challenged with PPV 7909 at 35 dpi after single-dose immunization. The immunized guinea pigs did not present clinical signs, whereas decreased appetite and activity were observed in the PBS group. The DNA in spleens and livers of challenged guinea pigs were extracted, and RT-PCR was performed to measure viral loads. As shown in Fig. 6a, low virus gene quantity in the spleen was detected in group 3 (25 μg), whereas the PBS control group showed remarkably higher viral loads than the other groups. Viral loads showed the same trend in liver samples of challenged guinea pigs (Fig. 6b). These results indicated that the PPV-VP2 VLP vaccine has the potential to protect guinea pigs against PPV infection.

Viral loads of guinea pigs inoculated with PPV vaccines and challenged with pathogenic PPV strain 7909. Female guinea pigs in each group were immunized with 2.8 μg, 8.3 μg, and 25 μg of the VLP vaccine, inactivated PPV vaccine, and PBS. Four guinea pigs from each group were challenged with 100 μL of 106.8 TCID50/mL of PPV 7909 at 35 dpi after a single-dose immunization, and were then monitored for 10 days. Spleens and livers were isolated, and the genomes were extracted to measure the content of PPV by RT-PCR. a Viral loads in the spleens from challenged guinea pigs. b Viral loads in the livers from challenged guinea pigs. Error bars indicated the SEM (n = 4). Statistical significance was determined at p < 0.05 (*), p < 0.01 (**), p < 0.001 (***), p < 0.0001 (****)
Safety test of PPV-VP2 VLP vaccine in pigs
To estimate the safety of the PPV-VP2 VLP vaccine, groups 1, 2, and 3 of pigs were inoculated with 2 ml, 5 ml, and 10 ml vaccine containing 28 μg, 70 μg, and 280 μg of the VP2 protein, respectively. All immunized pigs were monitored daily for 28 days, and no clinical symptoms were observed in the immunized pigs, including elevated body temperature, loss of appetite, or diarrhea. This suggests that the PPV-VP2 VLP vaccine prepared in this study does not have observable side effects.
Antibody response in pigs
To further investigate the correlation between the dose and the duration of PPV-specific antibodies, three additional groups of 150-day-old pigs were inoculated with 28 μg, 83 μg, and 250 μg of the PPV-VP2 VLP vaccine, and antibody response was continuously tracked for 180 days. As shown in Fig. 7 a and b, HI and VN antibody titers increased rapidly at 14 dpi, and then gradually increased and maintained high titers until 180 dpi. Moreover, HI and VN titers of pigs in the VLPs groups were significantly higher compared with the animals that received the commercial inactivated vaccine and PBS groups. This implies that the PPV-VP2 VLP vaccine provides better immune protection than the commercial vaccine. Interestingly, HI and NA titers of group 2 (83 μg) were higher than those of group 3 (250 μg), indicating that the PPV-VP2 VLP vaccine–stimulated pigs producing specific antibody titers would not always increase in conjunction with the antigen dose.

PPV-specific immune response in pigs. A total of 150-day-old pigs (n = 4) in each group were immunized with 28 μg, 83 μg, and 250 μg of the VLP vaccine, inactivated PPV vaccine, and PBS. a HI antibody from 150-day-old pigs. b IPMA assay for VN antibody from 150-day-old pigs
Discussion
PPV-VP2 VLPs have been expressed in several expression systems, including yeast, insect, mammalian cells, and also E. coli (Antonis et al. 2006). Eukaryotic expression systems like insect and mammalian cells produce highly bioactive recombinant proteins, but their application is limited by various technical problems, such as low yield, high cost, and the potential risk of contamination. Compared with eukaryotic expression systems, E. coli is more straightforward, cheaper, and it is easier to manipulate and scale-up, but has a problem regarding proper protein folding. In a previous study, our team identified and solved this problem by co-expressing the chaperone Tf2 with PPV-VP2 (Ji et al. 2017). In this work, we optimized the PPV-VP2 gene, screened vectors, and introduced a highly efficient chaperone Tf16 to increase the production and bioactivity of PPV-VP2 protein with His-tag. Results from SDS-PAGE and HA tests indicated that yields and bioactivity were significantly improved compared with the system previously reported.
High-density fermentation technology of E. coli has advantages in cost effectiveness, reduced culture volume, reduced waste of water, and reduced investment in equipment. For these reasons, E. coli is widely applied for the large-scale production of recombinant proteins. In the present study, a high-density fermentation strategy was deployed to co-express the PPV-VP2 protein with chaperone Tf16 in E. coli. During the entire fermentation process, keeping sufficient nutrient concentrations and balancing the carbon-nitrogen ratio were ensured to achieve optimum fermentation (Smith and Holtzapple 2010). Except for the primary and secondary seeds, a modified fermentation medium was used in later seeds, which was much richer in nutrients compared with the LB medium, resulting in greater cell mass and allowing higher recombinant protein expression levels. By contrast, the addition of Mg2+ was better for cell growth in E. coli. Glycerol and yeast extracts in the fed-batch medium were used to supply enough carbon and nitrogen in an optimal ratio during late fermentation. The total yield of wet weight E. coli cells was 124.86 g/L, and PPV-VP2 protein was about 2.5 g/L, which was much higher than that in baculovirus cell systems (25 mg/L) (Maranga et al. 2002). Results from SDS-PAGE tests showed that chaperone Tf16 protein was expressed simultaneously with the PPV-VP2 protein. The co-expressed PPV-VP2 protein showed a high HA activity that reached 222, indicating natural VLP characteristics. Additionally, TEM result demonstrated that the purified PPV-VP2 protein with an N-terminal His-tag self-assembled into VLPs in E. coli through high-density fermentation.
E. coli has a significant advantage in expressing the recombinant PPV-VP2 protein, but endotoxin removal is an essential issue in the manufacturing process. Endotoxin released by gram-negative bacteria contaminates protein solutions derived from bioprocesses (Petsch and Anspach 2000). Because of high toxicity effects, such as changing metabolic functions, raising body temperature, triggering of the coagulation cascade, and causing shock in humans and animals (Magalhaes et al. 2007), endotoxin removal is essential for vaccine safety. Various methods are employed for removing endotoxin, such as activated carbon powder (Gun’Ko et al. 2006), ion-exchangers or hydrophobic interaction chromatography (Hou et al. 2011; Lowe et al. 2012), Triton X-114 two-phase extraction (Duan et al. 2017; Teodorowicz et al. 2017), and gel filtration chromatography (London et al. 2014). Considering the needs of large-scale production of the PPV-VP2 protein, a new efficient and cost-effective method was established to remove the endotoxin and purify PPV-VLPs. By performing a single cycle of Triton X-114 two-phase extraction, endotoxin levels in the PPV-VP2 protein were reduced by more than 90%. Further reduction of the endotoxin could be achieved if more cycles of phase extraction were performed. After carrying out 3 cycles, 2% activated carbon powder was added to adsorb Triton X-114 and the remaining endotoxin. As shown in Table 1, the endotoxin in the purified PPV-VP2 protein was reduced to 2500 EU/mL, and HA titers reached 219. Importantly, HA activity was not affected during the entire purification process.
Vaccine adjuvant is a critical factor that stimulates specific components of either the humoral or cell-mediated immune response (Lahariya 2014). An ideal adjuvant is the one that can stimulate the humoral immune response early (onset) and promote the production of high antibody titers (strength/intensity) that would last long (duration) (Ibrahim Eel et al. 2015). It is reported that the MONTANIDE™ ISA 201 VG adjuvant, which could not induce a specific immune response used alone, generates a rapid, high, and long-lasting immune response and shows less interference from maternal antibodies than the aqueous vaccines (Dar et al. 2013; Khorasani et al. 2016). In this study, MONTANIDE™ ISA 201 VG, a W/O/W adjuvant, which is robust, stable, easy to inject, and induces stronger and longer lasting protection, was used to prepare the PPV-VLP vaccine. For the PPV-VP2 VLP vaccine to be effective, it should be able to generate HI and VN antibody responses at faster and higher rates as well as for a longer duration than the current vaccines. In the present study, we employed a PPV-susceptible animal model and used guinea pigs to assess the immune efficacy of the PPV-VP2 VLP vaccine further. HI and VN antibody titers in the PPV-VP2 VLP vaccine groups were significantly higher compared with the commercial inactivated vaccine and PBS group (Fig. 5). This finding suggested that VLP vaccines stimulated guinea pigs to produce an effective humoral immune response and a better immunity than the commercial vaccine used in this work. We subsequently analyzed the proliferation of lymphocytes from inoculated guinea pigs at 35 dpi. The high-dose VLP vaccine group in guinea pigs exhibited a higher SI index than the commercial vaccine and PBS group (Table 2), but all the data showed a weak T cell–mediated immune response. The results of the virus challenge test and HI and VN antibody titers suggested that the humoral immune response plays a significant role in immune protection against PPV, as previously reported (Antonis et al. 2006). Ten days after challenging with PPV, viral titers in spleens and livers of guinea pigs were measured by RT-PCR. Viral loads in guinea pigs from the VLP vaccine groups were significantly lower compared with the PBS and commercial vaccine group (Fig. 6). Thus, the PPV-VLP vaccine has greater potential against PPV virus infection than the commercial vaccine used in this work. The low dose group (2.8 μg) provided protective immunity against PPV infection, revealing adequate protection offered by the PPV-VP2 VLP vaccine.
To evaluate the safety of the PPV-VP2 VLP vaccine, the purified VP2 protein was mixed with the ISA 201 VG adjuvant and inoculated intramuscularly to pigs weighing 10–20 kg. No adverse reactions were observed in any of the immunized pigs, suggesting that the VLP vaccine was safe, and the process developed for large-scale purification of PPV-VP2 VLP vaccine was feasible.
Previous studies reported that mummification and even infection of fetuses could be prevented when HI titers in pregnant sows stayed above 80 (Antonis et al. 2006; Foerster et al. 2016; Jozwik et al. 2009). In this work, 150-day-old pigs were divided into five groups, and immunized with PPV-VLP vaccines, the commercial inactivated vaccine, and PBS. Twenty-eight days after a single-dose injection, HI titers reached 128, 2048, and 256, whereas the VN titers reached 608, 3444, and 2048 in group 1 (28 μg), group 2 (83 μg), and group 3 (250 μg), respectively (Fig. 7). More persuasively, all the HI and VN titers in the PPV-VLP vaccine group were higher than those in the commercial inactivated vaccine and PBS group. These results indicated that the PPV-VLP vaccine could induce a higher efficient humoral immune response and had the potential to prevent PPV infection than the commercial vaccine. In addition, the VLP vaccine in group 2 (83 μg) induced higher HI and VN antibody titers than group 3 (250 μg), probably because immunological paralysis was induced by the high dose of VP2 antigen in pigs. The 28 μg dose of PPV-VLPs could provide higher HI and VN antibody and may confer protective immunity against PPV infection. In this study, high titers of HI and VN antibodies were detected in guinea pigs and pigs, and antibody titers of pigs immunized with VLP vaccine were still above 1024, even at 180 dpi. This indicated that the VLP vaccine reported in this work induces a long period of immune protection in pigs.
In summary, we developed an efficient process to mass produce the PPV-VP2 protein in E. coli that includes high-density fermentation, endotoxin removal, and vaccine preparation. The PPV vaccine generated in this work was a highly effective PPV-VP2 VLP vaccine that could stimulate animals to produce a high intensity and long-term immune response against PPV infection. Animal experiments revealed that the PPV-VP2 VLP vaccine elicited a significantly higher specific humoral immune response against PPV in both guinea pigs and pigs compared with the commercial inactivated vaccine. No significant side effects were observed in safety tests with pigs. In conclusion, we report a process for producing a large-scale PPV-VP2 VLP vaccine that is highly effective and has the potential to be a new commercial vaccine against PPV infection.
References
Antonis AF, Bruschke CJ, Rueda P, Maranga L, Casal JI, Vela C, Hilgers LA, Belt PB, Weerdmeester K, Carrondo MJ, Langeveld JP (2006) A novel recombinant virus-like particle vaccine for prevention of porcine parvovirus-induced reproductive failure. Vaccine 24(26):5481–5490. https://doi.org/10.1016/j.vaccnie.2006.03.089
Chen HY, Li XK, Cui BA, Wei ZY, Li XS, Wang YB, Zhao L, Wang ZY (2009) A TaqMan-based real-time polymerase chain reaction for the detection of porcine parvovirus. J Virol Methods 156(1–2):84–88. https://doi.org/10.1016/j.jviromet.2008.10.029
Cotmore SF, Tattersall P (2013) Parvovirus diversity and DNA damage responses. Cold Spring Harb Perspect Biol 5(2):152–158. https://doi.org/10.1101/cshperspect.a012989
Dar P, Kalaivanan R, Sied N, Mamo B, Kishore S, Suryanarayana VV, Kondabattula G (2013) Montanide ISA 201 adjuvanted FMD vaccine induces improved immune responses and protection in cattle. Vaccine 31(33):3327–3332. https://doi.org/10.1016/j.vaccine.2013.05.078
Duan ZG, Zhu CH, Zhang J, Fan DD (2017) Endotoxin removal from human-like collagen using triton X-114 two-phase extraction and affinity chromatography resin. Chem Eng 45(10):6–11
Feng H, Hu G, Wang H, Liang M, Liang H, Guo H, Zhao P, Yang Y, Zheng X, Zhang Z (2014) Canine parvovirus VP2 protein expressed in silkworm pupae self-assembles into virus-like particles with high immunogenicity. PLoS One 9(1):e79575. https://doi.org/10.1371/journal.pone.0079575
Foerster T, Streck AF, Speck S, Selbitz HJ, Lindner T, Truyen U (2016) An inactivated whole-virus porcine parvovirus vaccine protects pigs against disease but does not prevent virus shedding even after homologous virus challenge. J Gen Virol 97(6):1408–1413. https://doi.org/10.1099/jgv.0.000446
Gun’Ko VM, Betz WR, Patel S, Murphy MC, Mikhalovsky SV (2006) Adsorption of lipopolysaccharide on carbon sieves. Carbon 44(7):1258–1262
Guo C, Zhong Z, Huang Y (2014) Production and immunogenicity of VP2 protein of porcine parvovirus expressed in Pichia pastoris. Arch Virol 159(5):963–970. https://doi.org/10.1007/s00705-013-1907-0
Hou Y, Jiang C, Shukla AA, Cramer SM (2011) Improved process analytical technology for protein a chromatography using predictive principal component analysis tools. Biotechnol Bioeng 108(1):59–68. https://doi.org/10.1002/bit.22886
Ibrahim Eel S, Gamal WM, Hassan AI, Mahdy Sel D, Hegazy AZ, Abdel-Atty MM (2015) Comparative study on the immunopotentiator effect of ISA 201, ISA 61, ISA 50, ISA 206 used in trivalent foot and mouth disease vaccine. Vet World 8(10):1189–1198. https://doi.org/10.14202/vetworld.2015.1189-1198
Ji P, Liu Y, Chen Y, Wang A, Jiang D, Zhao B, Wang J, Chai S, Zhou E, Zhang G (2017) Porcine parvovirus capsid protein expressed in Escherichia coli self-assembles into virus-like particles with high immunogenicity in mice and guinea pigs. Antivir Res 139:146–152. https://doi.org/10.1016/j.antiviral.2017.01.003
Jozwik A, Manteufel J, Selbitz HJ, Truyen U (2009) Vaccination against porcine parvovirus protects against disease, but does not prevent infection and virus shedding after challenge infection with a heterologous virus strain. J Gen Virol 90(Pt 10):2437–2441. https://doi.org/10.1099/vir.0.012054-0
Khorasani A, Madadgar O, Soleimanjahi H, Keyvanfar H, Mahravani H (2016) Evaluation of the efficacy of a new oil-based adjuvant ISA 61 VG FMD vaccine as a potential vaccine for cattle. Iran J Vet Res 17(1):8–12
Lahariya C (2014) A brief history of vaccines & vaccination in India. Indian J Med Res 139(4):491–511
London AS, Mackay K, Lihon M, He Y, Alabi BR (2014) Gel filtration chromatography as a method for removing bacterial endotoxin from antibody preparations. Biotechnol Prog 30(6):1497–1501. https://doi.org/10.1002/btpr.1961
Lowe AJ, Bardliving CL, Batt CA (2012) Methods for chromatographic removal of endotoxin. Methods Mol Biol 899(899):265–275. https://doi.org/10.1007/978-1-61779-921-1_17
Magalhaes PO, Lopes AM, Mazzola PG, Rangel-Yagui C, Penna TC, Pessoa A Jr (2007) Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci 10(3):388–404
Maranga L, Rueda P, Antonis AF, Vela C, Langeveld JP, Casal JI, Carrondo MJ (2002) Large scale production and downstream processing of a recombinant porcine parvovirus vaccine. Appl Microbiol Biotechnol 59(1):45–50. https://doi.org/10.1007/s00253-002-0976-x
Meszaros I, Olasz F, Csagola A, Tijssen P, Zadori Z (2017) Biology of porcine parvovirus (Ungulate parvovirus 1). Viruses 9(12). https://doi.org/10.3390/v9120393
Oh WT, Kim RY, Nguyen VG, Chung HC, Park BK (2017) Perspectives on the evolution of porcine parvovirus. Viruses 9(8). https://doi.org/10.3390/v9080196
Opriessnig T, Shen HG, Pal N, Ramamoorthy S, Huang YW, Lager KM, Beach NM, Halbur PG, Meng XJ (2011) A live-attenuated chimeric porcine circovirus type 2 (PCV2) vaccine is transmitted to contact pigs but is not upregulated by concurrent infection with porcine parvovirus (PPV) and porcine reproductive and respiratory syndrome virus (PRRSV) and is efficacious in a PCV2b-PRRSV-PPV challenge model. Clin Vaccine Immunol 18(8):1261–1268. https://doi.org/10.1128/CVI.05057-11
Petsch D, Anspach FB (2000) Endotoxin removal from protein solutions. J Biotechnol 76(2–3):97–119. https://doi.org/10.1016/s0168-1656(99)00185-6
Rymerson RT, Babiuk L, Menassa R, Vanderbeld B, Brandle JE (2003) Immunogenicity of the capsid protein VP2 from porcine parvovirus expressed in low alkaloid transgenic tobacco. Mol Breed 11(4):267–276
Smith AD, Holtzapple MT (2010) Investigation of nutrient feeding strategies in a countercurrent mixed-acid multi-staged fermentation: development of segregated-nitrogen model. Bioresour Technol 101(24):9700–9709. https://doi.org/10.1016/j.biortech.2010.07.072
Song C, Zhu C, Zhang C, Cui S (2010) Detection of porcine parvovirus using a taqman-based real-time pcr with primers and probe designed for the NS1 gene. Virol J 7:353–354. https://doi.org/10.1186/1743-422X-7-353
Sumana C, Angelica MS, Doris C, Mary S, Terika S, Brito LA, Pu Z, Gillis O, Mandl CW, Mason PW (2013) Generation of a parvovirus B19 vaccine candidate. Vaccine 31(37):3872–3878. https://doi.org/10.1016/j.vaccine.2013.06.062
Teodorowicz M, Perdijk O, Verhoek I, Govers C, Savelkoul HF, Tang Y, Wichers H, Broersen K (2017) Optimized Triton X-114 assisted lipopolysaccharide (LPS) removal method reveals the immunomodulatory effect of food proteins. PLoS One 12(3):e0173778. https://doi.org/10.1371/journal.pone.0173778
van Beurden SJ, Leroy B, Wattiez R, Haenen OL, Boeren S, Vervoort JJ, Peeters BP, Rottier PJ, Engelsma MY, Vanderplasschen AF (2011) Identification and localization of the structural proteins of anguillid herpesvirus 1. Vet Res 42:105. https://doi.org/10.1186/1297-9716-42-105
Wrathall AE, Wells DE, Cartwright SF, Frerichs GN (1984) An inactivated, oil-emulsion vaccine for the prevention of porcine parvovirus-induced reproductive failure. Res Vet Sci 36(2):136–143
Zhou H, Yao G, Cui S (2010) Production and purification of VP2 protein of porcine parvovirus expressed in an insect-baculovirus cell system. Virol J 7:366. https://doi.org/10.1186/1743-422X-7-366
Zhou Y, Ma X, Hou Z, Xue X, Meng J, Li M, Jia M, Luo X (2012) High cell density cultivation of recombinant Escherichia coli for prodrug of recombinant human GLPs production. Protein Expr Purif 85(1):38–43. https://doi.org/10.1016/j.pep.2012.06.016
Zhou Y, Lu Z, Wang X, Selvaraj JN, Zhang G (2018) Genetic engineering modification and fermentation optimization for extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol 102(4):1545–1556. https://doi.org/10.1007/s00253-017-8700-z
Funding
This work was supported by grants from the National Key R&D Program (2017YFD0501103) and “1125 talent gathering plan” of Zhengzhou, as well as the Special Fund for Scientific Research and Development of Henan Academy of Agricultural Sciences ([2017]76-21).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical statement
All animal experiments were approved by the Animal Experiment Committee of Henan Academy of Agricultural Sciences with the approval number (LLSC1009605 and LLSC1009618). All the animals received humane care in compliance with good animal practice according to the animal ethics procedures and guidelines of the Institutional Animal Care and Use Committee (IACUC). All efforts were made to alleviate and minimize animal suffering.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, J., Liu, Y., Chen, Y. et al. Large-scale manufacture of VP2 VLP vaccine against porcine parvovirus in Escherichia coli with high-density fermentation. Appl Microbiol Biotechnol 104, 3847–3857 (2020). https://doi.org/10.1007/s00253-020-10483-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-020-10483-5