
Abstract
Porcine circovirus type 2 (PCV2) is a ubiquitous virus with high pathogenicity closely associated with the postweaning multisystemic wasting syndrome (PMWS) and porcine circovirus diseases (PCVDs), which caused significant economic losses in the swine industry worldwide every year. The PCV2 virus-like particles (VLPs) are a powerful subunit vaccine that can elicit high immune response due to its native PCV2 virus morphology. The baculovirus expression system is the widely used platform for producing commercial PCV2 VLP vaccines, but its yield and cost limited the development of low-cost vaccines for veterinary applications. Here, we applied a nonconventional yeast Kluyveromyces marxianus to enhance the production of PCV2 VLPs. After codon optimization, the PCV2 Cap protein was expressed in K. marxianus and assemble spontaneously into VLPs. Using a chemically defined medium, we achieved approximately 1.91 g/L of PCV2 VLP antigen in a 5-L bioreactor after high cell density fermentation for 72 h. That yield greatly exceeded to recently reported PCV2 VLPs obtained by baculovirus-insect cell, Escherichia coli and Pichia pastoris. By the means of two-step chromatography, 652.8 mg of PCV2 VLP antigen was obtained from 1 L of the recombinant K. marxianus cell culture. The PCV2 VLPs induced high level of anti-PCV2 IgG antibody in mice serums and decreased the virus titers in both livers and spleens of the challenged mice. These results illustrated that K. marxianus is a powerful yeast for cost-effective production of PCV2 VLP vaccines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Porcine circovirus (PCV) is one of the smallest animal viruses belonging to the genus Circovirus, family Circoviridae, and consists of a circular, single-stranded DNA with 1.7–2 kb in size. PCV virion is a nonenveloped, icosahedral particles of approximately 20 nm in diameter (Gillespie et al. 2009; Tischer et al. 1982). Currently, there are three genotypes of porcine circovirus, PCV1, PCV2, and PCV3. PCV1 was first isolated and characterized in contaminant of PK/15 cells (Tischer et al. 1974). It was proved to be non-pathogenic virus, and infection with this virus was widespread in almost all swine-producing areas (Allan et al. 2012). PCV2 is generally considered a ubiquitous virus with high pathogenicity that is closely associated with the postweaning multisystemic wasting syndrome (PMWS) and other porcine circovirus diseases (PCVDs), such as enteric disease, respiratory disease, porcine dermatitis and nephropathy syndrome (PDNS), and reproductive failure, which caused large economic losses in the swine industry worldwide every year (Chae 2005; Cruz et al. 2018). The genome of PCV2 shares approximately 68% identity to that of PCV1 in nucleotide sequence (Hamel et al. 1998). While PCV3 is a new genotype of porcine circovirus first identified from piglets with multi-systemic inflammation and cardiac pathology in the USA in 2016 (Phan et al. 2016), two major open reading frames (ORFs) were arranged in PCV3 genome inversely, which encode a replicase (Rep) with only 48% amino acid identity to the Rep of PCV2, and a structural capsid (Cap) protein that shares 24% and 26% amino acid identities to the Caps of PCV1 and 2, respectively (Phan et al. 2016).
Genome of PCV2 contains 11 ORFs, but only four of them (ORF1, ORF2, ORF3, and ORF4) could be translated into relevant proteins with explicit functions (Franzo et al. 2016). ORF1 encodes two replicase enzymes, Rep and Rep’, that are indispensable for the replication and transcription of the virus. ORF2 encodes a major structural protein (Cap), the major protective antigen of PCV2, which is assigned for the phylogenetic analysis of the PCV2 genotypes. ORF3, firstly identified in 2005, encodes an unstructural protein existing in the nucleus of infected cells that can induce cell apoptosis (Liu et al. 2006; Liu et al. 2005). ORF4 mainly inhibits the activity of caspase and regulates differentiations of the CD4+ and CD8+T lymphocyte (Lv et al. 2015). According to the EU consortium on porcine circovirus disease, the PCV2 are assigned to three subtypes, PCV 2a, PCV 2b, and PCV2c. Similar to RNA viruses, PCV2 has a high evolution rate (Firth et al. 2009). In 2010, a new genotype, PCV2d, emerged in China in which its genetic distance was larger than that for subtype classification (p = 0.035) and is becoming predominant over PCV2a and PCV2b strains in North America, South America, Europe, and Asia (Opriessnig et al. 2013; Opriessnig et al. 2017).
Vaccination is the most effective strategy to decrease the mortality rate and improve the growth in PCVAD-affected pig populations under field conditions (Fachinger et al. 2008). Commercial PCV2 vaccines can be subdivided into three types, commonly referred to as inactivated vaccine, chimeric virus vaccine, and subunit vaccine. The inactivated vaccines or chimeric virus vaccine, such as chimeric PCV1–2 vaccine, are commonly produced by the infection of PK-15 cells, but the extremely slow growth of PCV caused low titers of antigen in these vaccines, which increased the dosage in vaccinating pigs and the cost as well (Cheung and Bolin 2002; Ren et al. 2016; Yang et al. 2013). The PCV2 virus-like particles (VLPs), assembled spontaneously by its capsid (Cap) protein, are a form of subunit vaccine that mimic the native PCV2 virus morphology and possess potential advantages over the live-attenuated vaccine and inactivated vaccine for their particulate structure, non-replicating, and safety (Ulmer et al. 2006; Wu et al. 2012). Several hosts, including insect-baculovirus, Saccharomyces cerevisiae, Pichia pastoris, Escherichia coli, and silkworm pupae, had been used for expression of the Cap protein to obtain PCV2 VLPs (Masuda et al. 2018; Nainys et al. 2014; Nawagitgul et al. 2000; Tu et al. 2013; Wu et al. 2012), but the production is still the major barrier to the cost-effective production. The present study describes an efficient preparation of PCV2 VLPs using a nonconventional yeast Kluyveromyces marxianus. K. marxianus is an aerobic, Crabtree negative yeast, and is one of the fastest-growing eukaryotes that can yield high biomass even on mineral medium (Groeneveld et al. 2009). This yeast is also generally recognized as safe (GRAS) and qualified presumption of safety (QPS) in the USA and European Union, respectively, and has been approved as a new food material by National Health and Family Planning Commission of the People’s Republic of China. Through codon optimization according to the preferred codon usage of K. marxianus, the PCV2d Cap protein was highly expressed in K. marxianus, with a yield of approximately 1.91 g/L in a 5-L bioreactor. The Cap protein was assembled spontaneously into VLPs, showing the potential of cost-effective production for PCV2 vaccines.
Materials and methods
Microorganisms and plasmids
The host strain K. marxianus FIM-1ΔURA3 used in this study was derived from K. marxianus FIM-1 (China General Microbiological Culture Collection Center, CGMCC No.10621), of which URA3 gene was disrupted by homologous recombination. The expression pUKDN115 vector was constructed from the pUKD-S-PIT plasmid by substitution of a multiple cloning sites (MCS) for the fragment that contained an α factor signal peptide sequence and human interferon α-2a gene (Cai et al. 2005).
Construction of the recombinant KM-PCV2 strain
The Cap gene of PCV2 JS11 strain (GenBank KC153106.1) was optimized using Optimizer software tool (http://genomes.urv.es/OPTIMIZER/) on the basis of codon usage for K. marxianus (Puigbo et al. 2007), and the optimized nucleotide sequence was deposited in the NCBI GenBank database under the accession number MH748571. After synthesis by Genewiz Corporation (Suzhou, China), the optimized Cap gene was amplified with the following primers, N115CapF (5′-TTTTTTTTGTTAGATGAATTCATGACATATCCAAGGAG-3′) and N115CapR (5′-CCTTAATCAGATCAAAGCTTTCACTTAGGGTTAAGTGG-3′), in order to add 20 nt of homologous sequence from the pUKDN115 vector at the 5′-end of adjacent fragments, respectively. The amplicon was ligated with the EcoR I and Hind III linearized pUKDN115 vector by Gibson assembly (Gibson et al. 2009). The ligated product was directly transformed into the FIM-1 ΔURA3 strain according to a lithium acetate method described previously (Antunes et al. 2000). Transformants were selected on SD plates (6.7 g/L yeast nitrogen base, 20 g/L glucose, and 20 g/L agar) and were confirmed by PCR using the N115CapF and N115CapR primers. Correct colony was designated as the KM-PCV2 strain.
Expression of the Cap protein in K. marxianus
The KM-PCV2 strain was inoculated into 50 mL of liquid YD medium (2% yeast extraction and 4% glucose) and cultured at 30 °C for 60 h. One milliliter of yeast cells was harvested by centrifugation, washed twice with 1-mL PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4), and suspended in 500-μL lysate buffer (50 mM HEPS, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, pH 7.5) supplemented with approximately 200-μL glass beads (G8772 Sigma-Aldrich, Missouri, USA). Cells were disrupted using the Bead-beater (FastPrep-24, MP, California, USA) at 6 m/s for 2 min. The cell lysates were clarified by centrifugation at 12,000 rpm, 4 °C for 20 min, and the supernatant was subjected to polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting analysis. Western blotting was performed using a Mouse Anti-PCV2 Cap mouse monoclonal antibody (Ingenasa, Madrid, Spain) and a horseradish peroxidase conjugated anti Mouse IgG (H/L) Goat polyclonal antibody (1:5000 dilution) (074-1806, KPL, USA) as previously described (Alegria-Schaffer2014). The immunoblots were imaged on a GeneGnome HR system (Syngene, Cambridge, UK) with an ECL prime Western blotting detection Kit (RPN2232, GE Healthcare, Illinois, USA).
Fed-batch fermentation of the KM-PCV2 strain
A 5-L fermentor (BXBIO, Shanghai, China) was applied for fed-batch fermentation of the KM-PCV2 strain. The bioreactor was equipped with sensors that permit monitoring the parameters including pH, temperature, agitation, and dissolved oxygen. The fermentor loaded with approximately 1.5 L of mineral medium and was sterilized at 115 °C for 20 min. The composition of mineral medium was prepared according to Hensing et al. (1994), using glucose as the carbon resource. Seed cultures were prepared by inoculating the KM-PCV2 strain in 150-mL SD liquid medium (6.7 g/L yeast nitrogen base, 20 g/L glucose). After incubation at 30 °C for 16 h, the culture broth was used to inoculate the bioreactor. Fermentation conditions were controlled to a temperature of 30 °C and a pH value of 5.5 regulated with 25% ammonium hydroxide. Dissolved oxygen (dO2) was maintained above 10% by cascading it with agitation rate at fixed ventilation of 3 L/min. After batch glucose was consumed, indicated by a rise of the dO2 value after 8–10 h, the cultures were fed with 60% glucose in the range of 20–35 mL/h by adjusting feeding rates. During fermentation, cell cultures were withdrawn at indicated time points for determination of cell density (OD600nm), wet cell weight (WCW), and quantification of PCV2 VLPs.
Purification of PCV2 VLPs
KM-PCV2 cells from fed-batch fermentation were havested by centrifugation at 5000 rpm for 10 min. After washing with deionized water twice, the cell pellets were suspended in phosphate buffer (50 mM Na2HPO4, 200 mM NaCl, pH 6.8), followed by disruption with a JN High Pressure Homogenizer (JNBIO, Guangzhou, China) under a condition of 1300 bar, 4 °C for 2 times. The homogenates were clarified by centrifugation at 12,000 rpm, 4 °C for 30 min. To achieve a resin for ion exchange chromatography (IEX), each 3 mL of cation resins, including POROS™ 50 HS (Thermo Fisher Scientific, Illinois, USA), Capto™ SP ImpRes (GE healthcare), Capto™ S ImpAct (GE healthcare), and SP Bestarose FF (Bestchrom, Shanghai, China), and anion resins, including Capto Q ImpRes (GE healthcare), Q Bestarose FF (Bestchrom), and Capto Q XP (GE healthcare), was packed into Poly-Prep® Chromatography Columns (Bio-Rad, Hercules, CA, USA), respectively. Each column was loaded with 20 mL of supernatant. After wash with 10-mL phosphate buffer, the absorbed proteins were eluted with a 5 mL of phosphate buffer plus 1 M NaCl, and fractions were analyzed by SDS-PAGE.
Scale-up IEX purification of PCV2 VLPs was performed on a XK 50/30 column (GE healthcare) packed with 400 mL of SP Bestarose FF resins. The column was equilibrated with phosphate buffer (50 mM Na2HPO4, 50 mM NaCl, pH 6.8). Supernatant samples were diluted with three volumes of phosphate buffer and loaded onto the column at a speed of 50 mL/min. After washing step, the binding proteins were eluted by phosphate buffer described above. Downstream purification of PCV2 VLPs was performed on an AKTA Purifier 100 (GE Healthcare, USA) using a HiPrep™ 26/60 Sephacryl® S-500 HR column (GE Healthcare, USA). The column was loaded with 4 mL of the IEX purified sample and eluted with PBS at a flow rate of 0.5 mL/min. The elution fractions were analyzed by SDS-PAGE, TEM, and size-exclusion HPLC (SE-HPLC). Protein concentration was determined using a BCA Protein Assay Kit (23250, Thermo Fisher Scientific).
SE-HPLC quantification of PCV2 VLPs
Quantification of PCV2 VLPs was performed on an Agilent Series 1100 System (Agilent, Waldbronn, Germany) consisting of a degasser, a quaternary pump, and a diode array detector as previously described (Steppert et al. 2017). Typically, 100 μL of sample was injected on a TSKgel G4000 SWXL column (300 mm × 7.8 mm i.d.) (Tosoh Bioscience, Stuttgart, Germany) coupled with a TSKgel SWXL guard column (40.0 mm × 6.0 mm i.d.) (Tosoh Bioscience) and eluted with 100 mM phosphate buffer pH 6.7 containing 100 mM Na2SO4. Absorbance was measured at 280 nm. The amounts of PCV2 VLPs were quantified using a standard curve that was obtained by performing SE-HPLC with serial dilutions of the purified PCV2 VLPs described above.
Transmission electronic microscopy
The formation of PCV2 VLPs was examined by transmission electron microscopy using the JEM-2100 Electron Microscope (JEOL Tokyo, Japan) (Bucarey et al. 2009). Samples were spotted onto carbon-coated copper grids. After adsorption for 5 min at room temperature, the copper grids were dried with filter papers followed by staining with 3% of phosphotungstic acid (PTA) for 5 min, and then imaged at an accelerating voltage of 120 kV.
Immunization of mice
To generate the PCV2 VLPs vaccine, the purified PCV2 VLPs was diluted with PBS buffer, followed by emulsifying with 10% MONTANIDE™ Gel 01 adjuvant (Seppic, Paris, France) to a final concentration of 40 μg/mL. Twenty-one of 6–8-week-old Balb/C mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., and randomly divided into three groups. Mice of group A (n = 8) were subcutaneously injected with 0.3 mL of the PCV2 VLPs vaccine. Similarly, mice of group B (n = 5) were injected with 0.3 mL of Ingelvac CircoFLEX® vaccine (Boehringer Ingelheim, Ingelheim, Germany). As a negative control, mice of group C (n = 8) were injected with 0.3 mL of PBS. After 14 days post-immunization (dpi), serums were collected from five mice randomly in each group weekly to detect antibodies against PCV2. At 28 dpi, three mice from groups A and C were euthanized to separate the spleen lymphocytes, and the rest of the mice were intraperitoneally and orally challenged with 0.25 mL PCV2-WH strain (107TCID50/mL), respectively. At 21 days post-challenge (dpc), the challenged mice were euthanized to detect the PCV2 virus titers in liver and spleen tissues (Kiupel et al. 2001).
Antibody detection by enzyme-linked immunosorbent assay
The 96-well Costar Assay Plate (Corning, NewYork, USA) was coated with the purified Cap protein that was expressed and purified from E. coli as previously described (Bucarey et al. 2009). The coated plates were blocked with PBS containing 0.05% Tween-20 and 5% non-fat milk, and then, 100 μL of serially diluted serum samples was added per well. After incubation at room temperature (RT) for 1 h, the plates were washed with PBS containing 0.05% Tween-20 (PBST) and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:10,000) was added, followed by incubation at 37 °C for 40 min. After washing five times with PBST, the plates were washed and visualized by addition of 3,3′,5,5′-tetramethylbenzidine solution (Tiangen, Beijing, China) for 10 min at RT. Colorimetric reaction was terminated by 50 μL of 2 M sulfuric acid. Absorbance at 450 nm was recorded using an Eon™ High Performance Microplate Spectrophotometer (BioTek, Winooski, USA).
Quantitative real-time PCR assays of PCV2 nucleic acid
The viral DNAs were extracted from liver and spleen tissues of the 21 dpc mice using a TIANamp Virus DNA/RNA Kit (Tiangen). qPCR was performed on a LightCycler 480 II Real-Time PCR System (Roche, Penzberg, Germany), using a SYBR Premix Ex TaqII (TaKaRa) with the primers, qF (5′-TGCCACATCGAGAAAGCGAA-3) and qR (5′-AAACGTTACAGGGTGCTGCT-3). Titers of PCV2 virus were quantified according to a standard curve obtained by serial dilutions of pMD18T-WH, in which it contained a single copy of PCV2 genomic DNA.
Results
Expression of PCV2 Cap in K. marxianus
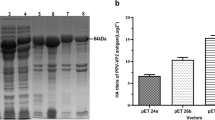
The optimized Cap gene, without any tag for detection and purification, was inserted into the multiple cloning sites (MCS) of the pUKDN115, generating the expression vector pUKDN115-PCV2 cap (Fig. 1a). The PCV2 Cap protein was intracellularly expressed under the inulinase gene promoter (Pinu) of K. marxianus. After being cultured in YD medium for 72 h, the KM-PCV2 cells were harvested and analyzed using SDS-PAGE and Western blot. As shown in Fig. 1b, an extra protein band with a size of approximately 28 kDa was found in the supernatant of the disrupted PCV2 cells, which was consistent with the predicted molecular weight of PCV2 Cap protein. This band was further verified by Western blotting using an anti-PCV2 Cap mouse monoclonal antibody (Fig. 1b), indicating that the PCV2 Cap protein was highly expressed intracellularly in a soluble form. The supernatant sample was subjected to the transmission electron microscopy (TEM) analysis, and result showed that the PCV2 cap proteins expressed in K. marxianus were assembled into VLPs spontaneously, with a diameter of approximately 20 nm (Fig. 1c).

Construction of the recombinant KM-PCV2 strain. a Schematic representation of the recombinant vector pUKDN115-PCV2 Cap. b SDS-PAGE and Western blotting assays for the PCV2 Cap expression in K. marxianus. Lane M: PageRuler Prestained Protein Ladder; lane 1: total cell lysate from K. marxianus FIM-1ΔURA3; lane 2: total cell lysate from K. marxianus FIM-1ΔURA3 transformed with pUKDN115 vector; lane 3: total cell lysate from the recombinant KM-PCV2 strain; lane 4: the supernatant of the KM-PCV2 strain cell lysate; lane 5: the precipitation of the KM-PCV2 strain cell lysate. c TEM image of the PCV2 VLPs assembled intracellularly in the recombinant KM-PCV2 strain. Scale bar, 100 nm
High-level expression of the PCV2 Cap in fed-batch fermentation by K. marxianus
To evaluate the yield of Cap protein, we conducted a high cell density fermentation of the KM-PCV2 strain using a defined synthetic medium in a 5-L reactor. Throughout the fermentation, glucose was limited to control the growth rate and to avoid the accumulation of ethanol and other byproducts. The highest cell density (OD600 nm) was obtained after 36-h fermentation, with a WCW of 500 g/L (Fig. 2a). Prolonging the fed-batch fermentation resulted in a decrease of biomass yield, whereas it increased the production of both PCV2 Cap proteins and VLPs (Fig. 2b, c). After 72 h, the amount of PCV2 VLPs reached up to 1.91 g/L (Fig. 2c), which was quantified by SE-HPLC using the purified PCV2 VLPs as a standard.

Fed-batch fermentation of the recombinant KM-PCV2 strain in a 5-L bioreactor. a Time course of the wet cell weight and the production of PCV2 VLPs. b SDS-PAGE analysis of the Cap protein expression in fed-batch fermentation at indicated times. Cultures were diluted with five volumes of PBS, and then cells were disrupted using the FastPrep-24 Bead-beater at 6 m/s for 2 min. Twenty microliters of each lysate was loaded onto 12% SDS-PAGE and stained by Coomassie brilliant blue G-250. c SE-HPLC quantification of the PCV2 VLP productions in fed-batch fermentation. Cell lysates were clarified by centrifugation at 12,000 rpm, 4 °C for 30 min, followed by filtration with 0.22-μm filters (Millipore, USA), and then 100 μL of each sample was injected on a TSKgel G4000SWXL column for SE-HPLC analysis. The amounts of PCV2 VLPs in samples were calculated according to the standard curve of the purified PCV2 VLPs
Purification of the PCV2 VLPs from K marxianus
Firstly, to purify of the PCV2 VLPs from the KM-PCV2 cells lysates, four cation resins along with three anion resins were tested for the IEX purification. As a result, all the tested IEX resins could absorb PCV2 VLPs in 50 mM phosphate buffer pH 6.8 containing 50 mM NaCl, but only the two resins, Capto™ SP ImpRes and SP Bestarose FF, seemed to be more suitable for the purification of PCV2 VLPs as to their high recoveries and purities (Fig. 3a, b). However, considering that the high pressure of the Capto™ SP ImpRes packed column would decrease the efficiency of IEX purification, the SP Bestarose FF resins were finally picked out to scale-up of the IEX purification (Fig. 3c). After purification by a XK 50/30 column (GE healthcare) packed with 400 mL of SP Bestarose FF resins, 256 mL of elution fraction containing 2.73 mg/mL PCV2 VLPs antigen was obtained from 1 L of fed-batch fermentation cultures (Table 1). To elevate the purity of PCV2 VLPs antigen, a polishing step purification was conducted using a HiPrep™ 26/60 Sephacryl® S-500 HR column. As shown in Fig. 4a, PCV2 VLPs were eluted at a retention time ranging from 60 to 100 mL. This result indicated that some of the PCV2 VLPs were aggregated in PBS buffer. The fractions eluted from 60 to 100 mL were pooled for SDS-PAGE, SE-HPLC, and TEM assays. From the SDS-PAGE, in addition to the PCV2 Cap protein, there was a minor protein with a molecular weight of approximately 55 kDa (Fig. 4a). This protein was not detected in SE-HPLC (Fig. 4b), but it could be hybridized by the anti-PCV2 Cap monoclonal antibody (Fig. 4d), indicating that it might be a dimer Cap protein. In SE-HPLC analysis, the PCV2 VLPs have two chromatographic peaks close together at retention times of 13.7 min and 15.5 min, respectively (Fig. 4b). Fractions of these two peaks were collected and analyzed by SDS-PAGE and TEM. SDS-PAGE showed that the two peaks both were the PCV2 Cap protein (Fig. 4c). From the TEM image, the component of HPLC peak at 13.7 min was the form of aggregated VLPs with multicopies, while the peak at 15.5 min is PCV2 VLPs with uniform spherical structure (Fig. 4e, f). Thus, according to the integration for areas of peak at 13.7 min and 15.5 min, the purity of the PCV2 VLPs from Sephacryl® S-500 HR gel filtration reached approximately 97%.

Purification of the PCV2 VLPs by ion-exchange chromatography. Four cation resins (a) and three anion resins (b) were tested for the purification of PCV2 VLPs. Twenty microliters of each fraction was analyzed by SDS-PAGE. Lane F: fractions of flow through; lane E: fractions of elution with 1 M NaCl. c Scale up purification of the PCV2 VLPs using SP Bestarose FF from the KM-PCV2 cell. Lane S: cell lysate sample; lane F: fractions of flow through; lane E1–3; fractions of elution with 1 M NaCl

a Gel filtration chromatography of the PCV2 VLPs. Four milliliters of the IEX purified sample were loaded onto a HiPrep™ 26/60 Sephacryl® S-500 HR column. Elution fractions that were collected at the retention times ranging from 50 to 110 mL were analyzed by 12% SDS-PAGE. b Determination of the purity of the purified PCV2 VLPs by SE-HPLC. Fractions ranging from 60 to 100 mL were pooled and concentrated to 4 mL with a 30 kDa Amicon® Ultra 15-mL filters (Millipore). A 100 μL of the concentrated sample was injected, and two elution peaks at retention times of 13.7 and 15.5 min were collected and subjected to SDS-PAGE (c), Western blot (d), and TEM (e, f) analysis
Immunogenicity of vaccine-immunized mice
The purified PCV2 VLP antigens were evaluated for their immunogenicity in mice. Vaccinated with a dose of approximately 12 μg PCV2 VLP antigen, anti-PCV2 IgG antibody in mice serums was detectable after 14 dpi, and thereafter, the titers continued to increase to a titer of 7300 before challenge at 28 dpi (Fig. 5a). These results indicated that our PCV2 VLP vaccine could elicit higher level of anti-PCV2 IgG antibody, which were comparable to that of the commercial PCV2 vaccine (Ingelvac CircoFLEX).

Analysis of the immunogenicity of PCV2 VLPs in mice. a Time courses of the anti-PCV2 IgG antibody titers in mice serums. Serums were collected from all grouped mice at indicated days post-immunization. b Quantification of PCV2 virus loads in livers and spleens of mice after challenge. Liver and spleen tissues were separated from the mice challenged with PCV2-WH strain after 21 days post challenge. Virus loads were determined by qPCR. Values were the means ± standard error of mean (SEM) from five independent experiments
At 28 dpi, the mice vaccinated with the PCV2 VLPs vaccine and the commercial PCV2 vaccine were challenged with PCV2-WH strain at a concentration of 107 TCID50/mL. After 21 dpc, the challenged mice were euthanized to detect the amounts of virus in livers and spleens using qPCR method. As shown in Fig. 5b, in the PCV2 VLPs vaccine-immunized mice, the average virus titers of PCV2 virus in the liver tissues and spleen tissues were 177 and 252 copies per mg, respectively, which were at the same level to that of the commercial PCV2 vaccine, but significantly lower than that of the unvaccinated mice (p < 0.01). These results indicated that vaccination with the PCV2 VLP vaccine could reduce the risk of PCV2 infection in mice.
Discussion
The PCV2 virus mainly infects and replicated in PK15 cell lines, with low titer yield of virions, which was difficult to obtain sufficient amounts of antigen for preparation of PCV2 vaccines (Misinzo et al. 2005). Cap protein of PCV2 has an intrinsic ability to self-assemble into VLPs that structurally resemble the authentic virions with high immunogenicity and can serve as effective and safe stand-alone vaccines (Bucarey et al. 2009; Marcekova et al. 2009). The baculovirus expression system is an effective platform for the production of VLPs, but the expression yield of this system limited the development of low-cost PCV2 VLPs vaccines for veterinary applications (Liu et al. 2015; Summers 2006). A recently promising work reported that the production of PCV2 VLPs increased to 198 mg/L from 56 mg/L using a new TB baculovirus expression cassette (Lopez-Vidal et al. 2015). In E. coli, the PCV2 Cap protein is difficult to express most likely due to the rare arginine codons at the 5′-end of its gene, but it possesses the nuclear localization signal region (NLS) that is essential for the self-assembly of VLPs (Liu et al. 2001; Wu et al. 2012). To tackle this obstacle, codon optimization or special host, e.g., E. coli BL21-CodonPlus (DE3)-RIPL cells, was usually introduced, and production levels of PCV2 VLPs became comparable to that of the baculovirus expression system (Marcekova et al. 2009; Trundova and Celer 2007; Wu et al. 2016).
Yeast is a particularly powerful platform for VLP production because it possesses post-translational modification, grows cheaply and rapidly, and readily ferments in high cell density (Lua et al. 2014). Up to date, only two yeast species, S. cerevisiae and P. pastoris, were used for the production of the PCV2 VLPs, with the highest yield of 174 mg/L (Bucarey et al. 2009; Nainys et al. 2014; Tu et al. 2013). The objective of this study was to assess the potential of the nonconventional yeast K. marxianus as a new platform for efficient production of PCV2 VLP vaccines. The Cap gene from the PCV2d genotype was used in this study based on the epidemiological studies on PCV2 infections in China and other countries (Jiang et al. 2017; Opriessnig et al. 2017). After codon optimization, the PCV2d Cap gene was highly expressed by the Pinu promoter of K. marxianus. In K. marxianus CBS 6556, this promoter could be induced by inulin and sucrose, strongly repressed by glucose and lactose, and slightly inhibited by ethanol (Rouwenhorst et al. 1988). However, such inducements or repressions were not noticed in our Fim-1 strain (data not shown). Comparing with inulin and sucrose, glucose is a more economic and efficient carbon resource for the KM-PCV2 strain, since it can be used to obtain high density of cells and high expression of Cap proteins, on condition that it is maintained below 1 g/L during the fed-batch fermentation. Under this condition, the KM-PCV2 strain could produce 1.91 g/L PCV2 VLPs after 72 h, much higher than that of other expression systems.
As reported by Zaveckas et al. (2015), PCV2 VLPs were not retained on Q Sepharose XL column during anion exchange chromatography in 50 mM Tris-HCl, pH 8.0 containing 20% (v/v) glycerol and 150 mM NaCl, but some host cell proteins and residual nucleic acids. An improved strategy for anion exchange chromatography, reported by Masuda et al., showed that the PCV2 VLPs was tightly bound to Hitrap Q HP resins after being precipitated by ammonium sulfate precipitation and resolved in 50 mM sodium phosphate plus 250 mM NaCl, pH 8.0 (Masuda et al. 2018). In our experiments, all of anion exchangers could absorb small quantity of the PCV2 VLPs in 50 mM sodium phosphate containing 50 mM NaCl, pH 6.8 without ammonium sulfate precipitation (Fig. 3b). This result may be attributed to a low pH value that aided the PCV2 VLPs in competition with other negatively charged proteins and nucleic acids. On the contrary, cation exchangers demonstrated higher efficiencies in recovering and purifying the PCV2 VLPs from K. marxianus cell lysates. By coupling with Sephacryl® S-500 HR gel filtration, the purity of the PCV2 VLPs reached approximately 97%.
Many viruses have evolved decoy epitopes to divert the host immune response and propagate effectively (Finlay and McFadden 2006). In PCV2, a decoy epitope composed of amino acid residues 169–180 is the most immune dominant among the antigenic determinants in Cap protein of PCV2b (Yu et al. 2016). These antigenic determinants are structurally buried inside the virus-like particle, and antibodies against them are non-neutralizing and cannot recognize the intact virus (Trible et al. 2012). Accordingly, the unfolded antigens with decoy epitopes in a subunit vaccine can affect the level of neutralizing antibody in an immunized animal (Jin et al. 2018). To test whether the PCV2 VLPs produced in K. marxianus contained unassembled or unfolded Cap protein, the cell lysates of the recombinant strain were fractionated by SE-HPLC, and then analyzed by Western blotting analysis with a swine anti-PCV2 Cap polyclonal antibody. As a result, only two chromatographic peaks, with retention times of 13.7 min and 15.5 min, were confirmed to contain Cap proteins, which represented the aggregated and uniform spherical PCV2 VLPs, respectively (data not shown), indicating that K. marxianus was a good platform for production of PCV2 VLP vaccines.
In SDS-PAGE analysis, a minor band with approximately 55 kDa in size was detected in the purified PCV2 VLP samples (Fig. 4a). This minor band also existed in recombinant Cap proteins from both E. coli and silkworm pupae (Masuda et al. 2018; Wu et al. 2016). To test whether it was a dimer form of Cap protein, this band was subjected to LC-MS/MS analysis on a Q Exactive mass spectrometer (Thermo Fisher Scientific) coupled to Easy nLC (Thermo Fisher Scientific) in positive ion mode. Alignment of MS/MS spectra with the Cap protein sequence indication confirmed that this band is a dimer of Cap protein (data not shown). The PCV2 VLPs produced by our K. marxianus strain, similarly in other expression systems such E. coli and silkworm pupae, tended to aggregate during the purification (Masuda et al. 2018; Wu et al. 2016). This is a common feature for VLPs that they are susceptible to aggregation during manufacture processes, and it is exacerbated in the case of storage in containers and cuvettes due to surface adsorption, commonly concomitant with the loss of vaccine efficacy (Chen et al. 2015; Ding et al. 2010; Shi et al. 2005). To evaluate the potency of our PCV2 VLPs vaccine, Balb/C mice were vaccinated with the purified PCV2 VLPs at a dose of 12 μg antigen per mouse. Fortunately, our PCV2 VLPs vaccine elicited high levels of anti-PCV2 IgG antibody; meanwhile, it reduced the virus titers both in liver and spleen tissues of PCV2 strain-challenged mice. These results indicated that the PCV2 VLPs produced by K. marxianus could be developed for a low-cost PCV2 vaccine for veterinary applications.
References
Alegria-Schaffer A (2014) Western blotting using chemiluminescent substrates. Methods Enzymol 541:251–259
Allan G, Krakowka S, Ellis J, Charreyre C (2012) Discovery and evolving history of two genetically related but phenotypically different viruses, porcine circoviruses 1 and 2. Virus Res 164:4–9
Antunes DF, de Souza CG, de Morais MA (2000) A simple and rapid method for lithium acetate-mediated transformation of Kluyveromyces marxianus cells. World J Microbiol Biotechnol 16:653–654
Bucarey SA, Noriega J, Reyes P, Tapia C, Saenz L, Zuniga A, Tobar JA (2009) The optimized capsid gene of porcine circovirus type 2 expressed in yeast forms virus-like particles and elicits antibody responses in mice fed with recombinant yeast extracts. Vaccine 27:5781–5790
Cai XP, Zhang J, Yuan HY, Fang ZA, Li YY (2005) Secretory expression of heterologous protein in Kluyveromyces cicerisporus. Appl Microbiol Biotechnol 67:364–369
Chae C (2005) A review of porcine circovirus 2-associated syndromes and diseases. Vet J 169:326–336
Chen Y, Zhang Y, Quan C, Luo J, Yang YL, Yu MR, Kong YJ, Ma GH, Su ZG (2015) Aggregation and antigenicity of virus like particle in salt solution-a case study with hepatitis B surface antigen. Vaccine 33:4300–4306
Cheung AK, Bolin SR (2002) Kinetics of porcine circovirus type 2 replication. Arch Virol 147:43–58
Cruz TF, Magro AJ, de Castro A, Pedraza-Ordonez FJ, Tsunemi MH, Perahia D, Araujo JP Jr (2018) In vitro and in silico studies reveal capsid-mutant porcine circovirus 2b with novel cytopathogenic and structural characteristics. Virus Res 251:22–33
Ding Y, Chuan YP, He LZ, Middelberg APJ (2010) Modeling the competition between aggregation and self-sssembly during virus-like particle processing. Biotechnol Bioeng 107:550–560
Fachinger V, Bischoff R, Ben Jedidia S, Saalmuller A, Elbers K (2008) The effect of vaccination against porcine circovirus type 2 in pigs suffering from porcine respiratory disease complex. Vaccine 26:1488–1499
Finlay BB, McFadden G (2006) Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124:767–782
Firth C, Charleston MA, Duffy S, Shapiro B, Holmes EC (2009) Insights into the evolutionary history of an emerging livestock pathogen: porcine circovirus 2. J Virol 83:12813–12821
Franzo G, Cortey M, Segales J, Hughes J, Drigo M (2016) Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol Phylogenet Evol 100:269–280
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–U341
Gillespie J, Opriessnig T, Meng XJ, Pelzer K, Buechner-Maxwell V (2009) Porcine circovirus type 2 and porcine circovirus-associated disease. J Vet Intern Med 23:1151–1163
Groeneveld P, Stouthamer AH, Westerhoff HV (2009) Super life - how and why 'cell selection' leads to the fastest-growing eukaryote. FEBS J 276:254–270
Hamel AL, Lin LL, Nayar GPS (1998) Nucleotide sequence of porcine circovirus associated with postweaning multisystemic wasting syndrome in pigs. J Virol 72:5262–5267
Hensing M, Vrouwenvelder H, Hellinga C, Baartmans R, Vandijken H (1994) Production of extracellular inulinase in high-cell-density fed-batch cultures of Kluyveromyces marxianus. Appl Microbiol Biotechnol 42:516–521
Jiang CG, Wang G, Tu YB, Liu YG, Wang SJ, Cai XH, An TQ (2017) Genetic analysis of porcine circovirus type 2 in China. Arch Virol 162:2715–2726
Jin JY, Park CH, Cho SH, Chung JH (2018) The level of decoy epitope in PCV2 vaccine affects the neutralizing activity of sera in the immunized animals. Biochem Biophys Res Commun 496:846–851
Kiupel M, Stevenson GW, Choi J, Latimer KS, Kanitz CL, Mittal SK (2001) Viral replication and lesions in BALB/c mice experimentally inoculated with porcine circovirus isolated from a pig with postweaning multisystemic wasting disease. Vet Pathol 38:74–82
Liu QG, Tikoo SK, Babiuk LA (2001) Nuclear localization of the ORF2 protein encoded by porcine circovirus type 2. Virology 285:91–99
Liu J, Chen I, Kwang J (2005) Characterization of a previously unidentified viral protein in porcine circovirus type 2-infected cells and its role in virus-induced apoptosis. J Virol 79:8262–8274
Liu J, Chen I, Du QY, Chua HK, Kwang J (2006) The ORF3 protein of porcine circovirus type 2 is involved in viral pathogenesis in vivo. J Virol 80:5065–5073
Liu YK, Zhang YY, Yao LG, Hao HF, Fu XJ, Yang ZQ, Du EQ (2015) Enhanced production of porcine circovirus type 2 (PCV2) virus-like particles in Sf9 cells by translational enhancers. Biotechnol Lett 37:1765–1771
Lopez-Vidal J, Gomez-Sebastian S, Barcena J, Nunez MD, Martinez-Alonso D, Dudognon B, Guijarro E, Escribano JM (2015) Improved production efficiency of virus-like particles by the baculovirus expression vector system. PLoS One 10:e0140039
Lua LHL, Connors NK, Sainsbury F, Chuan YP, Wibowo N, Middelberg APJ (2014) Bioengineering virus-like particles as vaccines. Biotechnol Bioeng 111:425–440
Lv QZ, Guo KK, Wang T, Zhang CC, Zhang YM (2015) Porcine circovirus type 2 ORF4 protein binds heavy chain ferritin. J Biosci (Bangalore) 40:477–485
Marcekova Z, Psikal I, Kosinova E, Benada O, Sebo P, Bumba L (2009) Heterologous expression of full-length capsid protein of porcine circovirus 2 in Escherichia coli and its potential use for detection of antibodies. J Virol Methods 162:133–141
Masuda A, Lee JM, Miyata T, Sato T, Hayashi S, Hino M, Morokuma D, Karasaki N, Mon H, Kusakabe T (2018) Purification and characterization of immunogenic recombinant virus-like particles of porcine circovirus type 2 expressed in silkworm pupae. J Gen Virol 99:917–926
Misinzo G, Meerts P, Bublot M, Mast J, Weingart HM, Nauwynck HJ (2005) Binding and entry characteristics of porcine circovirus 2 in cells of the porcine monocytic line 3D4/31. J Gen Virol 86:2057–2068
Nainys J, Lasickiene R, Petraityte-Burneikiene R, Dabrisius J, Lelesius R, Sereika V, Zvirbliene A, Sasnauskas K, Gedvilaite A (2014) Generation in yeast of recombinant virus-like particles of porcine circovirus type 2 capsid protein and their use for a serologic assay and development of monoclonal antibodies. BMC Biotechnol 14:100
Nawagitgul P, Morozov I, Bolin SR, Harms FA, Sorden SD, Paul PS (2000) Open reading frame 2 of porcine circovirus type 2 encodes a major capsid protein. J Gen Virol 81:2281–2287
Opriessnig T, Xiao CT, Gerber PF, Halbur PG (2013) Emergence of a novel mutant PCV2b variant associated with clinical PCVAD in two vaccinated pig farms in the US concurrently infected with PPV2. Vet Microbiol 163:177–183
Opriessnig T, Xiao CT, Halbur PG, Gerber PF, Matzinger SR, Meng XJ (2017) A commercial porcine circovirus (PCV) type 2a-based vaccine reduces PCV2d viremia and shedding and prevents PCV2d transmission to naive pigs under experimental conditions. Vaccine 35:248–254
Phan TG, Giannitti F, Rossow S, Marthaler D, Knutson TP, Li L, Deng X, Resende T, Vannucci F, Delwart E (2016) Detection of a novel circovirus PCV3 in pigs with cardiac and multi-systemic inflammation. Virol J 13:184
Puigbo P, Guzman E, Romeu A, Garcia-Vallve S (2007) OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucleic Acids Res 35:W126–W131
Ren LZ, Chen XR, Ouyang HS (2016) Interactions of porcine circovirus 2 with its hosts. Virus Genes 52:437–444
Rouwenhorst RJ, Visser LE, Vanderbaan AA, Scheffers WA, Vandijken JP (1988) Production, distribution, and kinetic-properties of inulinase in continuous cultures of Kluyveromyces marxianus CBS 6556. Appl Environ Microbiol 54:1131–1137
Shi L, Sanyal G, Ni A, Luo Z, Doshna S, Wang B, Graham TL, Wang N, Volkin DB (2005) Stabilization of human papillomavirus virus-like particles by non-ionic surfactants. J Pharm Sci 94:1538–1551
Steppert P, Burgstallera D, Klausberger M, Tover A, Berger E, Jungbauer A (2017) Quantification and characterization of virus-like particles by size-exclusion chromatography and nanoparticle tracking analysis. J Chromatogr 1487:89–99
Summers MD (2006) Milestones leading to the genetic engineering of baculoviruses as expression vector systems and viral pesticides. Adv Virus Res 68:3–73
Tischer I, Rasch R, Tochtermann G (1974) Characterization of papovavirus-and picornavirus-like particles in permanent pig kidney cell lines. Zentralbl Bakteriol Orig A 226:153–167
Tischer I, Gelderblom H, Vettermann W, Koch MA (1982) A very small porcine virus with circular single-stranded-DNA. Nature 295:64–66
Trible BR, Suddith AW, Kerrigan MA, Cino-Ozuna AG, Hesse RA, Rowland RR (2012) Recognition of the different structural forms of the capsid protein determines the outcome following infection with porcine circovirus type 2. J Virol 86:13508–13514
Trundova M, Celer V (2007) Expression of porcine circovirus 2 ORF2 gene requires codon optimized E. coli cells. Virus Genes 34:199–204
Tu YB, Wang YQ, Wang G, Wu JN, Liu YG, Wang SJ, Jiang CG, Cai XH (2013) High-level expression and immunogenicity of a porcine circovirus type 2 capsid protein through codon optimization in Pichia pastoris. Appl Microbiol Biotechnol 97:2867–2875
Ulmer JB, Valley U, Rappuoli R (2006) Vaccine manufacturing: challenges and solutions. Nat Biotechnol 24:1377–1383
Wu PC, Lin WL, Wu CM, Chi JN, Chien MS, Huang CJ (2012) Characterization of porcine circovirus type 2 (PCV2) capsid particle assembly and its application to virus-like particle vaccine development. Appl Microbiol Biotechnol 95:1501–1507
Wu PC, Chen TY, Chi JN, Chien MS, Huang CJ (2016) Efficient expression and purification of porcine circovirus type 2 virus-like particles in Escherichia coli. J Biotechnol 220:78–85
Yang X, Chen FW, Cao YH, Pang DX, Ouyang HS, Ren LZ (2013) Comparative analysis of different methods to enhance porcine circovirus 2 replication. J Virol Methods 187:368–371
Yu C, Li X, Liu JW, Diao WZ, Zhang LC, Xiao Y, Wei HF, Yu YL, Yu YQ, Wang LY (2016) Replacing the decoy epitope of PCV2b capsid protein with a protective epitope enhances efficacy of PCV2b vaccine. Vaccine 34:6358–6366
Zaveckas M, Snipaitis S, Pesliakas H, Nainys J, Gedvilaite A (2015) Purification of recombinant virus-like particles of porcine circovirus type 2 capsid protein using ion-exchange monolith chromatography. J Chromatogr B 991:21–28
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was supported by the projects of National Natural Science Foundation of China (31770094 and 91731310), National High Technology Research and Development Program of China (2014AA021301), Science and Technology Research Program of Shanghai (18391901800) and Open Research Funds of the State Key Laboratory of Genetic Engineering, Fudan University.
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All the mice experimental procedures were approved by the Animal Experiment Committee of Fudan University. All applicable international, national, and institutional guidelines for the care and use of animals were strictly followed.
Rights and permissions
About this article

Cite this article
Duan, J., Yang, D., Chen, L. et al. Efficient production of porcine circovirus virus-like particles using the nonconventional yeast Kluyveromyces marxianus. Appl Microbiol Biotechnol 103, 833–842 (2019). https://doi.org/10.1007/s00253-018-9487-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-9487-2