
Abstract
β-glucosidase (EC 3.2.1.21; BG) cleaves β-glucosidic linkages in disaccharide or glucose-substituted molecules. In an effort towards designing better BGs, we focused on the role of non-conserved residues across an otherwise homologous BG active site tunnel and designed mutants across the aglycone-binding site (V169C) and the gatekeeper residues (I246A) of the active site tunnel. We expressed in Escherichia coli, the Hore_15280 gene encoding a β-glucosidase (BG) in Halothermothrix orenii. The overexpressed and purified wild-type (B8CYA8) has a high specific activity of 345 μmol/min/mg on pNPGlc and a half-life of 1.13 h when assayed with pNPGlc at pH 7.1 and 70 °C. The specific activities of V169C and I246A were 1.7 and 1.2 times higher than that of wild-type (WT) enzyme with the model substrate pNPGlc, while the activity on the natural substrate cellobiose was slightly higher to the WT. The two mutants were kinetically stable with 4.4- to 11-fold longer half-life compared to the WT enzyme. When the two mutations were combined to generate the V169C/I246A mutant, the specific activity increased to nearly twofold higher than WT on both substrates and the half-life increased fivefold. The two single mutants also show enhanced saccharification of insoluble natural biomass on supplementation of Trichoderma viride cellulase cocktail. These enhanced properties suggest the need for a closer look at the active site tunnel of these enzymes, especially across residues that are not conserved towards improving catalytic efficiencies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellulose is the major component of plant cell walls, one of the most abundant polysaccharides found in nature and an inexhaustible source of bioenergy. The hydrolysis of pre-treated lignocellulosic biomass to produce sugars depends on a minimum of three enzymes, namely endoglucanases (EG), cellobiohydrolases (CBH), and β-glucosidase (BG), that work synergistically to degrade the cellulose (Chundawat et al. 2011; Woodward and Wiseman 1982). BGs (EC 3.2.1.21) degrade cello-oligosaccharides like cellobiose (Clb) to glucose (Datta 2016; Kuhad et al. 2011). β-glucosidase activity in the cellulase is very important because BG relieve product inhibition of CBH and EG by cellobiose (Sørensen et al. 2013).
Thermophilic cellulases have been suggested to have higher specific activities and higher stability (Sinha and Datta 2016; Yeoman et al. 2010). One such thermophile, Halothermothrix orenii is a moderate halophilic isolated from a Tunisian hypersaline lake and the first true halophilic thermophilic bacteria discovered (Cayol et al. 1994). H. orenii can use a large variety of sugar components as energy source and contains genes involved in the metabolism of cellobiose, glucose, xylose, and other sugars (Mavromatis et al. 2009) and therefore was a part of our program towards studying and improving extremophilic enzymes for biomass degradation. During the course of our study, the biochemical and structural characterization towards galacto-oligosaccharide synthesis by using the β-galactosidase (BGAL) activity of B8CYA8 was reported (Hassan et al. 2015, 2016). Subsequently, the same group engineered this enzyme to improve the lactose conversion and galacto-oligosaccharide production (Hassan et al. 2016). Our work is, however, focused on characterizing and improving the β-glucosidase activity of the enzyme.
The multiple sequence alignments across a large set of 72 thermophilic BGs selected across different clades of a phylogenetic analysis indicated that there were residues which were not conserved around the active site tunnel, in an otherwise homologous protein (Heins et al. 2014). This piqued our interest and we decided to initiate a study to understand the effect of these differences in the hope of improving the catalytic properties of this enzyme. The influence on enzyme activity by residues located far from the active site has been previously reported (Cao et al. 2015). We chose two such residues, Val 169 and Ile 246. Val 169 is located at the aglycone-binding site of the enzyme and Ile 246 is a residue at the entrance to the active site tunnel, henceforth to be referred to as a gatekeeper residue (Fig. 1). By mutating Val to a Cys, two hydrophobic residues are interchanged, without disturbing the overall hydrophobic nature of the active site tunnel. With the goal of decreasing steric clashes to allow more substrates into the active site, Ile 246 was substituted by Ala.

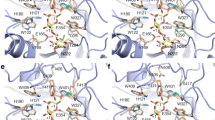
The important residues of the active site tunnel that play a role in catalysis. These residues are fall under three categories, from bottom of the tunnel to entry of the tunnel, glycone binding (W122, cyan); aglycone binding (V169, magenta); and gatekeeper regions (W168, E173, and I246, green). Catalytic acid/base residue and catalytic nucleophilic residue marked as yellow, glucose was shown in stick. Mutations have been created in one of the gate keeper residues, I246A and V169C at the aglycone region. This figure has been generated using Chimera 1.10.1 (Pettersen et al. 2004)
Here, we report the cloning, heterologous expression, and detailed enzymatic characterization of B8CYA8 (wild-type, WT) and its two mutants, V169C and I246A along with the double mutant, V169C/I246A. Detailed kinetic and thermostability studies across the wild-type and the three mutants, and the effect on real biomass help us in comparing the mutants with the wild-type. The results indicate the unique possibilities of further improvement and the use of variants of B8CYA8 as part of a thermophilic cellulase cocktail for saccharification of biomass.
Materials and methods
Chemicals
All chemicals used were reagent grade and bought from Sigma-Aldrich (St. Louis, USA). Restriction endonucleases, DNA ligase, and DNA polymerase were purchased from NEB (Ipswich, USA). Primers were synthesized by Eurofins (Bangalore, India). The active fractions post purification were pooled and concentrated using 30 kDa cutoff size Amicon-Ultra-15 membranes (EMD Millipore, Billerica, USA). Trichoderma viride cellulase (Cat # C9422) and Trametes versicolor laccase (Cat # 51639) was bought from Sigma-Aldrich (St. Louis, USA) and almond β-glucosidase (Cat # 82870) from SRL (Chennai, India).
Cloning and expression
The synthetic gene corresponding to the BG from H. orenii was constructed (Genbank ID KX808501) and assembled by Gene Art (Thermo Fisher Scientific, Waltham, USA). The gene was cloned into pBAD bacterial expression plasmid (Thermo Fisher Scientific, Waltham, USA) and the resulting plasmid named pSS1. The plasmid was transformed into the Escherichia coli Top 10F′ cells (Life Technologies, La Jolla, CA). An overnight culture of E. coli Top 10F′ (pSS1) in LB media containing ampicillin (100 μg/mL), was used to inoculate LB ampicillin media (typically 100 mL) and the bacterial culture was grown with constant shaking (225 rpm) at 37 °C. When the OD600 reached 0.5–0.6, L-arabinose was added to 0.002 % (w/v) and protein induction proceeded for 4.5 h. The cells were pelleted by centrifugation at 4000×g for 10 min at 4 °C and then stored at −20 °C until purification of the protein.
Mutants
The V169C and I246A mutations were introduced into pSS1 via a modified overlap extension PCR method. In brief, the two mutants V169C and I246A were generated via mega primer-based polymerase chain reaction (PCR) mutagenesis strategy (Brøns-Poulsen et al. 1998). Primers (V169C 5′-GACCTTCAAAAGCCACACACCACGGTTCGTTATG-3′, I246A 5′-CAGAAACCATGC ATTAGCGTAGTCATCCAGCAATG-3 and V169C/I246A were designed by using OligoAnalyzer (IDT Technology) and ApE (ApE Plasmid Editor, version 2.0.49 by M. Wayne Davis). The mutant genes were also cloned into pSS1 and transformed in E. coli expression strain TOP10F′. Each mutation was verified at the IISER Kolkata sequencing facility.
Protein purification
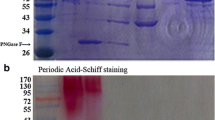
The protein was purified using a protocol similar to as detailed here (Goswami et al. 2016). The purity of B8CYA8 was confirmed by SDS–PAGE on a 10 % gel (Supplementary Fig. S1). The concentration of B8CYA8 was determined by measuring the absorbance at 280 nm and by calculating the extinction coefficient (ε280 = 106,230 M−1 s−1 with all free cysteines) using the modified Edelhoch and Gill/Von Hippel methods on Expasy (http://web.expasy.org/protparam/).
Enzyme activity assays
The specific activity of B8CYA8 was assayed at 70 °C in 100 mM HEPES buffer (pH 7.1) following a previously published protocol using substrate p-nitrophenyl-β-D-glucopyranoside (pNPGlc) and cellobiose (Clb), at saturating concentrations of each (Goswami et al. 2016). Clb hydrolysis produce two molecules of glucose and the calibration curve used was based on the glucose produced. The pH dependence of B8CYA8 was determined by measuring specific activities of the enzyme on p-nitrophenyl-D-glucopyranoside (pNPGlc) in the pH range of 4.0 to 10.0 at 70 °C after incubating the enzyme overnight at 4 °C in each buffer. The effect of temperature on enzyme activity for pNPGlc was measured from 25 to 80 °C while incubating in 100 mM HEPES buffer at pH 7.1. For the mutants, a similar protocol was followed except that the temperature dependence was measured using McIlvaine buffer, pH 6.5. The amount of enzyme to be used and the time of the assay were optimized for each enzyme based on the initial rate. Finally, specific activity was measured under the optimum conditions applicable to each enzyme.
Kinetic analysis
The kinetic parameters of B8CYA8 and the mutants were determined using pNPGlc and cellobiose as the substrates. The reaction conditions and the methods used to detect enzymatic activity were as described above. The reaction velocity was determined at different substrate concentrations from 0.1 up to 4 times K m for each substrate. The kinetic constants K m and k cat was calculated by a non-linear regression of the Michaelis-Menten equation using GraphPad PRISM version 5.0 (GraphPad Software, La Jolla, CA).
Thermostability assay
Enzymes were incubated in the appropriate buffer (100 mM HEPES buffer, pH 7.1 for wild-type enzyme and McIlvaine buffer at pHopt for each mutant) at 70 °C. At regular time intervals, aliquots were taken out, centrifuged, and assayed for residual activity. Half-life times were determined using the equation for a one-phase exponential decay in GraphPad PRISM. For assays in the presence of additives, the enzymes were first incubated with the additive for 30 min before the addition of substrate to start the assay. For each additive, blanks without enzyme were subtracted for any background absorbance.
Differential scanning fluorimetry: The experimental protocol and data transformation and analysis were performed using the DSF Analysis protocol essentially as previously described (Goswami et al. 2016; Niesen et al. 2007).
Insoluble substrate assay
Rice waste (husk) bought from rice processing mill, local market. The husk was steam pre-treated at 121 °C and 15 lb. pressure for 20 min followed by washing with filtered water and air dried. A slurry was made up of 5 % (w/v) dried rice husk measured in 100-mM sodium acetate buffer, pH 5 and all the reactions were carried out at 37 °C with different combinations of the following enzymes—T. viride cellulase (15 μg), T. versicolor laccase (15 μg), and β-glucosidase (1 μg). The hydrolysis reactions were carried out at pH 5, which is the pHopt of the commercial T. viride cellulase. A commercially available almond β-glucosidase (Sisco Research Laboratories, Madras, India) was used as a control for the recombinant wild-type and mutant enzymes. The reaction mixture was heat inactivated at 95 °C for 10 min, followed by measurement of the glucose generated with GOD-POD assay kit (Sigma-Aldrich, St. Louis, USA).
Results
Cloning, expression, and purification of β-glucosidase from H. orenii
The 1353-bp gene encoding the β-glucosidase from H. orenii was cloned into a pBAD vector, (plasmid named as pSS1) and expressed in E. coli. The enzyme was purified from crude extract obtained from harvested cells as a soluble protein via His-Trap affinity chromatography. The purification yielded about 75 mg of pure protein from a 1000-mL shake flask culture. Protein obtained at each step was analyzed by SDS-PAGE, and the final purified enzyme showed a single band with an apparent molecular mass of 53 kDa (Supplementary Fig. S1). This molecular mass agrees well with the predicted size of B8CYA8. When overloaded, no other bands were seen (data not shown) indicating the absence of any major impurity. The expression levels of the mutants were similar as the wild-type.
Characterization of B8CYA8 and mutants
B8CYA8 is active within the pH range of 5.0–7.5, retaining more than 85 % of its specific activity, when activity was determined after 24-h incubation at 4 °C. The WT enzyme and the mutants is most active (pHopt) at pH 7.1 and 6.5, respectively (Table 1). To find the temperature optimum (T opt) at this pH, the enzyme activity assays were performed between 30 to 80 °C with 20 mM pNPGlc. The T opt was found to be 70 °C for the wild-type and 72 °C for the single mutants (Table 1). The purified wild-type had a specific activity of 345 ± 25 μmol min−1 mg−1 on pNPGlc. The V169C shows a 1.7-fold increase in specific activity to 588 ± 44 μmol min−1 mg−1. I246A with a specific activity of 424.3 ± 15 μmol min−1 mg−1 is 1.23-fold more active than the WT. The two mutations combined into a double mutant show an even higher specific activity at 653 ± 22 μmol min−1 mg−1. Similar increases were observed with the natural substrate cellobiose with the specific activity higher than on pNPGlc in case of all mutants other than in V169C. The double mutant shows the highest activity at 712 ± 40 μmol min−1 mg−1 on Clb, which is highest among either the WT or the single mutants, against either of the two substrates.
The steady-state kinetic parameters of B8CYA8 were measured for pNPGlc and the natural substrate cellobiose and fit to the Michaelis-Menten equation by a simultaneous non-linear regression analysis fit, after an initial verification by Lineweaver-Burk analysis (Fig. 2). On pNPGlc, both I246A and V169C has an improved turnover number compared to the WT, and the combined double mutant shows even higher k cat (692 s−1) compared to the WT (339 s−1). Most BGs reported in literature exhibit a higher activity over pNPGlc even if higher activity on the real substrate cellobiose is what is most desired (Bhatia et al. 2002). However, in the case of B8CYA8, both WT and I246A shows higher turnover number on cellobiose compared to pNPGlc, while in V169C the turnover number for Clb is slightly lower than on pNPGlc. When the two mutations are combined, the turnover number on cellobiose is the highest (887 s−1) and increases 1.8-fold compared to WT (494 s−1). Very high k cat/K m has been reported to be one of the primary kinetic parameter for selecting the best BGs (Teugjas and Väljamäe 2013). The V169C/I246A double mutant is slightly more specific to cellobiose compared to the WT as seen by the higher k cat/K m (Table 2).

Dependence of the rate of B8CYA8 WT (filled circles), V169C (filled squares), I246A (filled triangles), and V169C/I246A (filled inverted triangles) β-glucosidase-catalyzed hydrolysis of pNPGlc and cellobiose, determined by visible spectrophotometry. The solid line indicates the best fit of the Michaelis-Menten equation. The kinetic parameters are listed in Table 2
Thermostability
To test the robustness of the enzyme, we measured the effect of different solvents on the enzyme specific activity (Supplementary Table S1). B9CYA8 was particularly tolerant to solvents. Thus in the presence of 5 % ethanol and 5 % methanol, the relative enzyme activity compared to activity measured in the absence of any alcohol is 97.8 and 88.3 %, respectively. The three mutants show higher relative activity compared to the activity in the absence of solvents. A similar stability is observed in the presence of 5 % DMSO wherein DMSO has no effect on enzyme stability. Ionic liquids (IL) are another common class of compounds that have been known to inactivate cellulase (Datta et al. 2010; Turner et al. 2003). Recently, it has been shown that highly thermostable cellulases are also tolerant to IL (Datta et al. 2010; Ferdjani et al. 2011). When we tested IL tolerance on two common ILs, [C2C1im][MeSO3] and [C2C1im][C1C1PO4] at a high concentration of 20 % (v/v), the WT enzyme showed a relative specific activity of 81.2 and 55.4 %, respectively, compared to specific activity without IL. The two mutants also showed similar tolerance to [C2C1im][MeSO3]. However, in the case of [C2C1im][C1C1PO4], the V169C/I246A double mutant shows 81 % activity compared to 60 % of the single mutants.
To measure the stability of these BGs at room temperature, an aliquot of the WT and mutants were kept at room temperature on the laboratory bench and the specific activity was measured at the T opt of the enzymes, by taking out aliquots at regular intervals. There was no change in specific activity up to 32 days, which was the time till which the measurements were made. The t 1/2 of these enzymes was measured at 70 °C. Half-life values for the wild-type and three mutants are listed in Table 3. V169C shows an enhanced half-life of 11.6 h at 70 °C while the I246A has a half-life of 5 h, with the V169C being more stable than the I246A, compared to the WT. The double mutant has a half-life of 5.8 h, with a more than fivefold increase compared to the WT. For the WT enzyme, the half-life hugely increases from 1.13 to 30.8 h with a 5 °C drop in the incubation temperature of the enzyme (data not shown). Thus there seems to be advantage in using this enzyme at a temperature slightly lower than the kinetic T opt without any penalty in terms of reduction of specific activity.
The mid-point of the melting curve from a differential scanning fluorescence (DSF) experiment is the temperature at which 50 % of the protein has denatured upon heating (T m) and is a measure of the protein’s inherent thermal stability. ΔT m of V169C, I246A, and V169C/I246A was measured and indicates a gain in stability by around 0.9 and 0.3 °C in V169C and V169C/I246A, respectively (Table 1). The increase in the melting temperature of the mutants compared to the WT indicates a gain in thermodynamic stability by these two mutants.
Biomass saccharification
To verify enzyme activity on real biomass saccharification, we chose the abundantly available rice husk. Rice husk was hydrothermally pre-treated for reducing the cellulose crystallinity. After drying the rice husk, hydrolysis reactions were carried out in the presence of commercial laccase and cellulase and also supplemented, and compared with, commercially available sweet almond BG. Laccase was also added for further de-lignifications of the rice husk. At pH 5 (pHopt for the commercial cellulase), both the WT enzyme and the mutants retain between 60 and 65 % of specific activity of respective enzymes (data not shown). The choice of temperature, 37 °C, was also dictated by the optimum temperature of the T. viride cellulase cocktail and clearly not at the optimum temperature of the BGs which display lower activity at 37 °C. As observed from Table 4, the mutant I246A has generated the highest amount glucose (2.57 ± 0.04 mM) as compared to wild-type (2.30 ± 0.12 mM), and mutant V169C (2.42 ± 0.09 mM) of glucose, respectively. When laccase was present in the reaction mixture, a slight increase of around 0.3 to 0.5 mM glucose generated was measured across the WT or I246A and V169C mutants. The pre-treated biomass contains lignin and therefore the increase was probably due to the laccase degradation of the lignin and aiding in the cellulase hydrolysis of the cellulose.
Discussions
Many examples of semi-rational based protein engineering, particularly in improving protein stability, based on the use of evolutionary information from homologous protein sequences through multiple sequence alignments (MSAs) and phylogenetic analyses have been previously reported in the literature (Lutz 2010). Based upon information gathered from protein structure, function, and sequence homology, we decided to preselect two sites for focused mutagenesis with limited amino acid diversity. These two mutants were selected based on the multiple sequence alignment with thermophilic BG sequences (shown in the alignment in Supplementary Fig. S2). These sequences were a subset of a previously reported phylogenetic analysis with care taken to select sequences across each clade to avoid bias from over-representation of similar sequences (Heins et al. 2014). We decided to pay attention to non-conserved residues near to the active site tunnel in order to understand their role in the hydrolysis as well as thermal stability of BGs and possible improvement (Fig. 1). Note that in spite of large sequence similarities across BGs, these enzymes exhibit wide variations in enzyme activity and stability, even across the smaller subset of thermophilic BGs. In general, the glycone binding site of active site tunnel is far more conserved across BGs than the aglycone binding site or the gate keeper regions at the entrance of the active site tunnel of BGs (de Giuseppe et al. 2014). The MSA showed that among others, aglycone binding residue V169 and gate keeper residue I246 is not conserved (Supplementary Fig. S2A, B). Hence, these two residues were chosen for the initial study—Val169 in aglycone binding site inside the active site tunnel, and gatekeeper residue I246, found at the entrance to the tunnel. Therefore the V169C and I246A mutants were constructed. We chose the thermophilic BG from H. orenii as our enzyme of study and report here the molecular cloning of the gene in E. coli, and the functional characterization of the product of the overexpressed B8CYA8 BG as well as the three mutants generated.
The WT and the mutants are tolerant to a wide range of pH and temperature and show high activity and thermal stability. The two to threefold gain in turnover numbers by the three mutants are significant improvements over the WT enzyme. The numbers are particularly high in the context of BG’s from thermophilic organisms. Thermoanaerobacterium thermosaccharolyticum BG with a T opt at 70 °C has the k cat of 104 s−1 on pNPGlc (Teugjas and Väljamäe 2013). The higher activity BGs reported in the literature are mostly from fungal origin. For example, Aspergillus niger BG has a turnover number of 2780 s−1 on cellobiose and 917 s−1 on pNPGlc at 40 °C which means that there is a lot of scope in improving the specific activity of these thermophilic BGs to be considered as a replacement of fungal BGs in larger-scale saccharifications (Yan et al. 1998).
B8CYA8 (WT) has typical (β/α)8 TIM-barrel fold adopted by retaining GH1 enzymes (Hassan et al. 2015). Based on the crystal structure, the active site appears at the end of a thin narrow tunnel at the bottom of which, the catalytic acid/base residue E166 and residue E354 acting as a nucleophile is located. When catalysis commences, the non-reducing end of the substrate is positioned at the bottom of the tunnel to maximize the interaction of hydroxyl groups of the non-reducing end with respective residues of the enzyme. BGs removes glycone moiety, i.e., glucose with hydrolysis of water from the bottom (Hrmova et al. 1998). We speculate that these steps are tightly regulated in enzymes like B8CYA8 and the mutants to achieve the very high turnover numbers. One of the primary requirements for such high turnover numbers is the absence of stable polar interaction across the active site tunnel such that the oxocarbonium ion-like transition step would be short lived. The absence of the polar interaction facilitates the spontaneous hydrolysis step, along with the lack of access of the product glucose to the catalytic residues to prevent inhibition of catalysis (Hassan et al. 2015). Most of the amino acid residues of B8CYA8 present across the glycone and aglycone binding sites and the gate keeper residues are highly hydrophobic. Some of these include W122, W409, and W401 at the −1 subsite, V169, W327 at +1 subsite, and W168 and I246 at +2 subsite. Some of these important residues are shown in Fig. 3a. From our crystal structure analysis, it is conjectured that there are similarly fewer polar contacts between water and the mutated residues. Only I246A has polar contact with water at the +2 subsite though there is a similar interaction in the WT enzyme too (Fig. 3b). In the −1 subsite, V169C does not have any kind of interaction with water either in WT as valine or as a cysteine in the mutant. Absence of stable, polar contacts as revealed from the glucose complexed structure might also explain the apolar environment across the active site tunnel which could promote the high rate of catalysis observed here (de Giuseppe et al. 2014). Also, the side chain of cysteine occupies lesser volume than that of valine and might allow greater access to substrates at this site. For the I246A mutant, the sterics probably play a similar role. The mutation of isoleucine to alanine in the case of I246A, reduces the side chain volume by almost 35 %, allowing more substrate to enter into the active site (Lee et al. 2012). The absence of substrate inhibition might also indicate the presence of a secondary binding site that binds substrate or product molecules, resulting in enhanced catalysis (Souza et al. 2013; Zanoelo et al. 2004).

Figure showing interaction of a few of the residues mentioned in Fig. 1 along with catalytic residues, glucose and water molecules (shown in balls and sticks) for wild-type (a) and in the mutant (b) showing both the mutations, the mutants were generated in silico using Pymol (The PyMOL Molecular Graphics System, Version 1.8 Schrodinger LLC. https://www.pymol.org/). a V169, one of the selected residue side chain is shown in green backbone. Very few water molecules are present at the site of catalysis. Aglycone binding residue W121 has one polar contact with water (2.8 Å) and gatekeeper residue I246 has one polar contact with water at same distance. b V169 is mutated to C169 containing a sulfhydryl group. The gatekeeper residue I246 is shown as mutated to I246A. Both mutants are colored in gray. Polar contacts remained at similar distances as in wild-type (2.8 Å). The coordinates for these figures were taken from PDB file 4PTX (Hassan et al. 2015)
One of the important conditions of efficient lignocellulosic biomass saccharification is the pre-treatment of biomass to reduce the crystallinity of cellulose and allow cellulase to access the cellulose. While we have used hydrothermal pre-treatment of rice husk followed by enzyme hydrolysis to obtain satisfactory sugar yields in comparison of commercial cellulases (vide infra), other methods like ionic liquid pre-treatment are important new and green methods to consider (Sheldon et al. 2002). ILs are green solvents typically consisting of an organic cation and either an organic or inorganic anion, and hold great promise for biomass pre-treatment (Swatloski et al. 2002). Most cellulases, and in particular β-glucosidases reported thus far are inactive in the presence of ionic liquids and (Heins et al. 2014). Recently, high thermostability of glycosidases have been correlated to biocatalytic reaction efficiency in water-miscible ILs (Ferdjani et al. 2011). We tested two ionic liquids of same cation but different anions, [C2C1im][MeSO3] (1-ethyl-3-methylimidazolium methane sulfonate) and [C2C1im][C1C1PO4] (1-ethyl-3-methylimidazolium dimethyl phosphate). The high relative specific enzyme activity indicates high tolerance on these two ionic liquids. One operational advantage of a thermostable β-glucosidase is the possibility of reducing contamination and improving the quality and yield (Liu et al. 2011). Both the WT and the two mutants have excellent stability against thermal denaturation. The gain of half-life at 70 °C for both mutants are more than twofold, with the V169C being more stable than I246A mutant. The high catalytic activity and stability at higher temperature are indeed important properties for enzymatic saccharification of biomass (Yeoman et al. 2010).
In biorefinery systems, agricultural crops or wood are utilized for direct bioconversion into chemicals, fuels, or other useful products. Rice husk is one of the most widely available agricultural wastes in many rice producing countries around the world including India. Globally, the total annual production of rice husk is 120 million tons (Gidde and Jivani 2007) and an important biomass for biofuel production. In the experiments reported here, the large increase in glucose production observed on cellobiose substrate was not observed with the biomass but it is important to note that the reactions were carried out at the T. viride cellulase optimal at pH 5 and at 37 °C, much lower than the pHopt and T opt of H. orenii BG. The comparison with the commercially available sweet almond BG data shows that even in this initial proof of concept results obtained under non-optimum conditions, our set of recombinant BGs work better and are active in a mix of insoluble substrate. More detailed and longer-term studies are needed to lineate the possible difference in behavior between the WT and the mutants on insoluble biomass using a thermophilic cocktail.
In conclusion, H. orenii encodes a thermophilic BG which is catalytically very active. Based on the sequence analysis of thermophilic BG’s three mutants, V169C and I246A, and a combined double mutant V169C/I246A were designed. The mutant variants exhibit increased specific activity on the natural substrate cellobiose. The improved variants have a longer half-life and are more thermostable. Both WT and the mutants show good synergism with the T. viride cellulase in degrading rice husk to glucose demonstrating catalysis as part of a cellulase cocktail. Our results indicate that the degree of conservation or diversity across residues around the active site, are important to understand and such studies might offer a simpler way towards enhancing the activities of these enzymes.
References
Bhatia Y, Mishra S, Bisaria VS (2002) Microbial β-glucosidases: cloning, properties, and applications. Crit Rev Biotechnol 22(4):375–407. doi:10.1080/07388550290789568
Brøns-Poulsen J, Petersen NE, Horder M, Kristiansen K (1998) An improved PCR-based method for site directed mutagenesis using megaprimers. Mol Cell Probes 12(6):345–348. doi:10.1006/mcpr.1998.0187
Cao L-C, Wang Z-J, Ren G-H, Kong W, Li L, Xie W, Liu Y-H (2015) Engineering a novel glucose-tolerant β-glucosidase as supplementation to enhance the hydrolysis of sugarcane bagasse at high glucose concentration. Biotechnol Biofuel 8:202. doi:10.1186/s13068-015-0383-z
Cayol JL, Ollivier B, Patel BK, Prensier G, Guezennec J, Garcia JL (1994) Isolation and characterization of Halothermothrix orenii gen. nov., sp. nov., a halophilic, thermophilic, fermentative, strictly anaerobic bacterium. Int J Syst Bacteriol 44(3):534–540. doi:10.1099/00207713-44-3-534
Chundawat SP, Beckham GT, Himmel ME, Dale BE (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu Rev Chem Biomol Eng 2:121–145. doi:10.1146/annurev-chembioeng-061010-114205
Datta S (2016) Recent strategies to overexpress and engineer cellulases for biomass degradation. Curr Metabol 4(1):14–22. doi:10.2174/2213235X03666150702155845
Datta S, Holmes B, Park JI, Chen Z, Dibble DC, Hadi M, Blanch HW, Simmons BA, Sapra R (2010) Ionic liquid tolerant hyperthermophilic cellulases for biomass pretreatment and hydrolysis. Green Chem 12(2):338–345. doi:10.1039/b916564a
de Giuseppe PO, Souza Tde A, Souza FH, Zanphorlin LM, Machado CB, Ward RJ, Jorge JA, Furriel Rdos P, Murakami MT (2014) Structural basis for glucose tolerance in GH1 β-glucosidases. Acta Crystallogr D Biol Crystallogr D70:1631–1639. doi:10.1107/S1399004714006920
Ferdjani S, Ionita M, Roy B, Dion M, Djeghaba Z, Rabiller C, Tellier C (2011) Correlation between thermostability and stability of glycosidases in ionic liquid. Biotechnol Lett 33(6):1215–1219. doi:10.1007/s10529-011-0560-5
Gidde MR, Jivani AP (2007) Waste to wealth- potential of rice husk in India—a literature review. In: Gidde M (ed) Proceedings of the international conference on cleaner technologies and environmental management. India, Allied Publishers Private Limited, Mumbai, Pondicherry, pp. 586–590
Goswami S, Gupta N, Datta S (2016) Using the β-glucosidase catalyzed reaction product glucose to improve the ionic liquid tolerance of β-glucosidases. Biotechnol Biofuels 9:72. doi:10.1186/s13068-016-0484-3
Hassan N, Nguyen TH, Intanon M, Kori LD, Patel BK, Haltrich D, Divne C, Tan TC (2015) Biochemical and structural characterization of a thermostable β-glucosidase from Halothermothrix orenii for galacto-oligosaccharide synthesis. Appl Microbiol Biotechnol 99(4):1731–1744. doi:10.1007/s00253-014-6015-x
Hassan N, Geiger B, Gandini R, Patel BK, Kittl R, Haltrich D, Nguyen TH, Divne C, Tan TC (2016) Engineering a thermostable Halothermothrix orenii β-glucosidase for improved galacto-oligosaccharide synthesis. Appl Microbiol Biotechnol 100(8):3533–3543. doi:10.1007/s00253-015-7118-8
Heins RA, Cheng X, Nath S, Deng K, Bowen BP, Chivian DC, Datta S, Friedland GD, D’Haeseleer P, Wu D, Tran-Gyamfi M, Scullin CS, Singh S, Shi W, Hamilton MG, Bendall ML, Sczyrba A, Thompson J, Feldman T, Guenther JM, Gladden JM, Cheng J-F, Adams PD, Rubin EM, Simmons BA, Sale KL, Northen TR, Deutsch S (2014) Phylogenomically guided identification of industrially relevant GH1 β-glucosidases through DNA synthesis and nanostructure-initiator mass spectrometry. ACS Chem Biol 9(9):2082–2091. doi:10.1021/cb500244v
Hrmova M, MacGregor EA, Biely P, Stewart RJ, Fincher GB (1998) Substrate binding and catalytic mechanism of a barley β-D-glucosidase/(1,4)-β-D-glucan exohydrolase. J Biol Chem 273(18):11134–11143. doi:10.1074/jbc.273.18.11134
Kuhad RC, Gupta R, Singh A (2011) Microbial cellulases and their industrial applications. Enzyme Res 2011:280696. doi:10.4061/2011/280696
Lee HL, Chang CK, Jeng WY, Wang AH, Liang PH (2012) Mutations in the substrate entrance region of β-glucosidase from Trichoderma reesei improve enzyme activity and thermostability. Protein Eng Des Sel 25(11):733–740. doi:10.1093/protein/gzs073
Liu D, Zhang R, Yang X, Xu Y, Tang Z, Tian W, Shen Q (2011) Expression, purification and characterization of two thermostable endoglucanases cloned from a lignocellulosic decomposing fungi Aspergillus fumigatus Z5 isolated from compost. Protein Expr Purif 79(2):176–186. doi:10.1016/j.pep.2011.06.008
Lutz S (2010) Beyond directed evolution—semi-rational protein engineering and design. Curr Opin Biotechnol 21(6):734–743. doi:10.1016/j.copbio.2010.08.011
Mavromatis K, Ivanova N, Anderson I, Lykidis A, Hooper SD, Sun H, Kunin V, Lapidus A, Hugenholtz P, Patel B, Kyrpides NC (2009) Genome analysis of the anaerobic thermohalophilic bacterium Halothermothrix orenii. PLoS One 4(1):e4192. doi:10.1371/journal.pone.0004192
Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2(9):2212–2221. doi:10.1038/nprot.2007.321
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612. doi:10.1002/jcc.20084
Sheldon RA, Lau RM, Sorgedrager MJ, van Rantwijk F, Seddon KR (2002) Biocatalysis in ionic liquids. Green Chem 4(2):147–151. doi:10.1039/B110008B
Sinha SK, Datta S (2016) β-Glucosidase from the hyperthermophilic archaeon Thermococcus sp. is a salt-tolerant enzyme that is stabilized by its reaction product glucose. Appl Microbiol Biotechnol 100(19):8399–8409. doi:10.1007/s00253–016-7601-x
Sørensen A, Lübeck M, Lübeck PS, Ahring BK (2013) Fungal β-glucosidases: a bottleneck in industrial use of lignocellulosic materials. Biomolecules 3(3):612–631. doi:10.3390/biom3030612
Souza FHM, Inocentes RF, Ward RJ, Jorge JA, Furriel RPM (2013) Glucose and xylose stimulation of a β-glucosidase from the thermophilic fungus Humicola insolens: a kinetic and biophysical study. J Mol Cat B: Enzym 94:119–128. doi:10.1016/j.molcatb.2013.05.012
Swatloski RP, Spear SK, Holbrey JD, Rogers RD (2002) Dissolution of cellulose with ionic liquids. J Am Chem Soc 124(18):4974–4975. doi:10.1021/ja025790m
Teugjas H, Väljamäe P (2013) Selecting β-glucosidases to support cellulases in cellulose saccharification. Biotechnol Biofuels 6(1):105. doi:10.1186/1754-6834-6-105
Turner MB, Spear SK, Huddleston JG, Holbrey JD, Rogers RD (2003) Ionic liquid salt-induced inactivation and unfolding of cellulase from Trichoderma reesei. Green Chem 5(4):443–447. doi:10.1039/b302570e
Woodward J, Wiseman A (1982) Fungal and other β-glucosidases—their properties and applications. Enzyme Microbial Technol 4(2):73–79. doi:10.1016/0141-0229(82)90084-9
Yan T-R, Lin Y-H, Lin C-L (1998) Purification and characterization of an extracellular β-glucosidase II with high hydrolysis and transglucosylation activities from Aspergillus niger. J Agric Food Chem 46(2):431–437. doi:10.1021/jf9702499
Yeoman CJ, Han Y, Dodd D, Schroeder CM, Mackie RI, Cann IK (2010) Thermostable enzymes as biocatalysts in the biofuel industry. Adv Appl Microbiol 70:1–55. doi:10.1016/S0065-2164(10)70001-0
Zanoelo FF, Polizeli Mde L, Terenzi HF, Jorge JA (2004) β-glucosidase activity from the thermophilic fungus Scytalidium thermophilum is stimulated by glucose and xylose. FEMS Microbiol Lett 240(2):137–143. doi:10.1016/j.femsle.2004.09.021
Acknowledgments
This work was supported in part by Rapid Grant for Young Investigators, Department of Biotechnology, Government of India, BT/PR6511/GBD/27/424/2012 (S.D.), Energy Bioscience Overseas Fellowship, Department of Biotechnology, Government of India, BT/NBDB/22/06/2011 (SD) and IISER Kolkata. SKS is supported by a Junior Research Fellowship from CSIR, Govt. of India, SG is supported by a Junior Research Fellowship from IISER Kolkata and SDas by an Inspire Fellowship, DST, Govt. of India. We thank Ms. Baishali Roy and Ms. Neha Gupta for their initial help in the cloning and expression studies. The comments of the editor and the anonymous reviewers are gratefully acknowledged.
Authors’ contributions
SD, SG, and SDas designed the research. SG, SKS, and SDas performed the experiments. SG and SD wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors
Additional information
Sushant K. Sinha and Shubhasish Goswami contributed equally to this manuscript and are co-first authors.
Electronic supplementary material
ESM. 1
(PDF 4981 kb)
Rights and permissions
About this article

Cite this article
Sinha, S.K., Goswami, S., Das, S. et al. Exploiting non-conserved residues to improve activity and stability of Halothermothrix orenii β-glucosidase. Appl Microbiol Biotechnol 101, 1455–1463 (2017). https://doi.org/10.1007/s00253-016-7904-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-016-7904-y