
Abstract
d-Phenyllactic acid is a versatile natural organic acid, which is used as an antiseptic agent, monomer of the biodegradable material poly-phenyllactic acid and in the synthesis chiral intermediate of pharmaceuticals. In this report, the novel NADH-dependent d-lactate dehydrogenase LrLDH was identified by screening a shotgun genome of Lactobacillus rossiae. To improve cofactor regeneration, the Exiguobacterium sibiricum glucose dehydrogenase EsGDH was overexpressed together with LrLDH in E. coli BL21(DE3)-pCDFDuet-1-gdh-ldh. The total enzyme activity in the fermentation broth of E. coli BL 21(DE3)-pCDFDuet-1-gdh-ldh peaked at 2359.0 U l−1 when induced by 10 g l−1 lactose at 28 °C and 150 rpm for 14 h. The biocatalytic reduction of sodium phenylpyruvate to d-phenyllactic acid was successfully carried out using whole cells of the engineered E. coli. Under the optimized biocatalysis conditions, 50 g l−1 sodium phenylpyruvate was completely converted to d-phenyllactic acid with a space-time yield and enantiomeric excess of 262.8 g l−1 day−1 and > 99.5%, respectively. To our best knowledge, it is the highest productivity reported to date, with great potential for the mass production of d-phenyllactic acid.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phenyllactic acid (PLA; 2-hydroxy-3-phenylpropanoic acid), is an organic acid that is widely distributed in honey and some fermented foods such as cheese, pickles, and milk (Mu et al. 2012a). It has an inhibitory effect against a wide range of fungi and bacteria by destroying the cell membrane (Lavermicocca et al. 2000; Lavermicocca and Valerio 2003; Dieuleveux et al. 1998a, b; Wang et al. 2018). Because the molecule contains a chiral carbon atom, PLA has two enantiomers, whereby D-PLA has shown better antimicrobial activity and gained more attention than L-PLA (Mu et al. 2012a; Valerio et al. 2004). Owing to its low molecular mass and high hydrophilicity, PLA can diffuse comparatively easily (Wang et al. 2016). Moreover, the excellent antimicrobial activity, good stability, board effective pH range, and good safety make D-PLA an ideal antiseptic agent, which is widely used in the food industry and as a green alternative to antibiotics added to livestock feed (Schnürer and Magnusson 2005; Wang et al. 2009, 2010). In addition, D-PLA can be used as a monomer for the synthesis of biocompatible and biodegradable poly-phenyllactic acid. Due to the presence of a bulky aromatic side chain, poly-phenyllactic acid displays enhanced toughness, improved thermostability, and outstanding ultraviolet absorption properties compared to poly lactic acid (Kawaguchi et al. 2014; Fujita et al. 2013; Kawaguchi et al. 2017). Furthermore, as chiral building blocks, D-PLA plays an important role in the synthesis of pharmaceuticals and fine chemicals such as englitazone, ragaglitazar, danshensu, and the anthelmintic agent PF1022A, among others (Urban and Moore 1992; Ebdrup et al. 2003; Xu et al. 2016; Weckwerth et al. 2000).
D-PLA is a versatile compound with promising applications, accompanied with great commercial demands, which inspired a growing research interest in D-PLA synthesis. Due to its mild reaction conditions, exquisite stereoselectivity, environmental friendliness, and energy-effective operation, biocatalytic asymmetric ketone reduction was identified as a reliable, scalable, and straightforward route to optically active alcohols (Wang et al. 2017a; Choudhury et al. 2014; He et al. 2014; Ni and Xu 2012). Natural microorganisms are low-cost and easily available biocatalysts, which are widely distributed in the environment. In recent years, numerous bacteria with the ability to produce D-PLA from phenylpyruvic acid have been identified such as Lactobacillus plantarum (Dallagnol et al. 2011; Zhang et al. 2014), Lactobacillus pentosus (Zhu et al. 2015), Leuconostoc mesenteroides (Li et al. 2014), Pediococcus acidilactici (Mu et al. 2012b), Pediococcus pentosaceus (Yu et al. 2015), and Bacillus coagulans (Zheng et al. 2011). However, the D-PLA yields obtained using these organisms were relatively low level, because of poor enzyme expression. Moreover, two PLA-producing enzymes with opposite stereoselectivity coexist in the cells, so that the optical purity of D-PLA produced by natural microorganisms was moderate to low. In the catabolism of lactic acid bacteria, PLA is produced from phenylpyruvic acid in a reaction catalyzed by lactate dehydrogenase (LDH) (Wang et al. 2016). Escherichia coli is a versatile host for heterologous protein expression and easy to manipulate. Using recombinant E. coli expressing a D-LDH encoding gene as biocatalyst can overcome the shortcomings of biotransformations catalyzed by natural microorganisms. The reductive activity of LDH depends on nicotinamide adenine dinucleotide (NADH), which acts as a hydride donor (Jia et al. 2018). Due to the high price of NADH, an efficient and economic internal cofactor recycling system is essential for large-scale synthetic applications (Wang et al. 2017b). Glucose dehydrogenase (GDH) is one of the enzymes most frequently used for cofactor regeneration due to its high activity, wide pH range, prominent stability, and tolerance of many organic solvents (Xu et al. 2007; Wichmann and Vasic-Racki 2005). In addition, GDH uses cheap glucose as hydride source and the byproduct gluconic acid does not interfere with the synthesis reaction (Zhou et al. 2015). As a result, numerous biocatalysis systems were constructed with cofactor regeneration based on GDH, which exhibited excellent catalytic efficiency. For example, Cui et al. (2018) achieved the efficient synthesis of methyl-(R)-3-hydroxybutyrate with a productivity of 265 g l−1 day−1 and enantiomeric excess above 99.9% using whole cells of engineered E. coli expressing carbonyl reductase and GDH for the in situ regeneration of the coenzyme. Tang et al. developed a coupled system consisting of carbonyl reductase and GDH and produced optically pure (S)-3-chloro-1-phenyl-1-propanol from 100 g l−1 3-chloro-1-phenyl-1-propanone with 100% conversation within 12 h (Tang et al. 2018). Wang et al. coupled the mutational aldo–keto reductase KmAKR with GDH to catalyze the asymmetric reduction of t-butyl 6-cyano-(5R)-hydroxy-3-oxohexanoate and produced up to 237.4 mmol l−1 t-butyl 6-cyano-(3R,5R)-dihydroxyhexanoate with a productivity of up to 372.8 g l−1 day−1 (Wang et al. 2017c; Yu et al. 2019).
Screening new biocatalysts is an important task in biosynthesis. To achieve high productivity of optically pure D-PLA, we endeavored to discover novel and efficient D-LDH enzymes. In this work, a novel strain of Lactobacillus rossiae was isolated which exhibited the ability to reduce sodium phenylpyruvate (PPA) to D-PLA. Subsequently, a D-LDH encoding gene (ldh) was identified through shotgun library screening and co-expressed in E. coli together with glucose dehydrogenase from Exiguobacterium sibiricum. A whole cell biotransformation for D-PLA synthesis with in situ cofactor regeneration was implemented and the application potential was investigated.
Materials and methods
Reagents
Sodium phenylpyruvate (PPA), kanamycin, streptomycin, and the Plasmid Extraction Kit were purchased from Sangon Biotech (Shanghai, China). d- and l-phenyllactic acids (PLA) were from Sigma-Aldrich (St. Louis, MO, USA). Tryptone and yeast extract were obtained from Oxoid (United Kingdom). Protein size markers, 2 × Protein SDS-PAGE loading buffer and the Fast DNA SPIN Kit for Soil were purchased from TAKARA (Beijing, China). The DNA Gel Extraction Kit, PCR Cleanup Kit and Plasmid Miniprep Kit were purchased from Axygen (Suzhou, China). Restriction enzymes were from Thermo Fisher Scientific Co., Ltd (Beijing, China). Oligonucleotides synthesis and DNA sequencing were provided by Sangon Biotech (Shanghai, China). Unless otherwise specified, all other reagents and chemicals used in this study were obtained from general commercial suppliers and used without further purification.
Escherichia coli BL21 (DE3) and the plasmids pCDFDuet-1 and pET28a used for recombinant protein expression were purchased from Novagen (Shanghai, China). The pMD-19-T vector for gene cloning was purchased from TAKARA (Beijing, China). E. coli BL21 (DE3)/pET-28b-gdh was stored in our laboratory.
LB medium (pH 7.0) contained 10.0 g l−1 tryptone, 5.0 g l−1 yeast extract and 10.0 g l−1 NaCl.
Cloning of ldh and the construction of E. coli-pET28a-ldh
The LDH gene (GenBank: KRL56345.1) with a length of 1014 bp was amplified by PCR from the genomic DNA of L. rossiae using the primers P1 and P2 listed in Table 1. PCR conditions were as follows: initial denaturation at 95 °C for 5 min, followed by 30 amplification cycles (95 °C for 30 s, 56 °C for 30 s, and 72 °C for 1.5 min), and a final extension at 72 °C for 10 min. The amplified PCR product was ligated into the pMD-19-T vector, double digested with Nco I and Xho I, and then inserted into the expression vector pET28a. The resulting expression vector pET28a-ldh was used to transform the competent E. coli BL21 (DE3) and the positive clone was verified by DNA sequencing.
Construction of the GDH/LDH co-expression strains
For the E. coli BL21 (DE3)/pCDFDuet-1-gdh-ldh strain, the ldh was amplified by PCR from genomic DNA of L. rossiae using the primers P2 and P3 listed in Table 1. The gdh gene from Exiguobacterium sibiricum (GenBank: AIZ68241.1) was amplified by PCR from pET28a-gdh using the primers P5 and P6 listed in Table 1. The PCR used the same temperature program as above. The amplified PCR products were inserted into the multiple cloning sites (MCS 1 and MCS 2) of the pCDFDuet-1 vector between BglII and XhoI and NcoI and NotI recognition sites (Fig. 1a). The resulting recombinant co-expression plasmid pCDFDuet-1-gdh-ldh was used to transform the competent cells of E. coli BL21 (DE3) to produce E. coli-gdh/ldh.

Schematic presentation of the co-expression plasmids contained ldh and gdh genes. a Plasmids pCDFDuet-1-gdh-ldh. b Plasmids pCDFDuet-1-ldh-gdh
For E. coli BL21 (DE3)/pCDFDuet-1-ldh-gdh, the ldh was amplified using the primers P1 and P4 and gdh was amplified using the primers P7 and P8 using the same PCR conditions as above. The amplified PCR products were inserted into pCDFDuet-1 vector between the NcoI and NotI, and NdeI and XhoI recognition sites (Fig. 1b). The resulting recombinant co-expression plasmid pCDFDuet-1-ldh-gdh was used to transform the competent cells of E. coli BL21 (DE3) to produce E. coli-ldh-gdh.
Cultivation of recombinant strains
For protein expression, E. coli/pET28a-ldh, E. coli/pET28a-gdh, E. coli-gdh-ldh, E. coli-ldh-gdh and E. coli/pCDFDuet-1 were incubated in LB medium containing 50 μg ml−1 kanamycin or streptomycin, at 37 °C and 200 rpm for 10 h. Then 4% (v/v) of the pre-culture was transferred to fresh, sterile LB medium containing 50 μg ml−1 kanamycin or streptomycin and cultivated under the same conditions. To initiate expression, lactose was added into the broth to a final concentration of 8 g l−1 when the culture reached an OD600 of 0.8. After induction at 28 °C and 150 rpm for 8 h, the cells were harvested by centrifugation at 8000×g for 10 min at 4 °C and washed twice with potassium phosphate buffer (100 mM, pH 7.0).
To examine the expression of heterologous proteins, cell pellets of the recombinant strains were suspended at a concentration of 20 g DCW l−1 in potassium phosphate buffer (100 mM, pH 7.0) and disrupted by sonication. The cell debris was removed by centrifugation at 10,000×g for 20 min at 4 °C and the resulting supernatant was analyzed by SDS-PAGE with 12% separating gel under denaturing conditions. One unit of biocatalyst activity was defined as the amount of cells (measured by dry cell weight) required for the generation of 1 μmol PLA per minute.
The optimum expression conditions for E. coli-gdh-ldh, including inducer concentration and induction time, were determined. The optimum inducer concentration was examined by adding lactose at concentrations from 2 to 14 g l−1 and culturing at 28 °C for 10 h. The optimum induction time was examined by harvesting the induced cells after 2–16 h. The cells were harvested and the activity and biomass were measured.
The catalytic efficiency of E. coli/pET28a-ldh, E. coli-gdh-ldh, and E. coli-ldh-gdh in the conversion of PPA into D-PLA was assayed by monitoring the generation of PLA using high-performance liquid chromatography (HPLC) as described below. The reaction mixture was composed of 100 mM potassium phosphate buffer (pH 7.0), 10 g l−1 PPA, 10 g l−1 glucose, and 10 g DCW l−1 cells in a total volume of 10 ml and incubated at 30 °C and 200 rpm for 15 min.
Optimization of reaction conditions for D-PLA production
The basic conditions for bioconversion were as follows: cells were resuspended in 10 ml of 100 mM potassium phosphate buffer containing glucose and 10 g l−1 PPA, and then incubated at 200 rpm for 15 min at an appropriate temperature (Scheme 1). To optimize the bioconversion conditions, reaction temperature (25–45 °C), pH (6.0–8.0; 100 mM potassium phosphate buffer), glucose concentration (0–30 g l−1), and whole-cell biocatalyst loading (5–30 g l−1) were investigated. Samples were withdrawn and the concentration and enantiomeric excess (e.e.) of D-PLA were assayed by HPLC as described below.

Scheme for the asymmetric preparation of D-PLA from PPA
Biotransformation by whole cells of E. coli-gdh-ldh
The biotransformation reactions were performed in 100 ml round bottom flasks with 50 ml of the reaction mixtures, which comprised potassium phosphate buffer (100 mM, pH 7.0), cells of E. coli-gdh-ldh at a total cell density of 15 g DCW l−1, PPA loading at concentrations ranging from 30 to 60 g l−1 and a twofold molar excess of glucose, without external cofactor addition. The reaction mixtures were incubated at 35 °C under magnetic stirring at 300 rpm and the pH was automatically adjusted to 7.0 by titrating 1.0 M Na2CO3 solution. Samples were withdrawn every 20 min and the concentration and e.e. of D-PLA were assayed by HPLC as described below.
Analytical methods
The concentrations of PLA and PPA were determined by HPLC on an LC-20A system (Shimadzu, Japan) equipped with an ODS HYPERSIL column (4.6 × 250 mm, 5 μm, Thermo, USA) and an SPD-10A VP Plus ultraviolet detector (Shimadzu, Japan) set at a wavelength of 210 nm. The column temperature was maintained at 40 °C. The mobile phase consisted of 1:4 (v/v) acetonitrile and 0.1% formic acid in water (v/v) at a flow rate of 1.0 ml min−1.
The optical purity of D-PLA was measured by HPLC on a CHIRALCEL OJ-RH column (4.6 × 150 mm, 5 μm; Daicel, Japan) maintained at 40 °C. The mobile phase was composed of acetonitrile, methanol, trifluoroacetic acid, and water at a volumetric ratio of 50:50:1.5:898.5 and run at a flow rate of 0.6 ml min−1.
The extent of reaction, optical purity of D-PLA, and productivity were indicated by Conversion (%), enantiomeric excess (e.e., %) and space-time yield (gD-PLA l−1 day−1), respectively, which were defined as follows:
where C0 is the initial molar concentration of substrate (PPA), Cs is the final molar concentration of substrate (PPA), CD and CL are the final molar concentrations of D-PLA and L-PLA respectively, Cp is the final molar concentration of product (PLA), and t is the reaction time.
Results and discussion
Co-expression of ldh and gdh in E. coli
In this work, a d-lactate dehydrogenase gene was identified by searching the whole genome sequence of L. rossiae in the NCBI database (https://www.ncbi.nlm.nih.gov/), and named as ldh. The coding sequence was amplified and inserted into the expressed vector pET-28a for heterologous expression in E. coli. The recombinant strain E. coli/pET28a-ldh displayed an excellent stereoselectivity, producing D-PLA with an e.e. > 99.5%. However, it had low catalytic efficiency and only 0.85 g l−1 D-PLA was generated within 15 min of reaction (Table 2). As LDHs are NADH-dependent oxidoreductases, cofactor recycling is one of the key limitations of biocatalytic D-PLA production. GDH is well known and frequently used for cofactor regeneration. Accordingly, whole cells co-expressing LDH along with GDH would be a preferable system for the cofactor recycling and D-PLA preparation. To establish a corresponding biocatalyst, the ldh from L. rossiae and gdh from E. sibiricum were amplified by PCR and cloned in two different relative positions into the pCDFDuet-1 vector with two MCS. The resulting recombinant plasmids pCDFDuet-1-gdh-ldh and pCDFDuet-1-ldh-gdh were subsequently used to transform to E. coli competent cells and the corresponding strains E. coli-gdh-ldh and E. coli-ldh-gdh were obtained. The strains were then induced with 8.0 g l−1 lactose at 28 °C for 8 h and harvested by centrifugation. Biotransformation reactions were performed using whole cells of E. coli/pET28a-ldh, E. coli-gdh-ldh, and E. coli-ldh-gdh, respectively. As indicated in Table 2, the D-PLA yields obtained using the two co-expression strains were much higher than that of E. coli/pET28a-ldh. It was therefore obvious that the coupling with EsGDH significantly improved the catalytic efficiency. Furthermore, the D-LDH titers produced by E. coli-gdh-ldh and E. coli-ldh-gdh within 15 min were 3.33 g l−1 and 2.47 g l−1, respectively and the former strain exhibited a better catalytic activity.
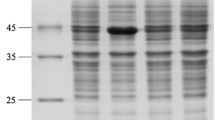
The cells of the strains were resuspended and disrupted by ultrasonication and the cleared lysates were examined by SDS-PAGE. As shown in Fig. 2, both E. coli-gdh-ldh and E. coli-ldh-gdh showed soluble expression of LDH and GDH. By comparing the amounts of both enzymes, it was obviously that more GDH, but less LDH was expressed in E. coli-gdh-ldh than in E. coli-ldh-gdh. This result suggested that the order of the two genes in pCDFDuet-1 can significantly affect the expression levels of the enzymes. E. coli-gdh-ldh had better relative expression levels of the two enzymes, which led to a relatively high PPA reduction activity (Table 2).

SDS-PAGE analysis. M: protein molecular weight marker; Lane 1–5: soluble fractions from lactose induced E. coli/pET28a-ldh, E. coli/pET28a-gdh, E. coli BL21-gdh-ldh, E. coli-ldh-gdh, and E. coli/pCDFDuet-1 respectively, induced by 8.0 g l−1 lactose at 28 °C for 8 h
Optimization of expression conditions for E. coli-gdh-ldh
To obtain the optimal protein expression conditions, the impact of lactose concentration and induction time on the specific biocatalyst activity and biomass of E. coli-gdh-ldh cells were studied. As shown in Fig. 3a, the specific activity increased at relatively low concentration of lactose and peaked at 10 g l−1, reaching 523.4 U g−1 DCW. Lactose addition at the tested concentrations was clearly beneficial to cell proliferation. Even though the cells kept proliferating, the maximal total activity 2219.2 U l−1 was obtained following induction with 10 g l−1 lactose for 10 h. Since the total activity is the decisive factor affecting biocatalytic efficiency, lactose with a concentration of 10 g l−1 was subsequently used for the induction. The optimal induction time was evaluated in a time range of 2–16 h. As shown in Fig. 3b, the specific activity and biomass were time dependent. The maximum total activity of 2359.0 U l−1 was obtained at 14 h. Therefore, E. coli-gdh-ldh was induced with 10 g l−1 lactose and allowed to express the recombinant enzymes at 28 °C and 150 rpm for 14 h to produce the optimized whole-cell biocatalyst for the following experiments.

Effect of induction conditions on specific activity and biomass. a Optimization of lactose concentration. Lactose were added as inducer at the range of 2–14 g l−1 and cultured at 28 °C for 10 h. b Optimization of induction time. The induction time was examined within a range of 2–16 h. All reactions were performed in triplicate, and error bars represent the standard error of mean
Optimization of biocatalysis conditions
E. coli-gdh-ldh cells were cultured, induced, and harvested as described above. To improve the efficiency of PLA production, the biocatalysis conditions, including reaction pH, temperature, molar ratio of glucose to PPA, and biocatalyst loading were investigated.
The reaction pH is a crucial parameter affecting enzyme activity and stability. Therefore, the effects of pH on PLA production from PPA using resting cells of E. coli-gdh-ldh were determined in a pH range from 6.0 to 8.0. As shown in Fig. 4a, the highest PLA production was achieved at pH 7.0, which indicated that neutral pH gives the best catalytic efficiency.

Optimization of biocatalysis conditions. Effect of initial pH (a), conversion temperature (b), ratio of glucose to PPA (c) and cell dosage of E. coli-gdh-ldh (d) on D-PLA production. Samples were quantitatively analyzed after biocatalysis for 15 min. All reactions were performed in triplicate, and error bars represent the standard error of mean
Temperature also plays an important role in biocatalytic reactions, because it affects the activity and stability of enzymes, as well as the solubility of substrates. Thus, the optimum temperature was examined in a range of 25–45 °C. The concentration of PLA rose, when the temperature was increased from 25 to 35 °C, but dropped sharply above 35 °C (Fig. 4b). The optimal temperature was therefore 35 °C.
Glucose was used as the substrate of GDH, which drives NADH regeneration. The effect of the molar ratio of glucose to PPA on PLA generation was also investigated as shown in Fig. 4c. At molar ratios below 2:1, the PLA production was significantly improved with increasing amounts of glucose added and reached the maximum value of 6.1 g l−1. However, further increasing the glucose concentration led to a dramatic decrease of PLA generation, probably because the addition of glucose increased the viscosity of the reaction solution, reducing the mass transfer efficiency.
The effect of biocatalyst loading on PLA production was also investigated. At biocatalyst concentrations below 15 g DCW l−1 in the reaction mixture, the reaction was significantly enhanced with the increase of cell dosage. However, biocatalyst loading exceeded 15 g DCW l−1 had no apparent effect on the PLA production (Fig. 4d) indicating that the catalyst was already saturated. As a result, an optimum biocatalyst loading of 15 g DCW l−1 was used for the following experiments.
Asymmetric synthesis of PLA
Based on the results of single-factor optimization experiments, asymmetric reduction of PPA at a substrate concentration of 30–60 g l−1 was conducted and the time courses of the bioconversions are illustrated in Fig. 5. PPA was completely converted within 100 min at a substrate concentration of 30 g l−1, 140 min at 40 g l−1, and 240 min at 50 g l−1 (Fig. 5a–c). However, further increasing the PPA concentration to 60 g l−1 led to a sharp decrease of D-PLA yield, suggesting that excess PPA loading brought about a detrimental substrate inhibition effect on enzyme activity (Fig. 5d). Substrate loading of 50 g l−1 afforded the highest D-PLA yield of 43.8 g l−1 after 240 min of the biocatalytic reduction, with an e.e. and space-time yield of > 99.5% and 262.8 g l−1 day−1, respectively. Table 3 summarizes the results and compares D-PLA production using different recombinant strains and processes reported in the literature. It is obvious that the batch catalytic process using resting cells co-expressing GDH and LDH in this work afforded the highest space-time yield for the production optically pure D-PLA.

Time course of D-PLA production from different concentration of PPA. The biotransformation reactions were carried out in 100 ml round bottom flasks with 50 ml of the reaction mixtures, which comprised potassium phosphate buffer (100 mM, pH7.0), cells of E. coli-gdh-ldh at a total cell density of 15 g DCW l−1, PPA loading at concentrations of 30 (a), 40 (b), 50 (c) and 60 g l−1 (d), respectively, and 2-fold glucose. The resulting mixture was maintained at 35 °C, 300 rpm in magnetic stirring apparatuses without external cofactor added, and the pH was automatically adjusted to 7.0. Samples were withdrawn every 20 min and the concentration and e.e. of D-PLA were assayed by HPLC. All reactions were performed in triplicate, and error bars represent the standard error of mean
Conclusions
The E. coli strain overexpressing only the NADH-dependent LDH catalyzes the asymmetric reduction of PPA to D-PLA with low efficiency. To improve the biotransformation efficiency, the cofactor regeneration enzyme GDH was introduced and co-expressed with LDH. Among the two recombinant strains with the two enzymes in different MCSs of the vector, E. coli-gdh-ldh showed a higher PPA reduction activity. After optimizing the expression and reaction conditions, asymmetric biocatalytic reduction of PPA for the production of D-PLA was performed with in situ NADH regeneration using the GDH intrinsic in the whole-cell biocatalyst and glucose as a co-substrate, which completely converted 50 g l−1 PPA, yielding D-PLA with a space-time yield of 262.8 g l−1 day−1 and e.e. of over 99.5%. This work provides a valuable whole-cell biocatalysis method for efficient D-PLA synthesis with great potential for industrial application.
References
Choudhury S, Baeg JO, Park NJ, Yadav RK (2014) A solar light-driven, eco-friendly protocol for highly enantioselective synthesis of chiral alcohols via photocatalytic/biocatalytic cascades. Green Chem 16(9):4389–4400
Cui YH, Wei P, Peng F, Zong MH, Lou WY (2018) Efficient biocatalytic stereoselective reduction of methyl acetoacetate catalyzed by whole cells of engineered E. coli. RSC Adv 8(18):9970–9978
Dallagnol AM, Catalan CAN, Mercado MI, Font de Valdez G, Rollan GC (2011) Effect of biosynthetic intermediates and citrate on the phenyllactic and hydroxyphenyllactic acids production by Lactobacillus plantarum CRL 778. J Appl Microbiol 111(6):1447–1455
Dieuleveux V, Van Der Pyl D, Chataud J, Gueguen M (1998a) Purification and characterization of anti-Listeria compounds produced by Geotrichum candidum. Appl Environ Microbiol 64(2):800–803
Dieuleveux V, Lemarinier S, Guéguen M (1998b) Antimicrobial spectrum and target site of D-3-phenyllactic acid. Int J Food Microbiol 40(3):177–183
Ebdrup S, Ingrid P, Rasmussen HB, Heinz-Josef D, Anette FJ, Mortensen SB, Jan F, Lone P, Lars N, Per S (2003) Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor alpha/gamma agonist ragaglitazar. J Med Chem 46(8):1306–1317
Fujita T, Hieu Duc N, Ito T, Zhou S, Osada L, Tateyama S, Kaneko T, Takaya N (2013) Microbial monomers custom-synthesized to build true bio-derived aromatic polymers. Appl Microbiol Biotechnol 97(20):8887–8894
He SB, Wang ZS, Zou Y, Chen SF, Xu XP (2014) Purification and characterization of a novel carbonyl reductase involved in oxidoreduction of aromatic beta-amino ketones/alcohols. Process Biochem 49(7):1107–1112
Jia BL, Pu ZJ, Tang K, Jia XM, Kim KH, Liu XL, Jeon CO (2018) Catalytic, computational, and evolutionary analysis of the d-lactate dehydrogenases responsible for d-lactic acid production in lactic acid bacteria. J Agric Food Chem 66(31):8371–8381
Kawaguchi H, Uematsu K, Ogino C, Teramura H, Niimi-Nakamura S, Tsuge Y, Hasunuma T, Oinuma KI, Takaya N, Kondo A (2014) Simultaneous saccharification and fermentation of kraft pulp by recombinant Escherichia coli for phenyllactic acid production. Biochem Eng J 88(6):188–194
Kawaguchi H, Ogino C, Kondo A (2017) Microbial conversion of biomass into bio-based polymers. Bioresour Technol 245:1664–1673
Lavermicocca P, Valerio FA (2003) Antifungal activity of phenyllactic acid against molds isolated from bakery products. Appl Environ Microbiol 69(1):634–640
Lavermicocca P, Valerio F, Evidente A, Lazzaroni S, Corsetti A, Gobbetti M (2000) Purification and characterization of novel antifungal compounds from the sourdough Lactobacillus plantarum strain 21B. Appl Environ Microbiol 66(9):4084–4090
Li L, Shin SY, Lee KW, Han NS (2014) Production of natural antimicrobial compound d-phenyllactic acid using Leuconostoc mesenteroides ATCC 8293 whole cells involving highly active d-lactate dehydrogenase. Lett Appl Microbiol 59(4):404–411
Mu WM, Yu SH, Zhu LJ, Zhang T, Jiang B (2012a) Recent research on 3-phenyllactic acid, a broad-spectrum antimicrobial compound. Appl Microbiol Biotechnol 95(5):1155–1163
Mu WM, Yu SH, Zhu LJ, Jiang B, Zhang T (2012b) Production of 3-phenyllactic acid and 4-hydroxyphenyllactic acid by Pediococcus acidilactici DSM 20284 fermentation. Eur Food Res Technol 235(3):581–585
Ni Y, Xu JH (2012) Biocatalytic ketone reduction: a green and efficient access to enantiopure alcohols. Biotechnol Adv 30(6):1279–1288
Schnürer J, Magnusson J (2005) Antifungal lactic acid bacteria as biopreservatives. Trends Food Sci Technol 16(1):70–78
Tang YP, Zhang GM, Wang Z, Liu D, Zhang LL, Zhou YF, Huang J, Yu FM, Yang ZS, Ding GF (2018) Efficient synthesis of a (S)-fluoxetine intermediate using carbonyl reductase coupled with glucose dehydrogenase. Bioresour Technol 250:457–463
Urban FJ, Moore BS (1992) Synthesis of optically-active 2-benzyldihydrobenzopyrans for the hypoglycemic agent englitazone. J Heterocycl Chem 29(2):431–438
Valerio F, Lavermicocca P, Pascale M, Visconti A (2004) Production of phenyllactic acid by lactic acid bacteria: an approach to the selection of strains contributing to food quality and preservation. FEMS Microbiol Lett 233(2):289–295
Wang JP, Yoo JS, Lee JH, Lee JH (2009) Effects of phenyllactic acid on production performance, egg quality parameters, and blood characteristics in laying hens. J Appl Poult Res 18(2):203–209
Wang JP, Lee JH, Yoo JS, Cho JH, Kim HJ, Kim IH (2010) Effects of phenyllactic acid on growth performance, intestinal microbiota, relative organ weight, blood characteristics, and meat quality of broiler chicks. Poultry Sci 89(7):1549–1555
Wang M, Zhu LF, Xu XL, Wang LM, Yin RC, Yu B (2016) Efficient production of enantiomerically pure d-phenyllactate from phenylpyruvate by structure-guided design of an engineered d-lactate dehydrogenase. Appl Microbiol Biot 100(17):7471–7478
Wang YJ, Shen W, Luo X, Liu ZQ, Zheng YG (2017a) Enhanced diastereoselective synthesis of t-Butyl 6-cyano-(3R,5R)-dihydroxyhexanoate by using aldo-keto reductase and glucose dehydrogenase co-producing engineered Escherichia coli. Biotechnol Prog 33(5):1235–1242
Wang YJ, Ying BB, Chen M, Shen W, Liu ZQ, Zheng YG (2017b) An NADPH-dependent Lactobacillus composti short-chain dehydrogenase/reductase: characterization and application to (R)-1-phenylethanol synthesis. World J Microbiol Biotechnol 33(7):144
Wang YJ, Ying BB, Shen W, Zheng RC, Zheng YG (2017c) Rational design of Kluyveromyces marxianus ZJB14056 aldo–keto reductase KmAKR to enhance diastereoselectivity and activity. Enzyme Microb Technol 107:32–40
Wang FT, Wu HH, Jin PP, Sun ZL, Liu F, Du LH, Wang DY, Xu WM (2018) Antimicrobial activity of phenyllactic acid against Enterococcus faecalis and its effect on cell membrane. Foodborne Pathog Dis 15(10):645–652
Weckwerth W, Miyamoto K, Iinuma K, Krause M, Glinski M, Storm T, Bonse G, Kleinkauf H, Zocher R (2000) Biosynthesis of PF1022A and related cyclooctadepsipeptides. J Biol Chem 275(23):17909–17915
Wichmann R, Vasic-Racki D (2005) Cofactor regeneration at the lab scale. Adv Biochem Eng Biotechnol 92:225–260
Xu ZN, Jing KJ, Liu Y, Cen PL (2007) High-level expression of recombinant glucose dehydrogenase and its application in NADPH regeneration. J Ind Microbiol Biotechnol 34(1):83–90
Xu GC, Zhang LL, Ni Y (2016) Enzymatic preparation of D-phenyllactic acid at high space-time yield with a novel phenylpyruvate reductase identified from Lactobacillus sp CGMCC 9967. J Biotechnol 222:29–37
Yu SH, Zhou C, Zhang T, Jiang B, Mu WM (2015) Short communication: 3-Phenyllactic acid production in milk by Pediococcus pentosaceus SK25 during laboratory fermentation process. J Dairy Sci 98(2):813–817
Yu H, Qiu S, Chen F, Cheng YN, Wang YJ, Zheng YG (2019) Improving the catalytic efficiency of aldo-keto reductase KmAKR towards t-butyl 6-cyano-(3R,5R)-dihydroxyhexanoate via semi-rational design. Bioorg Chem 90:103018
Zhang XQ, Zhang SL, Shi Y, Shen FD, Wang HK (2014) A new high phenyl lactic acid-yielding Lactobacillus plantarum IMAU10124 and a comparative analysis of lactate dehydrogenase gene. FEMS Microbiol Lett 356(1):89–96
Zheng ZJ, Ma CQ, Gao C, Li FS, Qin JY, Zhang HW, Wang K, Xu P (2011) Efficient conversion of phenylpyruvic acid to phenyllactic acid by using whole cells of Bacillus coagulans SDM. PLoS One 6(4):e19030
Zheng ZJ, Sheng BB, Gao C, Zhang HW, Qin T, Ma CQ, Xu P (2013) Highly stereoselective biosynthesis of (R)-alpha-hydroxy carboxylic acids through rationally re-designed mutation of d-lactate dehydrogenase. Sci Rep 3:3401
Zhou XT, Zhang RZ, Xu Y, Liang HB, Jiang JW, Xiao R (2015) Coupled (R)-carbonyl reductase and glucose dehydrogenase catalyzes (R)-1-phenyl-1,2-ethanediol biosynthesis with excellent stereochemical selectivity. Process Biochem 50(11):1807–1813
Zhu YB, Hu FG, Zhu YY, Wang LM, Qi B (2015) Enhancement of phenyllactic acid biosynthesis by recognition site replacement of D-lactate dehydrogenase from Lactobacillus pentosus. Biotechnol Lett 37(6):1233–1241
Zhu YB, Jiang ZY, Chen JH, Xu JM, Wang LM, Qi B (2017) Fusion of d-lactate dehydrogenase and formate dehydrogenase for increasing production of (R)-3-phenyllactic acid in recombinant Escherichia coli BL21 (DE3). J Biobased Mater Bioenergy 11(4):372–378
Acknowledgements
This work was financially supported by the Natural Science Foundation for Young Scholars of Zhejiang Province (LQ19B060003) and the Scientific Research Foundation of Taizhou University (2017PY035).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all the authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Luo, X., Zhang, Y., Yin, L. et al. Efficient synthesis of d-phenyllactic acid by a whole-cell biocatalyst co-expressing glucose dehydrogenase and a novel d-lactate dehydrogenase from Lactobacillus rossiae. 3 Biotech 10, 14 (2020). https://doi.org/10.1007/s13205-019-2003-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-019-2003-2