
Abstract
l-Hydroxyproline (Hyp) is a valuable intermediate for the synthesis of pharmaceuticals; consequently, a practical process for its production has been in high demand. To date, industrial processes have been developed by using l-Pro hydroxylases. However, a process for the synthesis of trans-3-Hyp has not yet been established, because of the lack of highly selective enzymes that can convert l-Pro to trans-3-Hyp. The present study was designed to develop a biocatalytic trans-3-Hyp production process. We speculated that ectoine hydroxylase (EctD), which is involved in the hydroxylation of the known compatible solute ectoine, may possess the ability to hydroxylate l-Pro, since the structures of ectoine and 5-hydroxyectoine resemble those of l-Pro and trans-3-Hyp, respectively. Consequently, we discovered that ectoine hydroxylases from Halomonas elongata, as well as some actinobacteria, catalyzed l-Pro hydroxylation to form trans-3-Hyp. Of these, ectoine hydroxylase from Streptomyces cattleya also utilized 3,4-dehydro-l-Pro, 2-methyl-l-Pro, and l-pipecolic acid as substrates. In the whole-cell bioconversion of l-Pro into trans-3-Hyp using Escherichia coli expressing the ectD gene from S. cattleya, only 12.4 mM trans-3-Hyp was produced from 30 mM l-Pro, suggesting a rapid depletion of 2-oxoglutarate, an essential component of enzyme activity as a cosubstrate, in the host. Therefore, the endogenous 2-oxoglutarate dehydrogenase gene was deleted. Using this deletion mutant as the host, trans-3-Hyp production was enhanced up to 26.8 mM from 30 mM l-Pro, with minimal loss of 2-oxoglutarate. This finding is not only beneficial for trans-3-Hyp production, but also for other E. coli bioconversion processes involving 2-oxoglutarate-utilizing enzymes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
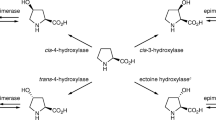
Owing to the growing importance of hydroxyproline (Hyp) as an ingredient in a wide range of pharmaceuticals, Pro hydroxylases capable of regio- and stereo-selective hydroxylation of the free-form l-Pro have been found in diverse microorganisms through extensive enzyme screening studies (Fig. 1). For example, trans-4-Hyp is a useful compound for the synthesis of carbapenem antibiotics and angiotensin-converting enzyme inhibitors (Remuzon 1996). trans-3-Hyp is also included as a constituent of microbial secondary metabolites (Hashizume et al. 2004; Schwartz et al. 1992; Sheehan et al. 1968; Suksamrarn et al. 2005). In naturally occurring Hyp isomers, the respective microbial l-Pro hydroxylases generate three Hyps (trans-4-Hyp, cis-4-Hyp, and cis-3-Hyp) from l-Pro in a regio- and stereo-selective manner (Hara and Kino 2009; Mori et al. 1997; Shibasaki et al. 1999a). However, finding an only trans-3-Hyp-forming Pro hydroxylase is considered to be the final research challenge, with both scientific significance and industrial application. trans-3-Hyp has attracted much attention in the field of synthetic chemistry as it serves as the scaffold for diverse pharmaceuticals, including peptide antibiotics (Davies et al. 2018; Kumar and Chandrasekhar 2012). Peterson et al. demonstrated slight l-Pro trans-3-hydroxylation activity in a fungal cell extract of Glarea lozoyensis, which is a trans-3-Hyp-containing pneumocandine B0 producer (Petersen et al. 2003), suggesting the involvement of a corresponding hydroxylase that catalyzed trans-3-Hyp formation. Recent genome sequence analysis of this fungus elucidated the pneumocandine biosynthetic gene cluster (Chen et al. 2013), and the trans-3-Hyp-generating hydroxylase (GloF) was found in this region. However, this enzyme enabled the simultaneous formation of both trans-4-Hyp and trans-3-Hyp, with the preferential formation of trans-4-Hyp with a molar yield of 8:1 (Houwaart et al. 2014). Recently, the orthologous enzyme, HtyE obtained from Aspergillus pachycristatus, has been reported; however, synthesis of trans-4-Hyp was still greater than trans-3-Hyp, with a molar yield of 2:1 (Mattay et al. 2018). This property seems unfavorable for yielding only trans-3-Hyp because chromatographic separations, at the very least, would likely be essential. Therefore, finding a bona fide enzyme for trans-3-Hyp synthesis remains a highly important target, as trans-3-hydroxylating enzymes would be useful to establish the enzymatic manufacturing of all known Hyp species.

Overview of diverse proline-related hydroxylases. a Simultaneous formation of cis-4-Hyp (major product) and cis-3-Hyp (minor product). b Simultaneous formation of trans-4-Hyp (major product) and trans-3-Hyp (minor product)
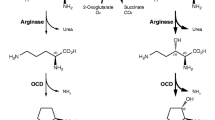
We recently proposed a unique three-step reaction strategy for the selective synthesis of trans-3-Hyp (Hara et al. 2016). In brief, l-arginine was hydroxylated by l-arginine 3-hydroxylase (Yin and Zabriskie 2004) to form (2S,3S)-3-hydroxyarginine. The product was then converted to (2S,3S)-3-hydroxyorinithine by arginase and further converted to trans-3-Hyp by ornithine cyclodeaminase. This synthetic route is significantly shorter and more convenient than chemical synthesis processes that require (de)protection, harsh conditions, and complex purification. Indeed, the route may be useful for the selective synthesis of trans-3-Hyp; however, the final enzyme in the pathway, ornithine cyclodeaminase, showed relatively low activity toward (2S,3S)-3-hydroxyornithine compared with the other enzymes and resulted in a reduced yield. Thus, a more efficient and straightforward approach capable of trans-3-Hyp generation, such as a one-step hydroxylation, is needed for practical trans-3-Hyp production.
Recent genomic analysis revealed that diverse halophilic or non-halophilic bacteria have the potential to synthesize 5-hydroxyectoine from ectoine (Czech et al. 2018); this biosynthetic route has been well investigated in Salibacillus salexigens (Bursy et al. 2007), Pseudomonas stuzeri (Seip et al. 2011), Streptomyces species (Bursy et al. 2008; Ikeda et al. 2014; Inbar and Lapidot 1988; Malin and Lapidot 1996; Prabhu et al. 2004), and Sphingopyxis alaskensis (Widderich et al. 2014). Of these, the X-ray crystallographic structures of EctD from Virgibacillus salexigens (Reuter et al. 2010) and S. alaskensis (Hoeppner et al. 2014) were solved. In general, ectoine and 5-hydroxyectoine are known as compatible solutes that serve as osmoprotectants and help organisms to survive under hyperosmotic environments, similar to glycine betaine, l-Pro, and trehalose (Pastor et al. 2010). Thus, these substances are so-called “chemical chaperones” that maintain proteins and cells against exogenous stresses, such as heat and osmotic stress. Owing to increased use of ectoine and hydroxyectoine in the healthcare and cosmetic industries (Lentzen and Schwarz 2006), there have been a number of reports on the functional analysis of hydroxyectoine biosynthesis, as well as applied studies of the use of hydroxyectoine in industrial processes (Kunte et al. 2014; Lentzen and Schwarz 2006). However, few studies have performed an in-depth investigation of the substrate range of EctD. The present study focused on the close relationship between the chemical structures of ectoine and l-Pro and their hydroxylated products 5-hydroxyectoine and trans-3-Hyp, especially in terms of the stereo configuration between the carboxy- and hydroxy-groups as trans-isomers. We thus hypothesized that EctD would accept l-Pro as a substrate, as well as ectoine, and that the hydroxylated product of l-Pro catalyzed by EctD would be trans-3-Hyp in this reaction scheme (Fig. 2).

The 2-OG-dependent hydroxylation reaction catalyzed by EctD. a Physiological reaction, b alternative reaction
This paper demonstrates that EctD hydroxylates l-Pro and the well-known EctD from Halomonas elongata was reevaluated as the standard enzyme for the formation trans-3-Hyp. Moreover, we gained a deeper insight into several EctD enzymes from non-halophilic actinobacteria, as only limited information on the substrate specificity of EctD is available in these organisms, except for the ectoine hydrolases of S. chrysomallus (Prabhu et al. 2004) and S. coelicolor A3(2) (Bursy et al. 2008). These two enzymes were previously reported to have no substrate specificity for l-Pro. We confirmed that trans-3-hydroxylating activity in EctD could promote the bioproduction of trans-3-Hyp.
Materials and methods
Chemicals
trans-3-Hyp, ectoine, 5-hydroxyectoine, and 2-methyl-l-Pro were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals were analytical grade and purchased from Wako Pure Chemical Industries (Osaka, Japan) or Tokyo Chemical Industry (Tokyo, Japan), unless otherwise mentioned.
Bacterial strains
We selected EctD from H. elongata as a model enzyme and other EctDs were chosen from four publicly available actinomycetes with a known genome sequence. H. elongata NBRC 15536T, Streptomyces coelicolor A3(2) (synonym S. violaceoruber NBRC 15146), S. avermitilis NBRC 14893T, S. cattleya NBRC 14057T, and Saccharopolyspora erythraea NBRC 13426T were obtained from the Biological Resource Center, NITE (Chiba, Japan). Escherichia coli JM109 was purchased from Nippon Gene (Tokyo, Japan), and E. coli Rosetta 2(DE3) was obtained from Merck (Darmstadt, Germany).
Construction of expression plasmids
To construct the expression plasmids for ectD genes fused with C-terminally His6-tag in E. coli, the five candidate genes from H. elongata (HeEctD, accession number HELO_4008), S. cattleya (ScaEctD, accession number SCATT_10620), S. erythraea (SerEctD, accession number SACE_0486), S. avermitilis (SavEctD, accession number SAV_6395), and S. coelicolor A3(2) (ScoEctD, accession number SCO1867) were amplified by polymerase chain reaction (PCR) from chromosomal DNA with the KOD-plus DNA polymerase (Toyobo, Osaka, Japan) for H. elongata or GC-rich PCR system (Roche, Basel, Switzerland), for other actinomycete strains, using the oligonucleotide primers listed in Table 1. PCR was conducted on an iCycler (Bio-Rad Laboratories, Hercules, CA, USA) using the following program: 94 °C for 2 min, and 25 cycles each of 94 °C for 15 s, 50 °C for 30 s, and 68 °C for 1 min. Subsequently, the amplified DNA was digested with NdeI and XhoI for ScaEctD, SerEctD, SavEctD, and ScoEctD. NdeI with HindIII for HeEctD were inserted into the same restriction sites of pET-21a(+) plasmid (Merck). The amplified ScaEctD gene, using the primer set listed in Table 2, was introduced directionally into the pQE-30 plasmid (Qiagen, Hilden, Germany) between the BamHI and HindIII sites. The resulting plasmids were introduced into E. coli JM109 and amplified; the structure was confirmed by restriction digestion and DNA sequencing.
Gene expression in E. coli and enzyme purification
To overexpress the ectD gene, E. coli Rosetta 2(DE3) harboring an expression vector was grown on 5-mL Luria-Bertani medium (Green and Sambrook 2012) containing 50 μg mL−1 ampicillin and 30 μg mL−1 chloramphenicol at 37 °C for 8 h at 280 rpm. Subsequently, 1 mL of the cell culture was transferred into 100 mL of the same medium, and cells were cultivated until the OD600 value reached approximately 0.5. Subsequently, isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM. The incubation was continued for another 8 h at 25 °C; after this, the cells were collected by centrifugation at 5000×g for 10 min at 4 °C. The cell pellet was stored at − 80 °C until use for enzyme purification. The enzyme purification procedure was carried out as described previously (Hara et al. 2015).
Enzyme assay
The hydroxylation activities of ectoine and l-Pro with purified EctD were assayed by determining the conversions of ectoine to 5-hydroxyectoine and l-Pro to trans-3-Hyp by using high-performance liquid chromatography (HPLC) in comparison with authentic standards. To test whether the enzymes accepted ectoine and l-Pro as substrates, the hydroxylation reaction was conducted as follows: a 200-μL reaction mixture containing 100 mM potassium phosphate buffer (pH 7.5), 5 mM ectoine or l-Pro, 10 mM 2-OG, 1 mM l-ascorbate, 0.5 mM FeSO4, and 20 μg of each purified enzyme was incubated at 30 °C for various intervals of up to 1 h. The reaction was terminated by heat denaturation (80 °C, 10 min), immediately followed by centrifugation to assay the supernatant.
The effects of pH and temperature on enzyme activity were determined by ectoine hydroxylation reaction under the above conditions with the exception of various pH values (5–10) and temperatures (10–60 °C).
To determine the effect of chemicals on enzyme activity, ScaEctD was used as a representative example, and 2 mM chelating reagents (EDTA and 1,10-phenanthroline), 2 mM metal ions (MgSO4, MnCl2, FeCl3, CoCl2, ZnSO4, CuSO4, and NiSO4), or 100 mM salts (NaCl and KCl) were added individually to the ectoine hydroxylation reaction mixture, as described above.
The steady-state kinetic analysis of EctD for ectoine and l-Pro was determined by varying concentrations of ectoine and l-Pro between 0 and 50 mM. Other conditions were the same as described above.
Preparation of E. coli cells for trans-3-Hyp production
E. coli endogenous sucAB gene encoding enzymes that generate succinyl-CoA from 2-oxoglutate were disrupted by the Quick and Easy E. coli Gene Deletion Kit (Gene Bridges, Heidelberg, Germany) as per the manufacturer’s instructions. The deletion cassette was constructed by PCR amplification using FRT-PGK-gb2-neo-FRT as the template DNA with the primer pair listed in Table 1.
E. coli harboring ScaEctD gene on pQE-30 plasmid was inoculated into 5 mL of LB medium, including 50 μg mL−1 ampicillin, and incubated aerobically at 37 °C for 16 h at 280 rpm. This culture was transferred into 1 L of fresh terrific broth (Green and Sambrook 2012) containing 50 μg mL−1 ampicillin and cultivated using a model MDL-200 jar fermentor (B.E. MARUBISHI, Tokyo, Japan). The following parameters were used: temperature, 37 °C; agitation, 600 rpm; and air flow rate, 1 L min−1. When the OD600 reached approximately 4, 1 mM IPTG was added to the medium, and the temperature was lowered to 25 °C. After cultivation for 12 h, the cells were harvested by centrifugation at 12,000×g for 5 min at 4 °C. The collected cells were stored frozen at − 80 °C until use.
For bioconversion of l-Pro, the thawed cells, with an OD600: 150 were suspended in the reaction mixture containing 30 mM l-Pro, 30 or 60 mM 2-OG, 10 mM l-ascorbate, and 5 mM FeSO4 in a total volume of 50 mL. The reaction was conducted in the enzyme catalysis device model BME (ABLE Corporation, Tokyo, Japan) at the following conditions: temperature, 20 °C; agitation, 1500 rpm; air flow rate, 200 mL min−1; pH, 7.0 adjusted by 1 M HCl. A small portion of the mixture was withdrawn at each time interval, and concentrations of amino acids and organic acids in the supernatant after centrifugation were determined by HPLC.
HPLC and MS analyses
l-Pro and Hyp were determined as described previously (Hara et al. 2016). Ectoine and 5-hydroxyectoine were determined by the Chromaster HPLC system (Hitachi High-Technologies, Tokyo, Japan) equipped with an Inertsil ODS-3 column (4.6 × 150 mm; GL Science, Tokyo, Japan) kept at 40 °C using a mobile phase consisting of 0.1% (v/v) perchloric acid, at a flow rate of 0.5 mL min−1 flow, and a detection wavelength of 210 nm. 2-OG and succinate were determined by the same system equipped with an Ultron PS-80H column (Shinwa Chemical Industries, Kyoto, Japan) kept at 60 °C using mobile phase consisting of 0.1% (v/v) perchloric acid at the 1 mL min−1 flow rate and 210 nm detection wavelength. FDAA-derivatized amino acids were detected by ESI-MS analysis in positive mode using an LCQ-Fleet system (Thermo Fisher Scientific, Waltham, MA, USA).
Results
Gene expression and purification of EctDs
The five candidate genes encoding HeEctD, ScaEctD, SerEctD, SavEctD, and ScoEctD were amplified by PCR and inserted into a pET-21a(+) vector at each compatible site between NdeI with XhoI or HindIII under the T7 promoter. The resulting plasmids were introduced into E. coli Rosetta 2(DE3). After IPTG-induced gene expression, SDS-PAGE analysis revealed that HeEctD, ScaEctD, SerEctD, and SavEctD were expressed in soluble forms as carboxy-terminal His6-tagged fusion proteins. These proteins were purified to homogeneity by Ni2+ affinity chromatography followed by gel filtration chromatography (Fig. 3). The molecular masses of the purified enzymes estimated by SDS-PAGE analysis were nearly identical to those of the calculated values, including His6-tag. However, ScoEctD was not entirely expressed in E. coli as either soluble or insoluble forms.

SDS-PAGE analysis of recombinant ectoine hydroxylases. Lane M, molecular standard marker; lanes 1–3, ScaEctD; lanes 4–6, SavEctD; lanes 7–9, SerEctD; lanes 10–12, HeEctD. Lanes 1, 4, 7, and 10, soluble proteins of cell extracts after centrifugation; lanes 2, 5, 8, and 11, insoluble proteins of disrupted cells; lanes 3, 6, 9, and 12, purified enzymes
Substrate specificity of EctDs
The substrate specificity of EctDs, except for ScoEctD, was assessed using the purified enzymes. When the physiological substrate ectoine was subjected to the hydroxylation reaction, all enzymes showed ectoine hydroxylation activity. The formation of 5-hydroxyectoine was clearly confirmed by HPLC analysis (Fig. 4a), and the reactions were linear up to 1 h. Subsequently, an enzymatic l-Pro hydroxylation reaction was conducted under the same conditions, and a dose-dependent decrease in l-Pro corresponding with an increase of an unidentified product was clearly confirmed in HeEctD and ScaEctD. According to chiral HPLC analysis, the retention time of the enzymatically produced substance was entirely identical to that of the authentic standard of trans-3-Hyp, and no other Hyp isomers were detected (Fig. 4b). Furthermore, we isolated the product and confirmed the molecular mass by electrospray ionization-mass spectrometry (ESI-MS) analysis, and the corresponding protonated ion of FDAA-derivatized trans-3-Hyp was detected (observed, 383.79; calculated, 384.11). Slight l-Pro hydroxylation activity was also detected under the standard conditions using SerEctD and SavEctD, whereas they had sufficient capacity for ectoine hydroxylation.

Identification of hydroxylated products by HPLC analysis. Arrows indicate the positions where authentic compounds eluted. a Ectoine hydroxylation, b l-Pro hydroxylation
To investigate the mechanism of l-Pro acceptance in EctD, molecular simulation was performed to predict the potential interactions between EctD and trans-3-Hyp (Fig. S1). The docking model showed that the hydroxy group on the C-3 carbon of trans-3-Hyp was apparently adjacent to the Fe3+ atom at a distance of 2.81 Å, allowing the coordination with other catalytic residues (H150, D152, and H251) and cosubstrate 2-OG.
In addition, other structurally relevant amino acids were tested using ScaEctD as a model, owing to its high catalytic activity. HPLC and ESI-MS analyses revealed that 3,4-dehydro-l-Pro, 2-methyl-l-Pro, and l-pipecolic acid were converted to putative oxygenated products (Fig. S2). The reaction product from 3,4-dehydro-l-Pro was confirmed by the comparison of authentic cis- and trans-3,4-epoxy-l-Pro synthesized in accordance with previous reports (Hara et al. 2014; Shibasaki et al. 1999b). Although it was difficult to isolate the products owing to extremely low yield, the protonated molecular masses of the FDAA-derivatized reaction products from 2-methyl-l-Pro and l-pipecolic acid were 398.13 and 398.12, respectively, which were greater than each substrate by 16, indicating the occurrence of ScaEctD-mediated hydroxylation.
Catalytic efficiency on ectoine and l-Pro
HeEctD and ScaEctD showed prominent hydroxylation activity toward l-Pro as well as ectoine (Fig. 4); thus, we performed further kinetic analysis for each substrate to obtain insights into their catalytic properties. The velocity was plotted against the substrate concentrations, and they were fitted to the Michaelis-Menten model to determine Km and kcat values (Fig. 5). As shown in Table 2, Km value of ScaEctD for ectoine was three times higher than that of HeEctD, and the kcat value was 2.5 times higher in ScaEctD than in HeEctD. In case of l-Pro, however, Km values in both enzymes were 10 times higher than that for ectoine. kcat values for l-Pro decreased in both enzymes when compared with those for ectoine. In addition, the comparison of catalytic efficiency (kcat/Km) demonstrated that HeEctD resulted in a slightly faster ectoine hydroxylation activity than ScaEctD, whereas ScaEctD exhibited slightly higher activity than HeEctD in l-Pro hydroxylation.

Michaelis–Menten plot for the hydroxylation of ectoine and l-Pro. a Ectoine hydroxylation by ScaEctD; bl-Pro hydroxylation by ScaEctD; c ectoine hydroxylation by HeEctD; dl-Pro hydroxylation by HeEctD
Effects of temperature, pH, and ligands
We determined the optimum reaction temperature, pH, metal ions, and salts of this enzyme. The highest activity was obtained at 30 °C, which significantly decreased at temperatures greater than 40 °C. The optimum pH was approximately neutral; the most suitable value was 7.5.
More than a 50% decrease in activity was observed in the presence of divalent metal ions, such as Mn2+, Co2+, Zn2+, Cu2+, and Ni2+, and the activity was entirely abolished by the addition of EDTA or 1,10-phenanthroline (Fig. S3). Several reported EctDs were affected by salts, such as KCl, NaCl, and potassium glutamate. In particular, KCl showed an activation effect (Widderich et al. 2014). Thus, we investigated the effect of KCl and NaCl. However, unlike the reported EctDs, salts did not facilitate enzyme activity in ScaEctD under the conditions used in this study (Fig. S3).
Production of trans-3-Hyp using ectD-expressing whole-cell E. coli
As mentioned above, we identified EctD exhibiting l-Pro hydroxylation activity to form trans-3-Hyp in the purified enzyme reaction. To confirm the feasibility of the whole-cell biocatalyst, we investigated trans-3-Hyp production using E. coli expressing the ScaEctD gene as a whole-cell biocatalyst. Jar-fermentor-cultivated E. coli was freeze-thawed to improve substrate permeability, and the bioconversion of l-Pro into trans-3-Hyp was performed in a 200-mL batch reactor with a 50-mL working volume. Considering 2-OG degradation by the E. coli endogenous enzyme, a two-fold higher concentration of 2-OG over l-Pro was utilized in the reaction mixture.
In our preliminary experiments, low reaction temperatures were favorable for long-term trans-3-Hyp production rather than optimum conditions, probably owing to time-dependent enzyme inactivation. Subsequently, the bioconversion was terminated within 1 h; only 12.4 mM trans-3-Hyp was produced with a conversion of 41% from 30 mM l-Pro (Fig. 6a). This result was probably caused by the depletion of 2-OG, suggesting a rate-limiting factor for sufficient trans-3-Hyp productivity.

Bioconversion of l-Pro into trans-3-Hyp using whole cells of ScaEctD-expressing E. coli. Circles, squares, triangles, and diamonds indicate trans-3-Hyp, l-Pro, 2-OG, and succinate, respectively. a 60 mM 2-OG in wild-type strain; b 60 mM 2-OG in sucAB disruptant; c 30 mM 2-OG in sucAB disruptant. The data represent the mean of three independent experiments, and error bars show the standard deviations
To overcome the limitation in the process, we searched for the enzyme involved in 2-OG degradation in E. coli based on the genome sequence. As such, SucAB, which encodes the 2-OG dehydrogenase complex, appeared to serve as a major 2-OG degradation enzyme and a member of the tricarboxylic acid cycle. Indeed, we confirmed the loss of 2-OG degradation activity in the absence of l-Pro (Fig. S4). Thus, using SucAB-deficient E. coli instead of wild-type JM109, we repeated the same bioconversion of l-Pro. As expected, trans-3-Hyp was successfully produced at up to 26.8 mM with a conversion of 89%, and the consumed 2-OG was comparable with the produced trans-3-Hyp (Fig. 6b). Finally, when an equal concentration of 2-OG and l-Pro was supplied in the bioconversion, 25.1 mM trans-3-Hyp with a conversion of 84% was yielded from 30 mM l-Pro (Fig. 6c).
Discussion
Regio- and stereo-selective synthesis of trans-3-Hyp is the last remaining challenge in the synthesis of naturally occurring l-Hyps. In the present study, to achieve a practical process for trans-3-Hyp production, we screened for enzymes catalyzing selective hydroxylation at the C3 position of l-Pro. EctD catalyzes the conversion of ectoine into 5-hydroxyectoine for osmotolerance. Although the substrate specificity of the enzyme was highly limited to ectoine in previous reports, we discovered l-Pro as a new substrate for EctD. The reaction product was identified as trans-3-Hyp. We also accomplished the bioconversion of l-Pro into trans-3-Hyp using E. coli expressing the ectoine hydroxylase gene. These findings will create a new approach for practical trans-3-Hyp production, which is involved in the enzymatic production of all naturally occurring l-Hyps.
To date, the following two studies have reported the substrate specificity of EctD. One study was conducted on EctD obtained from Salibacillus salexigens (Bursy et al. 2007). The native EctD of this strain was purified to homogeneity, and the substrate specificity was investigated in detail. Although this enzyme had comparative capacity to hydroxylate ectoine, no l-Pro hydroxylation activity was detected. Moreover, the EctD did not act on ectoine analogs, not even the seven-membered ring analog (4S)-2-methyl-4,5,6,7-tetrahydro-1H-1,3-diazepine-4-carboxylic acid (homoectoine) or five-membered ring analog 4,5-dihydro-2-methylimidazole-4-carboxylic acid (DHMICA). The other investigation was also reported by the same study group. The native EctD from S. coelicolor A3(2) was purified to homogeneity, and the substrate specificity was assessed (Bursy et al. 2008). Similar to the other enzyme, this EctD was able to hydroxylate ectoine, but it did not act on l-Pro. These reports suggested that the substrate specificities of EctDs were significantly narrow and strict. Generally, the known EctDs are highly specific to ectoine, and no such EctDs capable of hydroxylating l-Pro have been reported. Thus, our results demonstrate a novel substrate specificity of EctD.
For the application to trans-3-Hyp bioproduction, we carried out bioconversion using whole-cell E. coli expressing ScaEctD. However, trans-3-Hyp accumulation was not sufficient owing to the lack of 2-OG (Fig. 6a). We proposed an improved strategy using gene disruption of the endogenous SucAB (Fig. 6b, c). It would be generally advantageous to the 2-OG-dependent bioconversion using a whole-cell biocatalyst, especially for high cell densities. l-Pro was markedly degraded only in wild-type strain. This was also affected by the depletion of 2-OG, resulting in degradation of residual substrate by proline dehydrogenase (PutA, EC 1.5.5.2). When sufficient 2-OG existed in the reaction mixture, l-Pro hydroxylation occurred prior to degradation. In contrast, synthesized trans-3-Hyp did not decrease in all cases. This was attributed to the strict specificity of proline dehydrogenase (Ostrander et al. 2009). The productivity of trans-3-Hyp was still lower than industrial trans-4-Hyp production, probably due to low catalytic efficiency. l-Pro may be an inappropriate substrate for EctD, because this enzyme essentially prefers ectoine to l-Pro, as revealed by kinetic analysis. Although this should be overcome by enzyme engineering for better product yield in future industrial applications, our findings still elucidate the mechanisms of trans-3-selective hydroxylation of l-Pro.
In conclusion, we demonstrated that HeEctD has the ability to hydroxylate l-Pro as well as ectoine, and EctDs from actinomycetes exhibited the same characteristics to varying degrees. Among the EctDs, ScaEctD showed the highest l-Pro hydroxylation activity. This enzyme was applicable to trans-3-Hyp production by whole-cell bioconversion with genetically modified E. coli. Hence, this process has great potential for use in industrial production of trans-3-Hyp.
References
Bursy J, Pierik AJ, Pica N, Bremer E (2007) Osmotically induced synthesis of the compatible solute hydroxyectoine is mediated by an evolutionarily conserved ectoine hydroxylase. J Biol Chem 282(43):31147–31155. https://doi.org/10.1074/jbc.M704023200
Bursy J, Kuhlmann AU, Pittelkow M, Hartmann H, Jebbar M, Pierik AJ, Bremer E (2008) Synthesis and uptake of the compatible solutes ectoine and 5-hydroxyectoine by Streptomyces coelicolor A3(2) in response to salt and heat stresses. Appl Environ Microbiol 74(23):7286–7296. https://doi.org/10.1128/AEM.00768-08
Chen L, Yue Q, Zhang X, Xiang M, Wang C, Li S, Che Y, Ortiz-Lopez FJ, Bills GF, Liu X, An Z (2013) Genomics-driven discovery of the pneumocandin biosynthetic gene cluster in the fungus Glarea lozoyensis. BMC Genomics 14:339. https://doi.org/10.1186/1471-2164-14-339
Czech L, Hermann L, Stoveken N, Richter AA, Hoppner A, Smits SHJ, Heider J, Bremer E (2018) Role of the extremolytes ectoine and hydroxyectoine as stress protectants and nutrients: genetics, phylogenomics, biochemistry, and structural analysis. Genes (Basel) 9(4):E177. https://doi.org/10.3390/genes9040177
Davies SG, Fletcher AM, Linsdall SM, Roberts PM, Thomson JE (2018) Asymmetric syntheses of (2R,3S)-3-hydroxyproline and (2S,3S)-3-hydroxyproline. Org Lett 20(13):4135–4139. https://doi.org/10.1021/acs.orglett.8b01736
Green MR, Sambrook J (2012) Molecular cloning : a laboratory manual, 4th edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Hara R, Kino K (2009) Characterization of novel 2-oxoglutarate dependent dioxygenases converting l-proline to cis-4-hydroxy-l-proline. Biochem Biophys Res Commun 379(4):882–886. https://doi.org/10.1016/j.bbrc.2008.12.158
Hara R, Uchiumi N, Kino K (2014) Identification and characterization of 2-oxoglutarate-dependent dioxygenases catalyzing selective cis-hydroxylation of proline and pipecolinic acid from actinomycetes. J Biotechnol 172:55–58. https://doi.org/10.1016/j.jbiotec.2013.12.003
Hara R, Nakano M, Kino K (2015) One-pot production of l-threo-3-hydroxyaspartic acid using asparaginase-deficient Escherichia coli expressing asparagine hydroxylase of Streptomyces coelicolor A3(2). Appl Environ Microbiol 81(11):3648–3654. https://doi.org/10.1128/AEM.03963-14
Hara R, Kitatsuji S, Yamagata K, Kino K (2016) Development of a multi-enzymatic cascade reaction for the synthesis of trans-3-hydroxy-l-proline from l-arginine. Appl Microbiol Biotechnol 100(1):243–253. https://doi.org/10.1007/s00253-015-6992-4
Hashizume H, Hirosawa S, Sawa R, Muraoka Y, Ikeda D, Naganawa H, Igarashi M (2004) Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. - II. Structure elucidation. J Antibiot (Tokyo) 57(1):52–58
Hoeppner A, Widderich N, Lenders M, Bremer E, Smits SH (2014) Crystal structure of the ectoine hydroxylase: a snapshot of the active site. J Biol Chem 289(43):29570–29583. https://doi.org/10.1074/jbc.M114.576769
Houwaart S, Youssar L, Huttel W (2014) Pneumocandin biosynthesis: involvement of a trans-selective proline hydroxylase. ChemBioChem 15(16):2365–2369. https://doi.org/10.1002/cbic.201402175
Ikeda H, Kazuo SY, Omura S (2014) Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J Ind Microbiol Biotechnol 41(2):233–250. https://doi.org/10.1007/s10295-013-1327-x
Inbar L, Lapidot A (1988) The structure and biosynthesis of new tetrahydropyrimidine derivatives in actinomycin D producer Streptomyces parvulus. Use of 13C- and 15N-labeled l-glutamate and 13C and 15N NMR spectroscopy. J Biol Chem 263(31):16014–16022
Kumar TP, Chandrasekhar S (2012) Asymmetric syntheses of all stereoisomers of 3-hydroxyproline; a constituent of several bioactive compounds. Synthesis 44(18):2889–2894. https://doi.org/10.1055/S-0032-1316734
Kunte HJ, Lentzen G, Galinski EA (2014) Industrial production of the cell protectant ectoine: protection mechanisms, processes, and products. Curr Biotechnol 3(1):10–25. https://doi.org/10.2174/22115501113026660037
Lentzen G, Schwarz T (2006) Extremolytes: natural compounds from extremophiles for versatile applications. Appl Microbiol Biotechnol 72(4):623–634. https://doi.org/10.1007/s00253-006-0553-9
Malin G, Lapidot A (1996) Induction of synthesis of tetrahydropyrimidine derivatives in Streptomyces strains and their effect on Escherichia coli in response to osmotic and heat stress. J Bacteriol 178(2):385–395. https://doi.org/10.1128/jb.178.2.385-395.1996
Mattay J, Houwaart S, Huttel W (2018) Cryptic production of trans-3-hydroxyproline in echinocandin B biosynthesis. Appl Environ Microbiol 84(7):e02370–e02317. https://doi.org/10.1128/AEM.02370-17
Mori H, Shibasaki T, Yano K, Ozaki A (1997) Purification and cloning of a proline 3-hydroxylase, a novel enzyme which hydroxylates free l-proline to cis-3-hydroxy-l-proline. J Bacteriol 179(18):5677–5683
Ostrander EL, Larson JD, Schuermann JP, Tanner JJ (2009) A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry 48(5):951–959. https://doi.org/10.1021/bi802094k
Pastor JM, Salvador M, Argandona M, Bernal V, Reina-Bueno M, Csonka LN, Iborra JL, Vargas C, Nieto JJ, Canovas M (2010) Ectoines in cell stress protection: uses and biotechnological production. Biotechnol Adv 28(6):782–801. https://doi.org/10.1016/j.biotechadv.2010.06.005
Petersen L, Olewinski R, Salmon P, Connors N (2003) Novel proline hydroxylase activities in the pneumocandin-producing fungus Glarea lozoyensis responsible for the formation of trans 3- and trans 4-hydroxyproline. Appl Microbiol Biotechnol 62(2–3):263–267. https://doi.org/10.1007/s00253-003-1264-0
Prabhu J, Schauwecker F, Grammel N, Keller U, Bernhard M (2004) Functional expression of the ectoine hydroxylase gene (thpD) from Streptomyces chrysomallus in Halomonas elongata. Appl Environ Microbiol 70(5):3130–3132
Remuzon P (1996) Trans-4-hydroxy-l-proline, a useful and versatile chiral starting block. Tetrahedron 52(44):13803–13835. https://doi.org/10.1016/0040-4020(96)00822-8
Reuter K, Pittelkow M, Bursy J, Heine A, Craan T, Bremer E (2010) Synthesis of 5-hydroxyectoine from ectoine: crystal structure of the non-heme iron(II) and 2-oxoglutarate-dependent dioxygenase EctD. PLoS One 5(5):e10647. https://doi.org/10.1371/journal.pone.0010647
Schwartz RE, Sesin DF, Joshua H, Wilson KE, Kempf AJ, Goklen KA, Kuehner D, Gailliot P, Gleason C, White R, Inamine E, Bills G, Salmon P, Zitano L (1992) Pneumocandins from Zalerion arboricola. I. Discovery and isolation. J Antibiot (Tokyo) 45(12):1853–1866
Seip B, Galinski EA, Kurz M (2011) Natural and engineered hydroxyectoine production based on the Pseudomonas stutzeri ectABCD-ask gene cluster. Appl Environ Microbiol 77(4):1368–1374. https://doi.org/10.1128/AEM.02124-10
Sheehan JC, Mania D, Nakamura S, Stock JA, Maeda K (1968) The structure of telomycin. J Am Chem Soc 90(2):462–470
Shibasaki T, Mori H, Chiba S, Ozaki A (1999a) Microbial proline 4-hydroxylase screening and gene cloning. Appl Environ Microbiol 65(9):4028–4031
Shibasaki T, Sakurai W, Hasegawa A, Uosaki Y, Mori H, Yoshida M, Ozaki A (1999b) Substrate selectivities of proline hydroxylases. Tetrahedron Lett 40(28):5227–5230. https://doi.org/10.1016/S0040-4039(99)00944-2
Suksamrarn S, Suwannapoch N, Aunchai N, Kuno M, Ratananukul P, Haritakun R, Jansakul C, Ruchirawat S (2005) Ziziphine N, O, P and Q, new antiplasmodial cyclopeptide alkaloids from Ziziphus oenoplia var. brunoniana. Tetrahedron 61(5):1175–1180. https://doi.org/10.1016/J.Tet.2004.11.053
Widderich N, Hoppner A, Pittelkow M, Heider J, Smits SH, Bremer E (2014) Biochemical properties of ectoine hydroxylases from extremophiles and their wider taxonomic distribution among microorganisms. PLoS One 9(4):e93809. https://doi.org/10.1371/journal.pone.0093809
Yin X, Zabriskie TM (2004) VioC is a non-heme iron, alpha-ketoglutarate-dependent oxygenase that catalyzes the formation of 3S-hydroxy-l-arginine during viomycin biosynthesis. ChemBioChem 5(9):1274–1277. https://doi.org/10.1002/cbic.200400082
Funding
This study was supported in part by Grant-in-Aid for Young Scientists (B) (Grant Number 15K18677) and Grant-in-Aid for Scientific Research (C) (Grant Number 18K05400) from the Japan Society for the Promotion of Science (to RH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical statement
This article does not contain any studies involving human participants of animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1229 kb)
Rights and permissions
About this article

Cite this article
Hara, R., Nishikawa, T., Okuhara, T. et al. Ectoine hydroxylase displays selective trans-3-hydroxylation activity towards l-proline. Appl Microbiol Biotechnol 103, 5689–5698 (2019). https://doi.org/10.1007/s00253-019-09868-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-019-09868-y