
Abstract
Interleukin 6 (IL-6) is an important pathogenic factor in development of various inflammatory and autoimmune diseases and cancer. Blocking antibodies against molecules associated with IL-6/IL-6 receptor signaling are an attractive candidate for the prevention or therapy of these diseases. In this study, we developed a genetically modified strain of Lactococcus lactis secreting a single-chain variable fragment antibody against mouse IL-6 (IL6scFv). An IL6scFv-secretion vector was constructed by cloning an IL6scFv gene fragment into a lactococcal secretion plasmid and was electroporated into L. lactis NZ9000 (NZ-IL6scFv). Secretion of recombinant IL6scFv (rIL6scFv) by nisin-induced NZ-IL6scFv was confirmed by western blotting and was optimized by tuning culture conditions. We found that rIL6scFv could bind to commercial recombinant mouse IL-6. This result clearly demonstrated the immunoreactivity of rIL6scFv. This is the first study to engineer a genetically modified strain of lactic acid bacteria (gmLAB) that produces a functional anti-cytokine scFv. Numerous previous studies suggested that mucosal delivery of biomedical proteins using gmLAB is an effective and low-cost way to treat various disorders. Therefore, NZ-IL6scFv may be an attractive tool for the research and development of new IL-6 targeting agents for various inflammatory and autoimmune diseases as well as for cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Interleukin 6 (IL-6) exerts a pleiotropic activity on inflammation, immune response, and hematopoiesis through binding to an IL-6 receptor (IL-6R) (Kishimoto 2005). A transient acute phase response mediated by the IL-6/IL-6R signaling pathway in vivo, triggered by various environmental stimulants such as infection and tissue injury, plays an important role in host defense (Heinrich et al. 1990). However, dysregulated continual activation of this pathway is associated with the development of numerous chronic inflammatory disorders and autoimmune diseases as well as a malignant neoplasm (Tanaka et al. 2014). Clinical studies have been extensively performed to reveal the pathogenic role of IL-6 and therapeutic benefits of IL-6/IL-6R signaling blockade in rheumatoid arthritis (RA). Moreover, the employment of tocilizumab, a humanized anti-IL-6R, is currently recommended as a first-line biologic in therapeutic intervention for patients suffering from moderate-to-severe active RA in more than 100 countries (Tanaka and Kishimoto 2011; Tanaka et al. 2014). Besides this case, numerous clinical and preclinical evidences have suggested the availability of IL-6/IL-6R signaling blockade for other disorders such as juvenile idiopathic arthritis (De Benedetti et al. 2012; Yokota et al. 2008), Castleman’s disease (Katsume et al. 2002; Nishimoto et al. 2005), systemic lupus erythematosus (Illei et al. 2010; Shirota et al. 2013; Szepietowski et al. 2013), systemic sclerosis (Kitaba et al. 2012) (a completed clinical trial NCT01532869), and Crohn’s disease (Atreya et al. 2000; Ito et al. 2004; Jostock et al. 2001) as well as RA (Smolen et al. 2014).
Here, we propose a new inhibition strategy for IL-6/IL-6R signaling using genetically modified strains of lactic acid bacteria (gmLAB). gmLAB is a recombination microbe that produces various biomedical proteins and carried out as a mucosal delivery vehicle and a therapeutic agent in mouse models of inflammatory bowel diseases (IBDs) (Hanson et al. 2014; Motta et al. 2012; Shigemori et al. 2015), food allergy (Ai et al. 2014; Huibregtse et al. 2007), type 1 diabetes (Robert et al. 2014; Takiishi et al. 2012), and multiple sclerosis (Rezende et al. 2013). In addition, a phase I clinical trial in Crohn’s disease patients provided the evidence that IL-10-secreting gmLAB (LL-Thy12) is safe because there are only minor side effects and it was biologically contained (Braat et al. 2006). Therefore, gmLAB receive remarkable attention as an effective and low-cost live therapeutic agent for the inflammatory and autoimmune diseases. We supposed that gmLAB-producing IL-6 blocking antibody may be an attractive candidate for the treatment of various inflammatory and autoimmune diseases, especially the mucosa-associated illnesses such as IBD and asthma, as well as cancer of the mucosal tissue. To provide a tool for the research and development of new therapeutic agent targeting to IL-6/IL-6R signaling, we aimed to construct a genetically modified strain of Lactococcus lactis that secretes a single-chain variable fragment (scFv) antibody against mouse IL-6 (IL6scFv). We first validated the nisin-induced secretion of recombinant IL6scFv (rIL6scFv) in a gm-L. lactis strain harboring IL6scFv-secretion vector and the optimal conditions for the secretion of rIL6scFv. We also determined an immunoreactivity of rIL6scFv to mouse IL-6.
Materials and methods
Bacterial strains and growth conditions
L. lactis NZ9000 (NZ9000) that is a derivative of L. lactis subsp. Cremoris MG1363 and contains the regulatory genes nisR and nisK integrated into pepN gene (de Ruyter et al. 1996; Kuipers et al. 1998) was purchased from MoBiTec GmbH (Gottingen, Germany). NZ9000 was grown anaerobically at 30 °C in M17 broth (BD Difico, Becton, Dickinson and Company, Sparks, MD, USA) supplemented with 0.5 % glucose (GM17). Genetically modified strains of NZ9000 (gmNZ9000) were grown in GM17 with 10 μg/mL chloramphenicol (GM17cm). Escherichia coli M15 (QIAGEN K.K., Tokyo, Japan) (Villarejo and Zabin 1974) was grown in Luria-Bertani (LB) broth (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 20 μg/mL kanamycin (LB+kan) at 37 °C with intense shaking condition. If necessary, 50 μg/mL ampicillin was added to LB+kan broth (LB+kan+amp).
Construction of recombinant bacterial strains
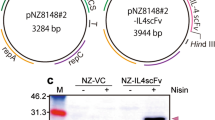
General molecular cloning techniques were performed essentially according to previously described methods (Sambrook and Russell 2001). All restriction enzymes were purchased from TaKaRa Bio Inc. (Kusatsu, Shiga, Japan). To construct gmNZ9000, a lactococcal plasmid, pNZ8148#2:SEC (Fig. 1a) which is a modified plasmid of pNZ8148 and contains the genes coding for a nisin-inducible promoter (P nis ), a signal peptide from the USP45 protein (SP usp45 ), a hexahistidine tag (His-tag), a factor Xa recognition site (FXa), a multiple cloning site (MCS), and a chloramphenicol acetyltransferase (cat), was employed as a secretion vector (Shigemori et al. 2014, 2015). A sequence coding for an IL6scFv (DDBJ accession number LC175907; http://getentry.ddbj.nig.ac.jp/getentry/na/LC175907/) was synthesized and subcloned into pEX-A by Eurofins Genomics K.K. (Tokyo, Japan) upon codon optimization for L. lactis subsp. Cremoris MG1363. A DNA fragment of IL6scFv was cut out from the subclone vector using restriction enzymes KpnI and HindIII and was then ligated with KpnI/HindIII-digested pNZ8148#2:SEC. The resulting plasmid (pNZ8148#2:SEC-IL6scFv; Fig. 1b) and an empty plasmid were separately electroporated into NZ9000 using a Gene Pulser Xcell electroporation system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) following the manufacturer’s instructions, generating NZ-IL6scFv or NZ-vector control (NZ-VC), respectively. pNZ8148#2:SEC-IL6scFv was analyzed by the DNA sequencing (performed by Eurofins Genomics K.K.) and was consistent with the predictive sequences (data not shown).

Schematic representations of lactococcal gene expression vectors. A lactococcal secretion plasmid, pNZ8148#2:SEC (a), was integrated a DNA fragment coding for IL6scFv gene in MCS to construct an IL6scFv-secretion vector, pNZ8148#2:SEC-IL6scFv (b). P nisin A promoter, SP sequence of the signal peptide from the USP45 protein, His-tag hexahistidine tag, MCS multiple cloning site, T terminator, rep replication gene, cat chloramphenicol acetyltransferase gene
Preparation of the protein samples from small-scale culture of gmNZ9000
To prepare the protein samples using to the expression analyses of rIL6scFv, gmNZ9000 were cultured in 2-mL culture system. The experimental procedures in this section were performed as previously described method (Shigemori et al. 2013, 2014). Briefly, a 1.9-mL aliquot of fresh GM17cm broth was inoculated 1:20 (v/v) with a 0.1-mL overnight culture of gmNZ9000 and was incubated at 30 °C. Gene expression in gmNZ9000 was induced by supplementation with nisin (MoBiTec GmbH) to culture medium at the point of an optical density at 600 nm (OD600) of around 0.4. After 0- to 48-h induction at 30 °C, OD600 in the cultures were measured, and bacterial cells and culture supernatants were separated by centrifugation. Cellular extracts and supernatant samples were then prepared using the lysozyme/sodium dodecyl sulfate (SDS) method or trichloroacetic acid precipitation, respectively. An equal volume of ×2 Laemmli Sample Buffer (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was added to each sample, and the mixture was then boiled for 5 min.
Preparation of the cellular extracts from large-scale culture of NZ-IL6scFv
Gene expression of rIL6scFv was induced with nisin in 50-mL or 4-L large-scale cultures according to the above-mentioned methods. After induction, cells from these 50-mL or 4-L cultures were collected by centrifugation at 3000×g and 4 °C for 20 min and were washed twice with ice-cold phosphate-buffered saline (PBS; 137 mM sodium chloride, 2.7 mM potassium chloride, 10 mM disodium hydrogen phosphate, 1.76 mM potassium dihydrogen phosphate, pH 7.4) or basic buffer, respectively, for immobilized metal ion affinity chromatography (IMAC0; 20 mM sodium phosphate, 500 mM sodium chloride, pH 8.0). The frozen-cellular pellet was grinded with aluminum oxide powder (Wako Pure Chemical Industries, Ltd.) (pellet:Al2O3 = 1:3) on a mortar for 15 min on ice. The homogenate was collected with ice-cold PBS or IMAC0 containing an ethylenediaminetetraacetic acid (EDTA) free-protease inhibitor cocktail tablet (Roche Diagnostics GmbH, Mannheim, Germany), and was clarified by two-time centrifugation at 20,000×g and 4 °C for 20 min. Cellular extracts prepared from 50-mL or 4-L cultures were further used for ELISA or His-tag purification, respectively.
SDS-polyacrylamide gel electrophoresis and western blotting
SDS-polyacrylamide gel electrophoresis (PAGE) was performed on a 15 % (v/v) polyacrylamide gel (Wang et al. 2015). The bands of separated proteins were visualized by gel staining with Coomassie Brilliant Blue (Polysciences Inc., Warrington, PA, USA) or transferred from the gel onto a polyvinylidene fluoride membrane (GE Healthcare, Buckinghamshire, UK) for western blotting. The protein-blotted membrane was blocked with skim milk and then was reacted with mouse anti-His tag antibody (Ab) (1/1000) (652,501; BioLegend, San Diego, CA, USA), followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG Ab (1/5000) (A4416; Sigma-Aldrich, St. Louis, MO, USA). The resulting blots were reacted with an ECL Prime Western Blotting Detection Reagent (GE Healthcare) and were detected using a lumino image analyzer (ImageQuant LAS 4000 mini, GE Healthcare). Densitometric analysis was performed using ImageJ software (Schneider et al. 2012) to quantify the detected bands.
Preparation of rHis-tagged green fluorescence protein from recombinant E. coli M15
To obtain a protein that could be used as a control for rIL6scFv, rHis-tagged green fluorescence protein (rGFP) was produced in E. coli M15 and was purified and characterized. A His-tagged GFP-expression vector was constructed by introduction of an EmGFP gene (714 bp, DDBJ accession number AB758275; http://getentry.ddbj.nig.ac.jp/getentry/ddbj/AB758275?filetype=html) coding for GFP into the E. coli expression vector, pQE-30 Xa (Qiagen K.K.), and was expressed in E. coli M15 as described in the manufacturer’s instructions. The resulting recombinant strain was designated as M15-GFP. rGFP produced by M15-GFP was purified using immobilized metal ion affinity chromatography techniques as described below. High purity (>99 %) and reactivity to anti-His tag Ab of purified rGFP was confirmed by SDS-PAGE or by two immunoassays (i.e., western blotting and ELISA), respectively (data not shown).
Purification of rIL6scFv from NZ-IL6scFv
The His-tagged proteins were purified by immobilized metal ion affinity chromatography techniques that were operated by a fast protein liquid chromatography system (AKTA pure 25, GE Healthcare). HisTrap HP column (1 mL; GE Healthcare) was equilibrated with 10 column volumes (CVs) of IMAC0 and was then loaded of the membrane (pore size 0.2 μm) filtered-cellular extracts using Superloop (50 mL; GE Healthcare). The column was washed with 10 CVs of IMAC0 containing 20 mM imidazole. The column-absorbed proteins were eluted by the linear gradient procedure (20 to 500 mM imidazole; 30 CVs). The flow rate of a series of events was controlled at 1 mL/min. The collected fractions (crude cellular extract, flow-through, wash, and elution) were analyzed by SDS-PAGE and western blotting according to the above-mentioned methods. The eluted fractions were dialyzed against PBS and were then concentrated by ultrafiltration (Centriprep 10 K device, Merck Millipore Ltd., Carrigtwohill Co., Cork, Ireland). The rIL6scFv concentration in the concentrate was determined by western blotting with anti-His tag Ab as described above. In this assay, rGFP purified from M15-GFP was used as a standard protein (data not shown).
ELISA for detection of His-tagged proteins
To determine the ability of rIL6scFv to bind to mouse IL-6, we employed a modified method of a standard ELISA technique (Voller et al. 1978). Briefly, flat-bottom immuno 96-well plates (Nunc MaxiSorp, Nunc A/S, Roskilde, Denmark) were coated by overnight incubation at 4 °C with 1 ng/mL recombinant mouse IL-6 (rmIL-6, 406-ML, R&D Systems, Inc., Minneapolis, MN, USA) that was diluted in PBS. The plates were washed three times with wash buffer (0.05 % Tween-20 in PBS) and were then saturated with Blocker (3 % bovine serum albumin in wash buffer) by incubation for 2 h at room temperature (RT). The plates were washed three times and were reacted with various concentrations of the rIL6scFv or rGFP or with cellular extracts (200 μg of protein) of nisin-induced gmNZ9000 strains by incubation for 2 h at RT. After five plate washes, the rmIL-6-binding proteins were reacted with anti-His tag Ab (1/1000) for 2 h at RT and were further incubated with HRP-conjugated goat anti-mouse IgG Ab (1/5000) for 1 h at RT. The plates were washed seven times, and the HRP substrate (3,3′,5,5′-tetramethylbenzidine (TMB); Nacalai Tesque Inc., Kyoto, Japan) was added to each well. After incubation with shaking for 15 min at RT, the enzymatic reaction was stopped with 2 N sulfuric acid, and absorbance was measured at 450 nm.
To measure the reactivity of cell extracts of gmNZ9000 strains with anti-His tag antibody and the amount of His-tagged peptides produced by gmNZ9000 strains, cellular extracts (20 μg of protein) of gmNZ9000 strains or various concentrations of rGFP that was used as a control protein were bound onto the plate by overnight incubation at 4 °C. After the plate was blocked, the plate-bound proteins were reacted with anti-His tag Ab and were further detected using the above-described methods.
Statistical analysis
Statistical analyses were performed using a statistical software package (ystat2004.xls, Igakutosho Shuppan, Tokyo, Japan). Student’s t test or one-way ANOVA followed by Dunnett’s test were used to determine the significance of the differences. Values are expressed as mean ± standard deviation (SD).
Results
Expression and secretion of rIL6scFv by NZ-IL6scFv
To generate rIL6scFv-secreting gmNZ9000, we engineered pNZ8148#2:SEC to construct the IL6scFv secretion vector, pNZ8148#2:SEC-IL6scFv (Fig. 1). This plasmid was designed to express the IL6scFv conjugating to SP usp45 and His-tag at N-terminus undercontrolled by P nis . The resulting plasmid was introduced into NZ9000 by electroporation, and the gene expression in cells and extracellular secretion of rIL6scFv were verified by SDS-PAGE (Fig. 2a, c) and western blotting with anti-His tag Ab (Fig. 2b, d). In SDS-PAGE analysis, it was confirmed that same amount of protein was loaded to each well (Fig. 2a, c). Western blotting detected two bands corresponding to the secretion precursor of rIL6scFv (34.0 kDa) and the secretion form of rIL6scFv (31.2 kDa) in cellular extracts of nisin-induced NZ-IL6scFv (Fig. 2b). It also showed that a single band corresponding to the secretion form of rIL6scFv was observed in the culture supernatant from nisin-induced NZ-IL6scFv (Fig. 2d). In contrast, considerable bands of rIL6scFv were not observed in both cellular extracts and culture supernatant from nisin-induced NZ-VC and non-induced two gmNZ9000 strains in western blotting (Fig. 2b, d).

Detection of rIL6scFv. Gene expression was induced in NZ-VC (VC) and NZ-IL6scFv (IL6scFv) with (+) or without (−) nisin. Cellular extracts (a, b) and culture supernatants (c, d) were analyzed with SDS-PAGE (a, c) or western blotting using an anti-His tag Ab (b, d). The black and pink arrows indicate the secretion precursor of rIL6scFv (34.0 kDa) and the secretion form of rIL6scFv (31.2 kDa), respectively. M molecular weight marker
Optimal conditions for rIL6scFv secretion in NZ-IL6scFv
To optimize the expression/secretion of rIL6scFv in NZ-IL6scFv, we tuned two parameters (i.e., nisin concentration and induction time) in the induction phase of gene expression. The turbidity of culture medium (OD600) at 3 h following the nisin induction was decreased in a nisin concentration-dependent manner (Fig. 3a). Particularly, cell growth was significantly inhibited in the presence of nisin more than 0.78 ng/mL as compared with a non-existent condition of nisin (0 ng/mL). In contrast, the western blot analysis showed that the expression/secretion level of rIL6scFv was gradually increased with rising in nisin concentration (Fig. 3b). However, the secretion precursor of rIL6scFv was observed in the supernatant from the culture induced with 1.56 ng/mL and over nisin (Fig. 3b). Next, we attempted optimization of the incubation time after nisin stimulation. An absorbance at OD600 in the culture increased in an induction time-dependent manner until 7 h and stabilized between 2.0 and 2.5 after 7-h incubation (Fig. 3c). The expression level of rIL6scFv per bacterial cell was high even when 1 h after nisin stimulation and was slightly decreased at 24 and 48 h (Cell in Fig. 3d). Secretion of rIL6scFv was elevated in a time-dependent manner (Sup in Fig. 3d). However, a clear band of the secretion precursor of rIL6scFv was detected in the supernatant from the culture induced for 12 h and more.

Optimization of the conditions for rIL6scFv secretion in NZ-IL6scFv. a, b NZ-IL6scFv was cultured with various concentrations of nisin (0 to 12.5 ng/mL). a After 3-h incubation at 30 °C, an OD600 in each culture was measured. *P < 0.05, **P < 0.01 vs. 0 ng/mL nisin. b Cellular extracts (Cell) and culture supernatants (Sup) were prepared from each culture and analyzed with western blotting using an anti-His tag Ab. c, d NZ-IL6scFv was cultured with 1 ng/mL nisin for 0 to 48 h. c An OD600 in the culture was measured at indicated time points. d Cellular extracts (Cell) and culture supernatants (Sup) were prepared from indicated time points and analyzed with western blotting using an anti-His tag Ab. a, c Data are the mean ± SD from three independent experiments. b, d The black and pink arrows indicate the secretion precursor of rIL6scFv (34.0 kDa) and the secretion form of rIL6scFv (31.2 kDa), respectively. Similar results were obtained from three independent experiments, and representative data are shown
Purification of rIL6scFv
The rIL6scFv was purified from cellular extracts of nisin-induced NZ-IL6scFv in a 4-L large-scale culture by immobilized metal ion affinity chromatography. The column-absorbed proteins were eluted using imidazole with the linear gradient condition, and the eluent was collected in three fractions (Fig. 4a). Western blotting showed that the rIL6scFv was detectable in all eluted fractions, and the highest amount of rIL6scFv was observed in F-2 (Fig. 4b). However, SDS-PAGE demonstrated that the F-1 and F-2 contained a considerable amount of irrelevant proteins (Fig. 4b); the F-3 was, therefore, used for the following experiment.

Purification of rIL6scFv. rIL6scFv was purified from cellular extracts prepared from nisin-induced NZ-IL6scFv by an immobilized metal ion affinity chromatography technique. a A chromatogram in the elution phase in the chromatography. The eluent was collected in three fractions (F-1 to F-3). b Fractions obtained from the chromatography were analyzed with SDS-PAGE (upper) and western blotting using an anti-His tag Ab (lower). The black and pink arrows indicate the secretion precursor of rIL6scFv (34.0 kDa) and the secretion form of rIL6scFv (31.2 kDa), respectively
Immunoreactivity of rIL6scFv
To examine whether rIL6scFv was biologically active, the immunoreactivity of rIL6scFv against rmIL-6 was assayed in an ELISA. As shown in Fig. 5a, in this assay, the purified recombinant proteins or cellular extracts from nisin-induced gmNZ9000 strains were reacted with the solid phase-immobilized rmIL-6, and the rmIL-6 binding protein was then detected with anti-His tag Ab. When the purified rIL6scFv was applied to this system, absorbance at 450 nm was increased in a protein concentration-dependent manner (Fig. 5b). On the other hand, when purified rGFP was applied, absorbance remained at baseline level for all examined rGFP concentrations (Fig. 5b). We further assessed the immunoreactivity of rIL6scFv against rmIL-6 using cellular extracts from nisin-induced gmNZ9000 strains. Using the ELISA method, we confirmed that the His-tagged peptides produced by both NZ-VC and NZ-IL6scFv were reactive with the anti-His tag Ab (data not shown). Absorbance at 450 nm was significantly higher when the NZ-IL6scFv extracts were applied to this ELISA system than when the NZ-VC extracts were applied (Fig. 5c). These results clearly demonstrated that the rIL6scFv produced by NZ-IL6scFv is immunoreactive.

Immunoreactivity assays of rIL6scFv. a A schematic representation of the assay based on an ELISA technique. A solid phase-immobilized rmIL-6 was reacted with the His-tagged proteins. The rmIL-6-binding proteins were then detected with an anti-His tag Ab. b Different concentrations of purified rIL6scFv (pink) and rGFP (green) were applied to the ELISA system to determine their rmIL-6 reactivity. c Cellular extracts from nisin-induced NZ-VC (VC) or NZ-IL6scFv (IL6scFv) were used as a source of His-tagged protein for the ELISA. **P < 0.01. Similar results were obtained from two or three independent experiments, and representative data are shown
Discussion
An scFv consists of the smallest paratope unit of an immunoglobulin molecule, in which the variable regions of heavy and light chains are joined together by a flexible peptide linker (Bird et al. 1988). This simple and small peptide allows production of fully functional antibodies in genetically modified bacteria. Particularly, gmLAB is an optimal platform in the productions of scFv for the purpose of the treatment of the disease. Kruger et al. (2002) reported that a construction of gmLAB (Lactobacillus zeae) produced scFv for streptococcal antigen I/II adhesin molecule of Streptococcus mutans. Interestingly, the oral delivery of scFv using gmLAB reduced caries in the rat model. Because the mucosa delivery of scFv using gmLAB does not require refinement and can evade a systemic side effect not to expect, it is therapeutic strategies for treatment of low cost. However, there are no reports of gmLAB-producing cytokine neutralizing scFv for the treatment of inflammatory disorders and autoimmune diseases. Therefore, in this study, we developed a gmNZ9000 strain that secretes rIL6scFv with nisin-controlled gene expression (NICE) system (Mierau and Kleerebezem 2005).
We constructed a secretion vector which has an expression cassette of a homologous signal peptide-IL6scFv fusion peptide undercontrolled by nisin A promoter. In western blotting, we demonstrated that a signal peptide-fused secretion precursor of rIL6scFv is intracellularly expressed in a gmNZ9000 strain transformed with the IL6scFv secretion vector and is subsequently secreted as a signal peptide-cleaved form to outer environment through a secretion machinery of the cell. To determine the optimal conditions for rIL6scFv expression/secretion, we examined the impact of nisin concentration and induction time with nisin on cell growth and production level of rIL6scFv. The expression/secretion level of rIL6scFv was increased in a nisin concentration-dependent manner. However, the secretion precursor of rIL6scFv was observed in culture supernatants from NZ-IL6scFv cultures stimulated with 1.56 ng/mL and over nisin. Given that the growth inhibition of NZ-IL6scFv was dependent on nisin concentration, it was suggested that the secretion precursor of rIL6scFv in the culture supernatants was passively leaked from the damaged cells by nisin. Next, we tuned the culturing time after nisin addition. The secretion levels in exponential phase (until 7 h) were increased in a time-dependent manner. In stationary/death phase (after 8 h), gradual decrease in intracellular expression of rIL6scFv and the leakage of the secretion precursor of rIL6scFv to outer environment were observed. Given comprehensive consideration to these results and discussions, we determined an optimal condition in the rIL6scFv expression/secretion as follows: nisin concentration around 1 ng/mL and induction time between 6 and 7 h. The intracellular expression and the secretion levels of rIL6scFv under optimal conditions were 322 and 7 ng/mL culture, respectively.
By reason that an scFv antibody is a small-size molecule, it has several advantages in clinical application including better tissue penetration, rapid blood clearance, and low immunogenicity as compared to a full-size antibody (Holliger and Hudson 2005). Therefore, an scFv with complete activity as an antibody is an attractive alternative to a full-size antibody in the clinical and diagnostic applications (Nelson 2010; Nelson and Reichert 2009). In this study, immunoreactivity of rIL6scFv was examined by ELISA. This assay clearly demonstrated that rIL6scFv produced by NZ-IL6scFv binds to rmIL-6. This fact gives promise that rIL6scFv and NZ-IL6scFv exert an anti-inflammatory property via neutralization of an rmIL-6 activity. Further studies including in vitro assays for determination of the inhibitory effect of rIL6scFv on the activation of IL-6/IL-6R signaling are needed to confirm this possibility.
IL-6/IL-6R signaling is considered to play an important role in the pathogenesis of chronic inflammatory and autoimmune diseases and of cancer in mucosal tissue (Hunter and Jones 2015; Rincon and Irvin 2012; Waldner and Neurath 2014). For example, in IBD including Crohn’s disease and ulcerative colitis, IL-6 has been associated with mucosal inflammation in the gastrointestinal tract that is mediated by various cellular mechanisms such as the regulation of T cell differentiation, activation, and proliferation (Hunter and Jones 2015; Waldner and Neurath 2014). A recent study in asthma patients demonstrated that IL-6 impaired lung function through exacerbation of mixed eosinophilic-neutrophilic bronchitis (Chu et al. 2015). In addition, IL-6-dependent activation of signal transducer and activator of transcription 3 (STAT3) promotes tumor cell proliferation and survival and has been implicated in tumorigenesis (Yu et al. 2009). Based on these findings, it has been suggested that IL-6/IL-6R signaling is a potential therapeutic target in those diseases. Indeed, several pre-clinical studies with model animals have demonstrated that systemic application of an antibody against IL-6 or the IL-6R could inhibit the development of inflammation and tumor in respiratory and gastrointestinal tracts (Chu et al. 2015; Song et al. 2014; Yamamoto et al. 2000). Therefore, direct delivery of rIL6scFv to the mucosa using NZ-IL6scFv might be a good strategy for the treatment of IBD, asthma, and cancer of the mucosal tissue, among other diseases. In vivo trials using model animals are needed to demonstrate the potential efficacy of NZ-IL6scFv for the prevention and therapy of those diseases.
In conclusion, we successfully developed a genetically modified strain of L. lactis NZ9000, NZ-IL6scFv, which secretes rIL6scFv with optimized culture conditions. Importantly, we clearly demonstrated that rIL6scFv from NZ-IL6scFv is immunoreactive since its ability to bind to rmIL-6. Therefore, NZ-IL6scFv may be an attractive tool for the research and development of new mucosal delivery vehicle of rIL6scFv- and IL-6-targeting agent for inflammatory and autoimmune diseases as well as cancer. This is the first report of gmLAB producing a functional anti-cytokine scFv. This finding becomes the base of low-cost therapeutic antibodies.
References
Ai C, Zhang Q, Ren C, Wang G, Liu X, Tian F, Zhao J, Zhang H, Chen YQ, Chen W (2014) Genetically engineered Lactococcus lactis protect against house dust mite allergy in a BALB/c mouse model. PLoS One 9(10):e109461. doi:10.1371/journal.pone.0109461
Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, Schutz M, Bartsch B, Holtmann M, Becker C, Strand D, Czaja J, Schlaak JF, Lehr HA, Autschbach F, Schurmann G, Nishimoto N, Yoshizaki K, Ito H, Kishimoto T, Galle PR, Rose-John S, Neurath MF (2000) Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in crohn disease and experimental colitis in vivo. Nat Med 6(5):583–588. doi:10.1038/75068
Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M (1988) Single-chain antigen-binding proteins. Science 242(4877):423–426
Braat H, Rottiers P, Hommes DW, Huyghebaert N, Remaut E, Remon JP, van Deventer SJ, Neirynck S, Peppelenbosch MP, Steidler L (2006) A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin Gastroenterol Hepatol 4(6):754–759. doi:10.1016/j.cgh.2006.03.028
Chu DK, Al-Garawi A, Llop-Guevara A, Pillai RA, Radford K, Shen P, Walker TD, Goncharova S, Calhoun WJ, Nair P, Jordana M (2015) Therapeutic potential of anti-IL-6 therapies for granulocytic airway inflammation in asthma. Allergy Asthma Clin Immunol 11(1):14. doi:10.1186/s13223-015-0081-1
De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, Cuttica R, Ravelli A, Schneider R, Woo P, Wouters C, Xavier R, Zemel L, Baildam E, Burgos-Vargas R, Dolezalova P, Garay SM, Merino R, Joos R, Grom A, Wulffraat N, Zuber Z, Zulian F, Lovell D, Martini A (2012) Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 367(25):2385–2395. doi:10.1056/NEJMoa1112802
de Ruyter PG, Kuipers OP, Beerthuyzen MM, van Alen-Boerrigter I, de Vos WM (1996) Functional analysis of promoters in the nisin gene cluster of Lactococcus lactis. J Bacteriol 178(12):3434–3439
Hanson ML, Hixon JA, Li W, Felber BK, Anver MR, Stewart CA, Janelsins BM, Datta SK, Shen W, McLean MH, Durum SK (2014) Oral delivery of IL-27 recombinant bacteria attenuates immune colitis in mice. Gastroenterology 146(1):210–221.e13. doi:10.1053/j.gastro.2013.09.060
Heinrich PC, Castell JV, Andus T (1990) Interleukin-6 and the acute phase response. Biochem J 265(3):621–636
Holliger P, Hudson PJ (2005) Engineered antibody fragments and the rise of single domains. Nat Biotechnol 23(9):1126–1136. doi:10.1038/nbt1142
Huibregtse IL, Snoeck V, de Creus A, Braat H, De Jong EC, Van Deventer SJ, Rottiers P (2007) Induction of ovalbumin-specific tolerance by oral administration of Lactococcus lactis secreting ovalbumin. Gastroenterology 133(2):517–528. doi:10.1053/j.gastro.2007.04.073
Hunter CA, Jones SA (2015) IL-6 as a keystone cytokine in health and disease. Nat Immunol 16(5):448–457. doi:10.1038/ni.3153
Illei GG, Shirota Y, Yarboro CH, Daruwalla J, Tackey E, Takada K, Fleisher T, Balow JE, Lipsky PE (2010) Tocilizumab in systemic lupus erythematosus: data on safety, preliminary efficacy, and impact on circulating plasma cells from an open-label phase I dosage-escalation study. Arthritis Rheum 62(2):542–552. doi:10.1002/art.27221
Ito H, Takazoe M, Fukuda Y, Hibi T, Kusugami K, Andoh A, Matsumoto T, Yamamura T, Azuma J, Nishimoto N, Yoshizaki K, Shimoyama T, Kishimoto T (2004) A pilot randomized trial of a human anti-interleukin-6 receptor monoclonal antibody in active Crohn’s disease. Gastroenterology 126(4):989–996 discussion 947
Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268(1):160–167
Katsume A, Saito H, Yamada Y, Yorozu K, Ueda O, Akamatsu K, Nishimoto N, Kishimoto T, Yoshizaki K, Ohsugi Y (2002) Anti-interleukin 6 (IL-6) receptor antibody suppresses Castleman’s disease like symptoms emerged in IL-6 transgenic mice. Cytokine 20(6):304–311
Kishimoto T (2005) Interleukin-6: from basic science to medicine—40 years in immunology. Annu Rev Microbiol 23:1–21
Kitaba S, Murota H, Terao M, Azukizawa H, Terabe F, Shima Y, Fujimoto M, Tanaka T, Naka T, Kishimoto T, Katayama I (2012) Blockade of interleukin-6 receptor alleviates disease in mouse model of scleroderma. Am J Pathol 180(1):165–176. doi:10.1016/j.ajpath.2011.09.013
Kruger C, Hu Y, Pan Q, Marcotte H, Hultberg A, Delwar D, van Dalen PJ, Pouwels PH, Leer RJ, Kelly CG, van Dollenweerd C, Ma JK, Hammarstrom L (2002) In situ delivery of passive immunity by lactobacilli producing single-chain antibodies. Nat Biotechnol 20(7):702–706. doi:10.1038/nbt0702-702
Kuipers OP, de Ruyter P, Kleerebezem M, de Vos WM (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J Biotechnol 64(1):15–21
Mierau I, Kleerebezem M (2005) 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl Microbiol Biotechnol 68(6):705–717. doi:10.1007/s00253-005-0107-6
Motta JP, Bermudez-Humaran LG, Deraison C, Martin L, Rolland C, Rousset P, Boue J, Dietrich G, Chapman K, Kharrat P, Vinel JP, Alric L, Mas E, Sallenave JM, Langella P, Vergnolle N (2012) Food-grade bacteria expressing elafin protect against inflammation and restore colon homeostasis. Sci Transl Med 4(158):158ra144. doi:10.1126/scitranslmed.3004212
Nelson AL (2010) Antibody fragments: hope and hype. MAbs 2(1):77–83
Nelson AL, Reichert JM (2009) Development trends for therapeutic antibody fragments. Nat Biotechnol 27(4):331–337. doi:10.1038/nbt0409-331
Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T (2005) Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 106(8):2627–2632. doi:10.1182/blood-2004-12-4602
Rezende RM, Oliveira RP, Medeiros SR, Gomes-Santos AC, Alves AC, Loli FG, Guimaraes MA, Amaral SS, da Cunha AP, Weiner HL, Azevedo V, Miyoshi A, Faria AM (2013) Hsp65-producing Lactococcus lactis prevents experimental autoimmune encephalomyelitis in mice by inducing CD4+LAP+ regulatory T cells. J Autoimmun 40:45–57. doi:10.1016/j.jaut.2012.07.012
Rincon M, Irvin CG (2012) Role of IL-6 in asthma and other inflammatory pulmonary diseases. Int J Biol Sci 8(9):1281–1290. doi:10.7150/ijbs.4874
Robert S, Gysemans C, Takiishi T, Korf H, Spagnuolo I, Sebastiani G, Van Huynegem K, Steidler L, Caluwaerts S, Demetter P, Wasserfall CH, Atkinson MA, Dotta F, Rottiers P, Van Belle TL, Mathieu C (2014) Oral delivery of glutamic acid decarboxylase (GAD)-65 and IL10 by Lactococcus lactis reverses diabetes in recent-onset NOD mice. Diabetes 63(8):2876–2887. doi:10.2337/db13-1236
Sambrook J, Russell D (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675
Shigemori S, Yonekura S, Sato T, Otani H, Shimosato T (2013) Expression of the immunoreactive buckwheat major allergenic storage protein in Lactococcus lactis. Appl Microbiol Biotechnol 97(8):3603–3611. doi:10.1007/s00253-012-4608-9
Shigemori S, Oshiro K, Wang P, Yamamoto Y, Wang Y, Sato T, Uyeno Y, Shimosato T (2014) Generation of dipeptidyl peptidase-IV-inhibiting peptides from β-lactoglobulin secreted by Lactococcus lactis. Biomed Res Int 2014:393598. doi:10.1155/2014/393598
Shigemori S, Watanabe T, Kudoh K, Ihara M, Nigar S, Yamamoto Y, Suda Y, Sato T, Kitazawa H, Shimosato T (2015) Oral delivery of Lactococcus lactis that secretes bioactive heme oxygenase-1 alleviates development of acute colitis in mice. Microb Cell Factories 14(1):189. doi:10.1186/s12934-015-0378-2
Shirota Y, Yarboro C, Fischer R, Pham TH, Lipsky P, Illei GG (2013) Impact of anti-interleukin-6 receptor blockade on circulating T and B cell subsets in patients with systemic lupus erythematosus. Ann Rheum Dis 72(1):118–128. doi:10.1136/annrheumdis-2012-201310
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B (2014) Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 73(9):1616–1625. doi:10.1136/annrheumdis-2013-205137
Song L, Smith MA, Doshi P, Sasser K, Fulp W, Altiok S, Haura EB (2014) Antitumor efficacy of the anti-interleukin-6 (IL-6) antibody siltuximab in mouse xenograft models of lung cancer. J Thorac Oncol 9(7):974–982. doi:10.1097/jto.0000000000000193
Szepietowski JC, Nilganuwong S, Wozniacka A, Kuhn A, Nyberg F, van Vollenhoven RF, Bengtsson AA, Reich A, de Vries DE, van Hartingsveldt B, Robinson DW Jr, Gordon R, Hsu B (2013) Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum 65(10):2661–2671. doi:10.1002/art.38091
Takiishi T, Korf H, Van Belle TL, Robert S, Grieco FA, Caluwaerts S, Galleri L, Spagnuolo I, Steidler L, Van Huynegem K, Demetter P, Wasserfall C, Atkinson MA, Dotta F, Rottiers P, Gysemans C, Mathieu C (2012) Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J Clin Invest 122(5):1717–1725. doi:10.1172/jci60530
Tanaka T, Kishimoto T (2011) Immunotherapy of tocilizumab for rheumatoid arthritis. J Clin Cell Immunol S6:001. doi:10.4172/2155-9899.S6-001
Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6(10):16. doi:10.1101/cshperspect.a016295
Villarejo MR, Zabin I (1974) β-galactosidase from termination and deletion mutant strains. J Bacteriol 120(1):466–474
Voller A, Bartlett A, Bidwell DE (1978) Enzyme immunoassays with special reference to ELISA techniques. J Clin Pathol 31(6):507–520
Waldner MJ, Neurath MF (2014) Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin Immunol 26(1):75–79. doi:10.1016/j.smim.2013.12.003
Wang Y, Yamamoto Y, Shigemori S, Watanabe T, Oshiro K, Wang X, Wang P, Sato T, Yonekura S, Tanaka S, Kitazawa H, Shimosato T (2015) Inhibitory/suppressive oligodeoxynucleotide nanocapsules as simple oral delivery devices for preventing atopic dermatitis in mice. Mol Ther 23(2):297–309. doi:10.1038/mt.2014.239
Yamamoto M, Yoshizaki K, Kishimoto T, Ito H (2000) IL-6 is required for the development of Th1 cell-mediated murine colitis. J Immunol 164(9):4878–4882
Yokota S, Imagawa T, Mori M, Miyamae T, Aihara Y, Takei S, Iwata N, Umebayashi H, Murata T, Miyoshi M, Tomiita M, Nishimoto N, Kishimoto T (2008) Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 371(9617):998–1006. doi:10.1016/s0140-6736(08)60454-7
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9(11):798–809. doi:10.1038/nrc2734
Acknowledgments
We are grateful to Yasuhiro Yasaki, Kohichi Sudo, and Fu Namai (Faculty of Agriculture, Shinshu University) for their excellent support in purification of recombinant proteins.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by a Grant-in-Aid for the Japan Society for the Promotion of Science Fellows (No. 14J06317) to SS and by a grant from Sumitomo Electric Industries Group CSR Foundation (No. 2012#7) to TSh.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Shigemori, S., Ihara, M., Sato, T. et al. Secretion of an immunoreactive single-chain variable fragment antibody against mouse interleukin 6 by Lactococcus lactis . Appl Microbiol Biotechnol 101, 341–349 (2017). https://doi.org/10.1007/s00253-016-7907-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7907-8