
Abstract
The pure stereoisomers of 1,2-propanediol (1,2-PDO) could be used as starting materials to synthesize high value-added specialty chemicals and chiral pharmaceutical products. As the stereoisomers of 1,2-PDO cannot be obtained by traditional chemical synthesis processes, biotechnological processes have gained increasing attention. However, to our knowledge, the production of S-1,2-PDO directly from glucose has not been previously reported. In this study, we demonstrate a novel artificial pathway to convert l-lactic acid to S-1,2-PDO and its integration into the genome of Escherichia coli strain BW25113∆poxB with synchronous deletion of genes responsible for branch metabolic pathways from glucose. l-lactate production was increased by replacing the native d-lactate dehydrogenase with the l-lactate dehydrogenase from Bacillus coagulans. The methylglyoxal bypass pathway was blocked to avoid synthesis of a racemic mixture of d- and l-lactate and prevent the accumulation of methylglyoxal, a toxic intermediate. To further improve the yield of S-1,2-PDO, a novel cofactor regeneration system was introduced by combining pyruvate decarboxylase and acetaldehyde-CoA dehydrogenase II to simultaneously regenerate NADH and the CoA donor of acetyl-CoA for the lactate conversion pathway. Finally, 13.7 mM S-1,2-PDO with >99 % enantiomeric purity was directly produced from glucose by disrupting the major carbon-competing pathways and strengthening the lactate transformation pathway. This study demonstrates the first attempt to synthesize S-1,2-PDO by direct fermentation of glucose.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
1,2-Propanediol (1,2-PDO) is a three-carbon diol with a stereogenic center at the central carbon atom, which exists in two forms: R-1,2-PDO and S-1,2-PDO. Racemic 1,2-PDO is a commodity chemical with a global demand of around 1.36 million tons/year (Shelley 2007). The major uses of racemic 1,2-PDO are in unsaturated polyester resins, liquid laundry detergents, pharmaceuticals, cosmetics, antifreeze, and deicers (Cameron et al. 1998). The pure stereoisomers of 1,2-PDO have numerous potential uses as chiral synthons in organic synthesis (Simon et al. 1987), specifically for the synthesis of specialty chemicals such as optically active propylene oxide and polymers, and they may be useful in the manufacture of chiral pharmaceutical products. However, their use is limited because of the high price and availability only in laboratory-scale quantities (Cameron et al. 1998).
The current commercial route to 1,2-PDO is via chemical methods from hydration of propylene or via high-pressure and high-temperature hydrogenolysis of sugars; however, these methods are expensive, environmentally harmful, and result in a racemic mixture (Altaras and Cameron 1999). With the ongoing world population increase and the relatively limited non-renewable resources, development of environmentally green processes for chemical production is of increasing importance (Jain and Yan 2011; Sun et al. 2015). In recent years, some attractive biological processes have been developed for commercial production of high-quality 1,2-PDO from renewable resources at low cost. To this end, anaerobic and facultative anaerobic microorganisms are interesting entities, as 1,2-PDO is the major product of their metabolism (Herrera 2004). More importantly, the pure stereoisomers of 1,2-PDO could be obtained by fermentation processes.
Many microorganisms can ferment sugars to 1,2-PDO through two main metabolic pathways. The first fermentation route to 1,2-PDO involves glycolytic metabolism of common sugars such as glucose and xylose. The glycolytic intermediate dihydroxyacetone phosphate (DHAP) is dephosphorylated to methylglyoxal, reduced to R-lactaldehyde and hydroxyacetone, and further reduced to R-1,2-PDO (Bennett and San 2001). The second direct fermentation route to 1,2-PDO from deoxy sugars such as l-rhamnose and l-fucose has been well characterized (Boronat and Aguilar 1981). This pathway involves the cleavage of the 6-deoxyhexose into DHAP and S-lactaldehyde, which is then reduced to S-1,2-PDO. l-fucose and l-rhamnose are metabolized through parallel pathways in organisms like Escherichia coli (Hacking and Lin 1976; Boronat and Aguilar 1981), Bacteroides ruminicola (Turner and Roberton 1979), Bacillus macerans (Weimer 1984), Salmonella typhimurium (Badía et al. 1985), and various yeasts (Suzuki and Onishi 1968). However, this fermentation process is not commercially feasible due to the high cost of l-fucose and l-rhamnose, as even the less expensive l-rhamnose sells for over $300/kg (Altaras and Cameron 1999).
Due to the limited availability of deoxyhexoses and insufficient knowledge, current studies exclusively focus on the methylglyoxal pathway to produce R-1,2-PDO, mainly by Clostridia strains (Cameron and Cooney 1986; Sanchez-Rivera et al. 1987) or recombinant E. coli strains (Altaras and Cameron 2000; Berríos-Rivera et al. 2003; Clomburg and Gonzalez 2011; Jain et al. 2015a). Although several direct fermentation routes for the production of R-1,2-PDO are documented and many experimental trials have been reported, production is limited and successful schemes have not yet been developed.
Unlike R-1,2-PDO, which could be produced by wild or engineered strains (Bennett and San 2001), the production of S-1,2-PDO directly from glucose has not been reported to our knowledge. Herein, we report the production of enantiomerically pure S-1,2-PDO from glucose by expressing an artificial biosynthetic pathway in E. coli strain BW25113. The genes responsible for branch metabolic pathways from glucose were knocked out, and the metabolism of glucose to l-lactate was strengthened by substitution of the endogenous d-lactate dehydrogenase in E. coli with that from the l-lactate hyperproducer Bacillus coagulans 2-6 (Wang et al. 2014). A novel NADH and acetyl-CoA simultaneous regeneration system was also included to further improve the productivity.
Materials and methods
Strains, plasmids, and cultivation conditions
Bacterial strains and plasmids used in this study are shown in Table 1. E. coli DH5α (Tiangen, Beijing, China) was used for general cloning, while E. coli BL21 (DE3) and E. coli Origami (DE3) (Tiangen, Beijing, China) were used for protein expression and whole-cell catalysis. E. coli BW25113∆poxB was used as the host strain for metabolic engineering for direct fermentation of glucose to S-1,2-PDO. E. coli strains used for genetic engineering were cultured in Luria-Bertani (LB) broth supplemented with tryptone (10 g/L), yeast extract (5 g/L), and NaCl (5 g/L), pH 7.0 at 37 °C. Antibiotic selection in E. coli cultures was conducted with ampicillin (100 μg/mL) and kanamycin (34 μg/mL).
General gene cloning and protein expression
To construct the artificial biosynthetic pathway from lactate to 1,2-PDO, several genes were selected from a database based on the availability of experimentally verified biochemical characteristics. The gene for CoA transferase (pct) was codon optimized to promote high expression in E. coli based on the gene sequence from Clostridium propionicum DSM 1682 (GenBank: AJ276553.1) with one point mutation (V193A) to alleviate the inhibitory effects on cell growth with overexpression of this enzyme in E. coli (Yang et al. 2010). The enzyme catalyzing transformation of lactoyl-CoA to lactaldehyde was selected as CoA-dependent succinate semialdehyde dehydrogenase (PdcD) from Yersinia enterocolitica subsp. enterocolitica 8081 (GenBank: CAL12775.1). The candidate enzymes catalyzing transformation of lactaldehyde to 1,2-PDO included 3-hydroxypropionate dehydrogenase (MmsB) from Bacillus cereus ATCC 14579 and l-1,2-PDO oxidoreductase (FucO) and NADPH-dependent aldehyde reductase (YqhD) from E. coli MG1655. The gene of pdcD was codon optimized before expression in E. coli, while fucO and yqhD were amplified directly from the genomic DNA of E. coli MG1655. The gene of mmsB was directly synthesized according to the gene sequence in GenBank (accession number: AE016877.1). The genes of pct, pdcD, mmsB, and fucO were inserted into vector pET-28a, and yqhD was inserted into vector pET-duet1. The primers used in this study are listed in Table 2. All constructed vectors mentioned above were transformed into E. coli BL21 (DE3) to investigate their enzymatic activities after expression. The codon-optimized gene sequences of pct and pdcD were deposited at the NCBI database under GenBank accession numbers of KT199425 and KT199426, respectively.
Protein purification and enzyme assays
The enzymes were purified by nickel affinity chromatography according to the manufacturer’s protocol (Tiandz, Beijing, China). The resultant effluent was filtered and loaded onto a HisTrap HP 5-mL column for desalination (GE Healthcare). The enzyme was eluted with phosphate-buffered saline (PBS, pH 7.0) at a flow rate of 5 mL/min. The fractions containing the enzyme were detected by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). After purification, all enzyme activities were determined. The activity of Pct was determined by high-performance liquid chromatography (HPLC) after the reaction between lactic acid and acetyl-CoA. The activities of PdcD, FucO, YqhD, and MmsB were determined by measuring the initial rate of NADH oxidation at 340 nm in 400 μL of 100 mM PBS, 100 mM substrate, 0.2 mM NADH, and 20 μg enzyme. The reaction was initiated by the addition of enzyme. One unit was defined as the amount of enzyme converting 1 μM NADH per min. Specific activity is expressed as units per milligram protein.
Verification of the artificial biosynthetic pathway from lactate to 1,2-PDO
To verify and optimize the combination of the selected enzymes, plasmid vectors pBAD43 and pET-duet1 were used to construct the biosynthetic pathway to test the 1,2-PDO production capacity. pct was cloned into pBAD43 under the control of PBAD promoter. pdcD was inserted into the first multiple clone site of pET-duet1 under the control of T7 promoter. mmsB, fucO, and yqhD were constructed into the second multiple clone site of pET-duet1 and also under the control of T7 promoter.
E. coli Origami (DE3) strains harboring the above recombinant plasmids were incubated in LB medium containing 100 μg/mL ampicillin at 37 °C with shaking at 160 rpm. When the culture reached an optical density of 0.6–0.8 at 600 nm, 0.05 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and 0.2 % arabinose were added to induce gene expression. After induction at 18 °C for 12 h, cells were harvested by centrifugation at 4 °C. The harvested cells were washed with sterilized saline solution three times. After adjusting the cell biomass to the same levels (the values of OD600 were 25), whole-cell catalysis was performed to produce 1,2-PDO at 20 °C without shaking for 24 h. l-lactic acid and glucose were added as substrates.
Integration of the S-1,2-PDO biosynthetic pathway into the E. coli genome
Methods for seamless chromosomal deletion, gene replacement, or integration were described previously using Red recombinase technology (Datsenko and Wanner 2000; Jantama et al. 2008). When replacing a gene in the genome, the gene sequence used for replacement was added in front of the FRT sites. PCR products were then transformed into E. coli-competent cells carrying plasmid pKD46. Single colonies were picked from plates containing kanamycin and inoculated into LB medium with kanamycin. Colony PCR was performed to verify that the genes in the genome had been replaced or knocked out. Competent cells of verified E. coli were prepared for transforming plasmid pCP20 to eliminate the kanamycin resistance gene. The primers used are listed in Table 2.
Disruption of d-lactate production and strengthening l-lactate production
To increase the l-lactate production, the d-lactate dehydrogenase genes ldhA was replaced by B. coagulans 2-6 l-lactate dehydrogenase (Lldh) (Wang et al. 2014), resulting in strain PD6 (PD3∆ldhA::Lldh). Additionally, the mgsA gene was deleted to avoid any production of d-lactate through the methylglyoxal bypass pathway and avoid accumulation of the toxic methylglyoxal intermediate. The resultant strain was designated PD7 (PD6∆mgsA).
Further improvement of performance by cofactor regeneration
The pyruvate decarboxylase gene (Zppdc) was cloned from Zymobacter palmae, as this enzyme exhibited the highest specific activity and the lowest K m for pyruvate compared to the other reported pyruvate decarboxylases (Raj et al. 2002). The NAD-binding acetaldehyde-CoA dehydrogenase II gene (mhpF) was cloned from E. coli MG1655 (Lee et al. 2006). A combination of the pyruvate decarboxylase and acetaldehyde-CoA dehydrogenase II could catalyze pyruvate to acetyl-CoA, which simultaneously regenerates NADH and acetyl-CoA. Zppdc and mhpF were cloned into plasmid pBAD43. The formic acid dehydrogenase gene (fdh) was also cloned from Candida boidinii for regeneration of NADH only (Slusarczyk et al. 2000).
Fermentative production of S-1,2-PDO by engineered strains
Modified NBS medium was used for testing the production capacity of different engineered E. coli strains, which consisted of (per liter) 20 g glucose, 1 g yeast extract, 3.5 g KH2PO4, 6.55 g K2HPO4, 3.5 g (NH4)2HPO4, 0.246 g MgSO4, 0.011 g CaCl2, 5 mg thiamine-HCl, and 0.154 g betaine-HCl, pH 7.0. Overnight LB cultures of engineered bacteria were reinoculated and induced with 0.2 mM IPTG or 0.2 % arabinose. All flasks were shaken at 250 rpm at 37 °C. Cell growth was constantly measured, and bacterial cultures were sampled in 24 h.
Analytical methods
1,2-PDO and lactate were measured using an HPLC system (Agilent 1260 series) equipped with an organic acid analytical column (Aminex HPX-87H, Bio-Rad) and a differential refractive index detector. The mobile phase was 6 mM H2SO4 at a flow rate of 0.5 mL/min. The column temperature was 55 °C with the detector temperature of 35 °C. The enantiomeric purity of 1,2-PDO was determined by gas chromatography (GC; Shimadzu, GC-2010) with a chiral capillary column (Beta DEXTM 120 capillary column, SUPELCO). The fermentation broth was concentrated by rotary evaporation and then extracted with n-butanol. One microliter sample was injected into the column at a temperature of 63 °C with an injector temperature of 250 °C. The products were detected with a flame ionization detector at 275 °C.
Results
Demonstration of S-1,2-PDO biosynthetic pathway from l-lactic acid
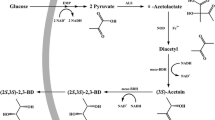
Yim et al. (2011) reported a designed pathway from succinate to 1,4-butanediol via two consecutive reduction processes, which has proven to be the high-priority pathway from organic acids to diols. Based on that, the pathway for 1,2-PDO production from lactic acid was designed as shown in Fig. 1. The catalyzed enzyme candidates were chosen from a database based on their experimentally verified biochemical characteristics. Briefly, Pct first catalyzed the transformation of l-lactic acid to l-lactyl-CoA, and PdcD catalyzed the subsequent transformation of l-lactyl-CoA to l-lactaldehyde. Finally, l-lactaldehyde could be catalyzed to S-1,2-PDO by MmsB, FucO, or YqhD, as previously reported (Cameron et al. 1998; Clomburg and Gonzalez 2011).

The artificial biosynthetic pathway from glucose to S-1,2-propanediol
As there are three candidates for the last step, the catalytic characteristics of the enzymes were first investigated. The specific activity of MmsB (1.15 ± 0.03 U/mg) was much higher than those of FucO (0.76 ± 0.09 U/mg) and YqhD (0.16 ± 0.02 U/mg) using NADH as the coenzyme. To further test the differences in enzymatic activity in vivo, the production of S-1,2-PDO from glucose catalyzed by Pct, PdcD, and each of the three enzymes individually was monitored. The results show that the production titer of S-1,2-PDO catalyzed by the combination of Pct, PdcD, and MmsB was the highest, and the addition of glucose further increased the production (Fig. 2a). The product was confirmed as 1,2-PDO by GC/mass spectrometry (MS) analysis (Fig. 2b). Thus, the artificial pathway for S-1,2-PDO production from l-lactic acid was well demonstrated.

Demonstration of S-1,2-propanediol biosynthesis from l-lactic acid. a The production titer of S-1,2-propanediol (1,2-PDO) from l-lactic acid by different combinations of enzymes. PM combination of CoA transferase (Pct) with succinic-semialdehyde dehydrogenase (PdcD) and 3-hydroxypropionic acid dehydrogenase (MmsB); PY combination of Pct, PdcD, and aldehyde reductase (YqhD); PF combination of Pct, PdcD, and propanediol oxidoreductase (FucO); G with glucose addition. b GC/MS analysis of the product. STD standard
Integration of the S-1,2-PDO biosynthetic pathway into the E. coli genome with synchronous deletion of genes responsible for branch metabolic pathway
After demonstration of the transformation pathway, the artificial pathway was integrated into the genome to construct a genetically stable strain for S-1,2-PDO production. To improve the yield and conversion rate of S-1,2-PDO, the genes responsible for undesired lactate utilization (lldD) and acetate (ackA-pta) and ethanol (adhE) formation must be knocked out. To simplify the process, the genes for Pct, PdcD, and MmsB were integrated into the genome of E. coli BW25113∆poxB with synchronous deletion of genes responsible for the above branch metabolic pathway from glucose. The pdcD gene was integrated into the chromosome at the adhE (encoding aldehyde/alcohol dehydrogenase) site. Similarly, the mmsB gene was integrated at the lldD (encoding l-lactate dehydrogenase, which catalyzes l-lactate to pyruvate) site, and the pct gene was integrated at the ackA-pta operon (encoding acetate kinase A-phosphotransacetylase) site. All the genes were driven by additionally added T5 promoters. The resultant strain was designated PD3 (E. coli BW25113∆poxB, ∆adhE::pdcD, ∆lldD::mmsB, ∆ackA-pta::pct). The initial PD3 strain could produce about 1.9 mM 1,2-PDO by direct fermentation of glucose.
Construction of a homo-l-lactate-producing strain
To initiate the metabolic engineering of PD3 to a homo-l-lactic acid producer, the fermentative E. coli ldhA in PD3 was first deleted, and the other d-lactate dehydrogenase (dld) was replaced by Bacillus coagulans 2-6Lldh, resulting in strain PD5 (PD3∆ldhA, ∆dld::Lldh). Only very small amounts of d-lactic acid were detected in the fermentation broth of strain PD5, while the production titer of l-lactic acid remained low (Fig. 3).

Stereospecificity of produced lactic acid by the engineering E. coli strains. PD5 deletion of d-lactate dehydrogenase (ldhA) and replacement of d-lactate dehydrogenase (dld) with B. coagulans l-lactate dehydrogenase (Lldh), PD6 replacement of ldhA with Lldh, PD7 deletion of the mgsA gene in PD6
This result demonstrates that ldhA is the primary route for lactate production in E. coli BW25113 and that dld expression is regulated at low levels. Thus, Lldh could not be expressed at high levels in PD5 to produce enough lactic acid for subsequent 1,2-PDO production. Therefore, we integrated B. coagulans Lldh in the ldhA locus of strain PD3, which is driven by the native ldhA promoter, to produce the resulting strain PD6 (PD3∆ldhA::Lldh). The production of l-lactic acid was significantly increased in strain PD6 with a small amount of d-lactic acid (Fig. 3), which d-lactic acid should be produced through the methylglyoxal detoxification pathways (Baldomà and Aguilar 1987; Booth et al. 2003). The mgsA gene was deleted to avoid production of any d-lactate through the methylglyoxal bypass pathway, and the resultant strain PD7 (PD6∆mgsA) produced >99.9 % enantiomerically pure l-lactic acid (Fig. 3). Strain PD7 could produce ∼4.1 mM 1,2-PDO from glucose. Furthermore, FrdA, a component of fumarate reductase, is responsible for fumarate synthesis. To further intensify the metabolic flux to lactic acid, we deleted frdA from strain PD7. As expected, the production titer of S-1,2-PDO by the resultant strain PD11 (PD7∆frdA) increased to 5.9 mM.
Improving 1,2-PDO production by cofactor and CoA regeneration system
The cofactor NADH is important for the designed S-1,2-PDO production pathway since the enzymes in the final two reduction steps in the pathway are NADH dependent. The NADH regeneration system was first introduced into the PD12 strain by overexpression of the fdh gene from C. boidinii (Slusarczyk et al. 2000). However, the additional NADH did not increase the production titer, as shown in Fig. 4. l-Lactate is first activated by CoA in the artificial pathway and we hypothesized that, in addition to NADH, CoA should be also a key factor in 1,2-PDO production. Therefore, pyruvate decarboxylase and acetaldehyde-CoA dehydrogenase II were combined to catalyze pyruvate to acetyl-CoA, simultaneously regenerating NADH and acetyl-CoA. In flask cultures with shaking, S-1,2-PDO production by the resultant strain PD13 slightly increased from 5.9 to 6.5 mM (Fig. 4). Furthermore, additional CoA transferase (Pct) was engineered into strain PD13 since this enzyme catalyzes lactate activation, in which CoA is incorporated. As expected, the production titer of S-1,2-PDO by the resultant strain PD14 was significantly increased from 6.5 to 13.7 mM. The enantiomeric purity of the resulting 1,2-PDO was analyzed by GC equipped with a chiral capillary column. S-1,2-PDO accumulated in the broth at >99 % enantiomeric purity, and no peak for R-1,2-PDO could be detected in the chromatogram (Fig. 5).

Production titers of S-1,2-propanediol by different engineered strains. The strains were cultivated in NBS medium at 37 °C for 24 h. The genotypes of the engineered strains are provided in Table 1

Enantiomeric purity of S-1,2-propanediol produced by the final engineered strain PD14. a The GC profile of standard mixture of R-1,2-propanediol and S-1,2-propanediol. b The GC profile of extract sample from strain PD14 cultivation broth
Discussion
The ongoing depletion of fossil fuels will increase the cost of manufacturing these chemicals in the near future and poses a threat to the environment by releasing vast quantities of carbon dioxide. With the emergence of metabolic engineering, better solutions are emerging to meet the global demand (Jain et al. 2015b) and have proven efficient for the production of indigenous metabolites. However, manufacturing non-native products requires the construction of novel synthetic pathways by expanding native metabolism through combinatorial metabolic engineering of microorganisms. Over the last decade, the biosynthesis of various high-value and commodity chemicals has been established by engineering E. coli (Herrera 2004).
The stereoisomers of 1,2-PDO are high-value chemicals with numerous potential uses as chiral synthons in organic synthesis (Simon et al. 1987). The biosynthesis of R-1,2-PDO has been achieved in E. coli by engineering the glycolysis pathway (Bennett and San 2001; Jain et al. 2015a, 2015b); however, the production of S-1,2-PDO directly from glucose has not been reported. Lactobacillus buchneri and Lactobacillus parabuchneri are reported to be able to degrade lactic acid to acetic acid with concomitant production of 1,2-PDO and trace ethanol under anoxic conditions (Oude Elferink et al. 2001; Nishino et al. 2003). Although a simple pathway was proposed, in which half of the lactate was transformed directly to the lactaldehyde intermediate and then to 1,2-PDO, the missing biochemical evidence makes this proposed process uncertain. Recently, Heinl et al. (2012) released the complete genome sequence of L. buchneri CD034, and one gene (aldA) was annotated as a possible enzyme catalyzing the reversible reaction between lactate and lactaldehyde. We synthesized this gene fragment in our study according to the sequence in GenBank (LBUCD034_0873). However, after successful expression in E. coli, only the activity of transforming lactaldehyde to lactate was detected both in vivo and in vitro after enzyme purification, which is consistent with the activity of lactaldehyde dehydrogenase from E. coli (data not shown) (Baldomà and Aguilar 1987). Thus, we were unable to explore an enzymatic pathway for directly catalyzing lactate to lactaldehyde.
Therefore, we focused on exploring an indirect pathway for lactate biotransformation. To achieve S-1,2-PDO production, we designed an artificial pathway from lactate to 1,2-PDO and demonstrated its success herein. As microbial fermentation of lactic acid from glucose is well established (Jiang et al. 2013; Li et al. 2013; Peng et al. 2013; Wang et al. 2014), microbial synthesis of 1,2-PDO can be reasonably achieved from glucose. Although the initial titer was low (∼1.9 mM), this strain was the first to directly produce S-1,2-PDO from glucose fermentation to our knowledge. Since the artificial reduction steps do not change the stereochemistry at the C2 position of lactate, the final stereospecificity of produced 1,2-PDO relies on the stereochemistry of the lactate intermediate. E. coli BW25113 produces d-lactic acid as the primary product in mineral salt medium using glucose; therefore, the native d-lactate production pathway must be fully eliminated. Besides the lactate production pathway from pyruvate by lactate dehydrogenase, d-lactic acid production also could be achieved through the methylglyoxal pathway, which is not desirable for enantiomerically pure S-1,2-PDO production. E. coli has several native methylglyoxal detoxification pathways that can lead to the production of both d- and l-lactate (Booth et al. 2003), compromising the enantiomeric purity of the product. In addition, methylglyoxal is a very toxic metabolite whose accumulation could severely impair metabolism and lead to cell death (Totemeyer et al. 1998). A previous report indicated that submillimolar concentrations of methylglyoxal could decrease E. coli cell viability, which was considered a major hurdle to achieving high-titer and high-yield biosynthesis of R-1,2-PDO through the methylglyoxal pathway (Niu and Guo 2015). Therefore, we blocked the methylglyoxal bypass pathways and engineered a homo-l-lactate-producing strain. The improved strain produced ∼4 mM S-1,2-PDO from glucose. To further intensify the metabolic flux to S-1,2-PDO, we disrupted fumarate reductase, which increased the S-1,2-PDO titer to 5.9 mM, about threefold greater than that for the starting strain.
The production of 1,2-PDO from lactate also requires the consumption of two NADH molecules, and we hypothesized that the development of a NADH regeneration system would serve as a driving force for 1,2-PDO production. However, only the exogenous NADH regeneration system could not stimulate the production of S-1,2-PDO, even when cells were fed additional formate. In the designed lactate transformation pathway, CoA is also needed to activate lactic acid in the initial step. Since the NADH supplement alone could not drive the metabolic flux toward S-1,2-PDO, we hypothesized that CoA might be needed, as the pyruvate dehydrogenase is normally inhibited under anaerobic conditions, leading to limiting amounts of acetyl-CoA. To overcome the bottleneck, we established a novel NADH and acetyl-CoA simultaneous regeneration system to replenish the NADH and acetyl-CoA consumed in the S-1,2-PDO pathway. This approach has the advantage of not requiring supplementation of other media components, making it a more suitable strategy for large-scale microaerobic production of reduced chemicals. As expected, the combined effects of disrupting the major carbon-competing pathways and strengthening the lactate transformation pathway by the additional CoA transferase enhanced the S-1,2-PDO titer from 1.9 to 13.7 mM. The metabolic engineering strategies are summarized in Fig. 6.

Summary of the sequential metabolic engineering process for S-1,2-propanediol production from glucose in E. coli BW25113. The crosses mean gene deletions and the thick arrow means the replacement of the native d-lactate dehydrogenase gene (ldhA) in E. coli by B. coagulans 2-6Lldh gene
To date, reports of the biological production of enantiomerically pure S-1,2-PDO are rare. The anaerobic fermentation route to S-1,2-PDO from l-rhamnose or l-fucose is economically unfeasible due to the highly expensive substrate and rather low titer (Boronat and Aguilar 1981; Altaras and Cameron 1999). During our research, Niu and Guo (2015) first published an artificial pathway from lactic acid to 1,2-PDO without systematically engineering microbial strains, in which the same pathway was designed with different selected enzyme candidates. After 72 h of cultivation, 1.7 g/L (∼22 mM) S-1,2-PDO was produced by transforming an l-lactic acid substrate with additional supplemented glucose in the plasmid-expressing system. In our study, 13.7 mM S-1,2-PDO with an enantiomeric purity >99 % was produced by direct fermentation of glucose in 24 h by metabolic engineered strains. This is the first report of an approach to biological production of non-native high-value S-1,2-PDO from glucose in E. coli. Although the current titer is not at desired levels, the metabolic engineering approach in this study provides the direction to further improve the performance of recombinant strains. For example, the lactic acid transformation pathway to S-1,2-PDO should be further strengthened by regulating its expression in hosts through combinational optimization of promoters and ribosome binding sites (Zelcbuch et al. 2013) to construct genetically stable strains with industrial potential.
In conclusion, we demonstrated a novel step toward the future biological manufacture of S-1,2-PDO from cheap carbon sources by engineered E. coli strains. The designed artificial biological pathway for S-1,2-PDO production and the implementation of rational metabolic engineering strategies enabled the engineered strain to produce enantiomerically pure S-1,2-PDO from glucose. Pathway-level optimization of S-1,2-PDO production to further enhance the productivity is under investigation in our laboratory.
References
Altaras N, Cameron D (1999) Metabolic engineering of a 1,2-propanediol pathway in Escherichia coli. Appl Environ Microbiol 65:1180–1185
Altaras N, Cameron D (2000) Enhanced production of (R)-1,2-propanediol by metabolically engineered Escherichia coli. Biotechnol Prog 16:940–946
Badía J, Ros J, Aguilar J (1985) Fermentation mechanism of fucose and rhamnose in Salmonella typhimurium and Klebsiella pneumoniae. J Bacteriol 161:435–437
Baldomà L, Aguilar J (1987) Involvement of lactaldehyde dehydrogenase in several metabolic pathways of Escherichia coli K12. J Biol Chem 262:13991–13996
Bennett GN, San KY (2001) Microbial formation, biotechnological production and applications of 1,2-propanediol. Appl Microbiol Biotechnol 55:1–9
Berríos-Rivera SJ, San KY, Bennett GN (2003) The effect of carbon sources and lactate dehydrogenase deletion on 1,2-propanediol production in Escherichia coli. J Ind Microbiol Biotechnol 30:34–40
Booth IR, Ferguson GP, Miller S, Li C, Gunasekera B, Kinghorn S (2003) Bacterial production of methylglyoxal: a survival strategy or death by misadventure? Biochem Soc Trans 31:1406–1408
Boronat A, Aguilar J (1981) Metabolism of L-fucose and L-rhamnose in Escherichia coli: differences in induction of propanediol oxidoreductase. J Bacteriol 147:181–185
Cameron DC, Altaras NE, Hoffman ML, Shaw AJ (1998) Metabolic engineering of propanediol pathways. Biotechnol Prog 14:116–125
Cameron DC, Cooney CL (1986) A novel fermentation: the production of (R)-1,2-propanediol and acetol by Clostridium thermosaccharolyticum. Bioresour Technol 4:651–654
Clomburg J, Gonzalez R (2011) Metabolic engineering of Escherichia coli for the production of 1,2-propanediol from glycerol. Biotechnol Bioeng 108:867–879
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645
Hacking AJ, Lin ECC (1976) Disruption of the fucose pathway as a consequence of genetic adaptation to propanediol as a carbon source in Escherichia coli. J Bacteriol 126:1166–1172
Herrera S (2004) Industrial biotechnology—a chance at redemption. Nat Biotechnol 22:671–678
Heinl S, Wibberg D, Eikmeyer F, Szczepanowski R, Blom J, Linke B, Goesmann A, Grabherr R, Schwab H, Pühler A (2012) Insight into the completely annotated genome of Lactobacillus buchneri CD034, a strain isolated from stable grass silage. J Biotechnol 161:153–166
Jantama K, Zhang X, Moore JC, Shanmugam KT, Svoronos SA, Ingram LO (2008) Eliminating side products and increasing succinate yields in engineered strains of Escherichia coli C. Biotechnol Bioeng 101:881–893
Jain R, Yan Y (2011) Dehydratase mediated 1-propanol production in metabolically engineered Escherichia coli. Microb Cell Fact 10:97
Jain R, Sun X, Yuan Q, Yan Y (2015a) Systematically engineering Escherichia coli for enhanced production of 1,2-propanediol and 1-propanol. ACS Synth Biol 4:746–756
Jain R, Huang J, Yuan Q, Yan Y (2015b) Engineering microaerobic metabolism of E. coli for 1,2-propanediol production. J Ind Microbiol Biotechnol 42:1049–1055
Jiang X, Xue YF, Wang AY, Wang LM, Zhang GM, Zeng QT, Yu B, Ma YH (2013) Efficient production of polymer-grade L-lactate by an alkaliphilic Exiguobacterium sp. strain under nonsterile open fermentation conditions. Bioresour Technol 143:665–668
Lee SJ, Ko JH, Kang HY, Lee Y (2006) Coupled expression of MhpE aldolase and MhpF dehydrogenase in Escherichia coli. Biochem Biophys Res Commun 346:1009–1015
Li Y, Wang LM, Ju JS, Yu B, Ma YH (2013) Efficient production of polymer-grade D-lactate by Sporolactobacillus laevolacticus DSM442 with agricultural waste cottonseed as the sole nitrogen source. Bioresour Technol 142:186–191
Nishino N, Yoshida M, Shiota H, Sakaguchi E (2003) Accumulation of 1,2-propanediol and enhancement of aerobic stability in whole crop maize silage inoculated with Lactobacillus buchneri. J Appl Microbiol 94:800–807
Niu W, Guo J (2015) Stereospecific microbial conversion of lactic acid into 1,2-propanediol. ACS Synth Biol 4:378–382
Oude Elferink SJ, Krooneman J, Gottschal JC, Spoelstra SF, Faber F, Driehuis F (2001) Anaerobic conversion of lactic acid to acetic acid and 1,2-propanediol by Lactobacillus buchneri. Appl Environ Microbiol 67:125–132
Peng LL, Wang LM, Che CC, Yang G, Yu B, Ma YH (2013) Bacillus sp. strain P38: an efficient producer of L-lactate from cellulosic hydrolysate, with high tolerance for 2-furfural. Bioresour Technol 149:169–176
Raj KC, Talarico LA, Ingram LO, Maupin-Furlow JA (2002) Cloning and characterization of the Zymobacter palmae pyruvate decarboxylase gene (pdc) and comparison to bacterial homologues. Appl Environ Microbiol 68:2869–2876
Shelley S (2007) A renewable route to propylene glycol. Chem Eng Prog 103:6–9
Simon ES, Whitesides M, Cameron DC, Weitz DJ, Cooney CL (1987) A combined microbial/chemical synthesis of (+)-(R)-methyloxirane having high enantiomeric excess. J Org Chem 52:4042–4044
Sanchez-Rivera F, Cameron DC, Cooney CL (1987) Influence of environmental factors in the production of 1,2-propanediol by Clostridium thermosaccharolyticum. Biotechnol Lett 9:449–454
Slusarczyk H, Felber S, Kula MR, Pohl M (2000) Stabilization of NAD-dependent formate dehydrogenase from Candida boidinii by site-directed mutagenesis of cysteine residues. Eur J Biochem 267:1280–1289
Sun X, Shen X, Jain R, Lin Y, Wang J, Sun J, Yan Y, Yuan Q (2015) Synthesis of chemicals by metabolic engineering of microbes. Chem Soc Rev 44:3760–3785
Suzuki T, Onishi H (1968) Aerobic dissimilation of L-rhamnose and the production of L-rhamnonic acid and 1, 2-propanediol by yeasts. Agric Biol Chem 32:888–893
Totemeyer S, Booth NA, Nichols WW, Dunbar B, Booth IR (1998) From famine to feast: the role of methylglyoxal production in Escherichia coli. Mol Microbiol 27:553–562
Turner KW, Roberton AM (1979) Xylose, arabinose, and rhamnose fermentation by Bacteroides ruminicola. Appl Environ Microbiol 38:7–12
Wang LM, Cai YM, Zhu LF, Guo HL, Yu B (2014) Major role of NAD-dependent lactate dehydrogenases in high optically pure L-lactic acid production by thermophilic Bacillus coagulans. Appl Environ Microbiol 80:7134–7141
Weimer PJ (1984) Fermentation of 6-deoxyhexoses by Bacillus macerans. Appl Environ Microbiol 47:263–267
Yang TH, Kim TW, Kang HO, Lee SH, Lee EJ, Lim SC, Oh SO, Song AJ, Park SJ, Lee SY (2010) Biosynthesis of polylactic acid and its copolymers using evolved propionate CoA transferase and PHA synthase. Biotechnol Bioeng 105:150–160
Yim H, Haselbeck R, Niu W, Pujol-Baxley C, Burgard A, Boldt J, Khandurina J, Trawick JD, Osterhout RE, Stephen R, Estadilla J, Teisan S, Schreyer HB, Andrae S, Yang TH, Lee SY, Burk MJ, Van Dien S (2011) Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol 7:445–452
Zelcbuch L, Antonovsky N, Bar-Even A, Levin-Karp A, Barenholz U, Dayagi M, Liebermeister W, Flamholz A, Noor E, Amram S, Brandis A, Bareia T, Yofe I, Jubran H, Milo R (2013) Spanning high-dimensional expression space using ribosome-binding site combinatorics. Nucleic Acids Res 41:e98
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (2011CBA00800), the Key Deployment Projects of Chinese Academy of Sciences (KGZD-EW-606), the National Natural Science Foundation of China (21466007), and the Project of Guangxi Provincial Science & Technology Development, China (14125008-2-22). BY is supported by the Youth Innovation Promotion Association, Chinese Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors confirm that principles of ethical and professional conduct have been followed in this research and in the preparation of this manuscript.
Conflict of interest
The authors do not have potential conflict of interest to disclose.
Additional information
Lingfeng Zhu and Xiangchen Guan contributed equally to this work.
Rights and permissions
About this article

Cite this article
Zhu, L., Guan, X., Xie, N. et al. Fermentative production of enantiomerically pure S-1,2-propanediol from glucose by engineered E. coli strain. Appl Microbiol Biotechnol 100, 1241–1251 (2016). https://doi.org/10.1007/s00253-015-7034-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7034-y