
Abstract
Successful bioremediation of oil pollution is based on a comprehensive understanding of the in situ physicochemical conditions and indigenous microbial communities as well as the interaction between microorganisms and geochemical variables. Nineteen oil-contaminated soil samples and five uncontaminated controls were taken from six major oilfields across different geoclimatic regions in China to investigate the spatial distribution of the microbial ecosystem. Microbial community analysis revealed remarkable variation in microbial diversity between oil-contaminated soils taken from different oilfields. Canonical correspondence analysis (CCA) further demonstrated that a suite of in situ geochemical parameters, including soil moisture and sulfate concentrations, were among the factors that influenced the overall microbial community structure and composition. Phylogenetic analysis indicated that the vast majority of sequences were related to the genera Arthrobacter, Dietzia, Pseudomonas, Rhodococcus, and Marinobacter, many of which contain known oil-degrading or oil-emulsifying species. Remarkably, a number of archaeal genera including Halalkalicoccus, Natronomonas, Haloterrigena, and Natrinema were found in relatively high abundance in some of the oil-contaminated soil samples, indicating that these Euryarchaeota may play an important ecological role in some oil-contaminated soils. This study offers a direct and reliable reference of the diversity of the microbial community in various oil-contaminated soils and may influence strategies for in situ bioremediation of oil pollution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The petroleum industry in China has grown rapidly with the increasing population and growing demand for raw oil as an energy source. The resulting exploration, production, transportation, and storage of raw oil have introduced extensive oil contamination to terrestrial ecosystems throughout China. Therefore, there is an urgent need to manage and remediate these oil-contaminated ecosystems. But, traditional mitigation technologies such as chemical and physical treatments are difficult to apply due to the inaccessibility and high volumes of the released oils. A greener and more sustainable method is to utilize indigenous microorganisms that are capable of oil degradation. Oil-degrading microorganisms are ubiquitously distributed in the environment, and successful bioremediation of terrestrial and marine oil spills has been reported in a number of studies (Atlas 1995; Brandt et al. 2002; Hazen et al. 2010; Kostka et al. 2011; Pritchard and Costa 1991; Song et al. 1990; Sun and Cupples 2012; Sun et al. 2010; Swannell et al. 1996). Although oil bioremediation appears to be promising, a comprehensive analysis of the in situ microbial degradation of petroleum components is still needed. Culture dependent and independent methods have been extensively applied in studies assessing the microbial degradation of petroleum components (Grassia et al. 1996; Gray et al. 2010, 2011; Liang et al. 2010; Orphan et al. 2000; Ren et al. 2011; Viñas et al. 2005). Nevertheless, characterization of indigenous microbial communities is still challenging due to complex in situ physicochemical conditions. Additionally, traditional molecular techniques are often restricted to the analysis of a small number of clones and thus underestimate the indigenous microbial community. The emergence of advanced molecular techniques, especially high-throughput techniques, provides an opportunity to better address microbial diversity in oil-contaminated environments. As an example, GeoChip, a high-throughput functional gene array, was used to evaluate the microbial functional genes involved in oil contaminant degradation (Liang et al. 2010). Pyrosequencing is another high-throughput method that can provide high numbers of DNA reads (Margulies et al. 2005), making it possible to detect microbes of low abundance in the environment. Pyrosequencing has been used to characterize in situ microbial communities in oilfields (Bell et al. 2011; Dos Santos et al. 2011). For instance, microbial communities in production and injection waters from Algerian oilfields were described. Many unclassified bacterial and archaeal sequences have been found in oil-contaminated sites through pyrosequencing (Lenchi et al. 2013). Bacterial communities were also investigated in permafrost soils along the China-Russia crude oil pipeline by pyrosequencing. The results revealed an immensely higher microbial diversity than anticipated (Yang et al. 2012). These pyrosequencing-based studies demonstrated that high-throughput sequencing provide a more complete picture of indigenous microbial communities than traditional molecular techniques.
Drilling and production activities associated with the petroleum industry result in the contamination of nearby surface and groundwater (Kharaka et al. 2005; O'Rourke and Connolly 2003; Osborn et al. 2011). This contamination can include various petroleum hydrocarbons (e.g., benzene, toluene, ethylbenzene, and xylene (BTEX) and polycyclic aromatic hydrocarbons (PAHs)), metals, salts, and other contaminants. The accumulation of substantial amounts of these materials in soil and groundwater can lead to severe environmental, economic, and health consequences (Olsgard and Gray 1995). A comprehensive understanding of the different microbial communities and how their activities can be enhanced is important for optimizing conditions for biodegradation and identifying amendments that can stimulate in situ oil bioremediation. Although diverse bacteria capable of degrading petroleum components have been isolated and characterized, the vast majority of oil-degrading bacteria remain undiscovered due to the limitations of culture-dependent and culture-independent techniques. In this study, we collected samples of oil-contaminated soils from six large oilfields across different geoclimatic regions within China. High-throughput sequencing of 16S ribosomal RNA (rRNA) was utilized to profile indigenous microbial communities in each soil sample. This study aims to provide novel data sets that can be used in conjunction with contamination profiles and geochemical parameters to better understand microbial processes at contaminated sites.
Materials and methods
Site information and sampling strategy
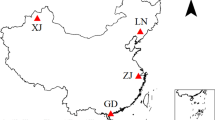
Soil samples were obtained from oil-contaminated sites at six different oilfields (fields in areas where oil is extracted), each located in different geographical regions of China: Daqing (DQ) and Jilin (JL) oilfields in northeast China, Changqing (CQ) and Xinjiang (XJ) oilfields in northwest China, Shengli (DY) oilfield in the Yellow River area in north China, and Jiangsu (JS) oilfield in east China (Fig. 1). These regions vary in climate. DQ (46° 44′ N, 124° 55′ E) and JL (41° 21′ N, 124° 47′ E) both have a temperate continental monsoon climate. XJ (41° 10′ N, 83° 35′ E) has a temperate continental arid climate. CQ (36° 32′ N, 107° 20′ E) has a temperate continental monsoon climate. JS (32° 57′ N, 119° 02′ E) has a subtropical humid monsoon climate, and DY (37° 29′ N, 118° 15′ E) has a warm temperate continental semi-humid monsoon climate. Contaminated soils were collected 2–10 cm beneath the surface, adjacent to crude oil pumping wells in which contamination had occurred for decades. Uncontaminated control soil samples were collected from undisturbed pristine soils. Sampling sites for the same oilfield were located within an area of 0.8 km2 (a circle with a radius of 300 m). The sample site information and sample names are summarized in Table S1.

Map of six large oilfields in China: Daqing (DQ) and Jilin (JL) in the northeast, Xinjiang (XJ) and Changqing (CQ) in the northwest, Dongying (DY) in the north, and Jiangsu (JS) in the east
Chemical analysis
Soil samples from each site were homogenized by thorough mixing and then stored in a refrigerator at 4 °C until further processing. Then, they were air-dried for 48 h and passed through a 2-mm sieve to remove leaves, plant roots, and gravel. Next, 5 g of dry soil was placed into a 150 mL Erlenmeyer flask and mixed with 25 mL of distilled water (1:5 soil/water ratios). The mixture was shaken for 5 min and then left to equilibrate for 20 min. The pH was measured using a calibrated HACH HQ30d pH meter (HACH, Loveland, USA). The supernatant was filtered through a 0.45 μm filter membrane. Soil cation (K+, Ca2+, Na+, NH4 +, and Mg2+) and anion (F−, SO4 2−, NO3 −, and Cl−) concentrations were determined by ion chromatography (DIONEX ICS-1500, Sunnyvale, USA). Soil total organic carbon (TOC) and total nitrogen (TN) were determined by an elemental analyzer (Vario EL/micro cube, Hanau, Germany). Soil moisture was determined by drying a 5 g soil sample at 105 °C for 24 h to achieve a constant weight.
DNA extraction, PCR amplification, and Illumina sequencing
Total genomic DNA was extracted directly from soil samples using a FastDNA® spin kit (MP Bio, Santa Ana, USA) following the manufacturer’s protocol. The DNA concentration was then determined using a Nanodrop ND-2000 UV-Vis spectrophotometer (Thermo Scientific, Wilmington, USA). DNA was stored at −80 °C until further analysis. The primer set F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACVSGGGTATCTAAT-3′) was designed to amplify the V4 hypervariable region from the total genomic DNA. Sequencing libraries were prepared from 200 ng of the resulting amplicon for each sample. Sequencing libraries were generated using the Illumina Truseq™ DNA Sample Preparation Kit (Illumina, San Diego, USA) following the manufacturer’s recommendations. Agarose gel electrophoresis was performed (120 V, 40 min, 1.5 % agarose gel) to select DNA fragments. After purification through a spin column (QIAGEN, Dusseldorf, Germany), DNA fragments containing adapter molecules on each end were selectively enriched by PCR (10 cycles) using the Illumina PCR Primer Cocktail. 16S rRNA tag-encoded high-throughput sequencing was carried out on the Illumina MiSeq platform by Novogene (Beijing, China). The reads were deposited into the NCBI short reads archive database (SRR1562515). Pairs of reads from the original DNA fragments were merged based on the method described previously (Magoč and Salzberg 2011). Sequencing reads were assigned to each sample according to the unique barcode assigned to each sample during preparation. The sequence were analyzed via the Quantitative Insights Into Microbial Ecology (QIIME) software package and UPARSE pipeline (Caporaso et al. 2010). The reads were first filtered through QIIME quality filters using the default settings for Illumina processing. Then, the UPARSE pipeline was used to assign operational taxonomic units (OTUs) at a minimum cutoff of 97 % similarity. For each OTU, a representative sequence was selected and used to assign taxonomic composition by using the Ribosomal Database Project (RDP) classifier (Wang et al. 2007). The Chao 1, Shannon, and the Simpson indexes were determined for 11 libraries to estimate species richness as described previously (Schloss et al. 2009).
Statistical analysis
Canonical correspondence analysis (CCA) was performed to identify the chemical properties having the most significant influence on microbial communities. Physicochemical parameters with significant correlations were examined by a Monte Carlo permutation. The triplot was generated by CANOCO 4.5 (Biometrics Wageningen, The Netherlands), and the figures were generated by CanoDraw 4.0 (Biometrics Wageningen, The Netherlands). Geochemical parameters and relative abundances of dominant genera and phyla were used for cluster analysis. The “clustsig” package in R was used to perform the cluster analysis. The unweighted pair group method with arithmetic mean (UPGMA) was used to compare the physicochemical parameters from different samples. Bray-Curtis was used to compare the dominant genera from different samples. The open source software Cytoscape 2.816 was employed to visualize the 38 most abundant genera based on their relative abundances.
Results
Physicochemical characterization of the samples
The geological characteristics of the sampling sites are reported in Table S1. The physicochemical properties of the soil are reported in Table 1. A number of environmental parameters were measured in all of the soil samples, including the cation and anion concentrations. In addition, pH, moisture, TOC, and TN were also measured in all of the samples. Most of the samples were alkaline, with pHs greater than 7, except one sample from the JS oilfield (JS4, pH = 6.01). All of the samples from the XJ oilfield contained high concentrations of Cl−1, SO4 2−, and Na+. Two out of four samples from the DY oilfield contained high concentrations of Cl−1 and Na+. TOC concentrations varied across different oil-contaminated samples. For instance, TOC was high in all contaminated samples from the CQ, JS, and DY oilfields but moderate in those from the DQ, XJ, and JL oilfields. This difference may be attributed to the presence of volatile contaminants in these soils. It is notable that TOC in the unpolluted control samples of the CQ, JS, and DY oilfields was much lower than their contaminated counterparts, suggesting that TOC could be used as an indicator of oil pollutant levels. The soil geochemical profiles were grouped based on UPGMA cluster analysis. Specifically, TOC, an indicator of the oil pollutant level, along with a suite of other important geochemical parameters that could shape the microbial community composition (e.g., soil moisture, sulfate, and nitrate) were used for cluster analysis. The geographic location may have been a factor for grouping (Fig. 2). Meanwhile, unpolluted control soil samples were distantly related to their contaminated counterparts, indicating that the oil pollutant levels might be another factor for grouping.

Cluster analyses showing the comparison between the geochemical parameters of the sampling sites
Diversity of microbial communities in oil-contaminated sites
The Illumina MiSeq platform generated 340,042 quality sequences from 24 soil samples. The RDP classifier was used to assign these sequences to different OTUs using a 3 % nucleotide cutoff. The number of OTUs and the Chao 1, Shannon, and Simpson indexes are summarized in Table 2. On the basis of the OTU number, one soil sample from the DY oilfields (DY2) had the richest diversity, followed by three samples from the DQ oilfields. Samples from the CQ oilfield exhibited relatively less richness with three of these having the lowest OTU numbers. The OTU numbers ranged from 949 (CQ2) to 2516 (DY2) in different samples. The Chao 1, Shannon, and Simpson indexes of diversity were also determined to evaluate the biodiversity and phylotype richness in these samples (Table 2). The patterns of the Chao 1, Shannon, and Simpson values were very similar to the OTU numbers. The wide variation in these indexes shows that microbial communities in different oilfields, and even between samples in the same oilfields, were different.
Taxonomic profiles
The sequences in each sample were assigned to different taxonomic levels (from phylum to genus) based on the RDP classifier. Figure 3 summarizes the relative abundances of the phylum level for each sample. A total of 28 archaeal and bacterial phyla were identified in all samples. No single phylum was dominant across all samples; instead, various phyla dominated in different samples. Actinobacteria (15.7 % of total reads) and Acidobacteria (10.8 % of total reads) were the most predominant phyla in eight samples across different oilfields while Proteobacteria (19.5 % of total reads) was the most predominant phylum in five samples from four oilfields. The other dominant phyla were Firmicutes, Acidobacteria, Verrucomicrobia, Planctomycetes, and Bacteroidetes (Table S2). As reported in a previous study, the 515f/806r primer pair is nearly universal to archaea and bacteria (Walters et al. 2011). In accordance with this observation, the sequences assigned to archaeal phyla were also detected in the current study. Among the archaeal phyla, Euryarchaeota was the most dominant phylum in three samples (JS1, JS2, and CQ4) and was highly enriched in JS3 (11 %). However, Euryarchaeota was found at low levels (<1 %) in other samples. Crenarchaeota occurred at high levels (relative abundance >1 %) in only a small number of samples. The dominance of archaeal phyla in some samples indicated that archaea might play an important ecological role in some oilfields. It is noteworthy that 21 % of the analyzed sequences were not assigned to any known phyla, indicating that the surface soil harbors many not-yet-described bacterial phyla.

Taxonomic classification of the bacterial and archaeal reads retrieved from 24 samples at the phylum level from 16S rRNA Illumina sequencing
Core genera
The dominant phyla were further compared at the genus level to further uncover the differences between these microbial communities. A total of 587 different genera were identified in 24 samples. Different genera showed relatively high abundances (relative abundance >1 %) in different soil samples. Among these enriched genera, several were dominant in some samples, with relative abundances above 10 %. Arthrobacter showed a relative abundance of 22.5 % at site CQ1 and 8.7 % at site CQ5. However, in other samples taken from the same oilfield (CQ3 and CQ4), this genus only accounted for 0.7 and 1.1 %. Dietzia also exhibited very high levels across the CQ oilfield, accounting for 10.7 % at site CQ1 and 53 % at site CQ5. Additionally, archaeal phylotypes were highly abundant in some soil samples. For instance, Halalkalicoccus showed high relative abundances at site JS1 (15.7 %) and JS2 (3.4 %). Interestingly, Halalkalicoccus also demonstrated a high relative abundance at site CQ4 (9.5 %) but not in any other samples from the CQ oilfield. Marinobacter was dominant in two samples taken from different oilfields: JS3 (20.5 %) and XJ12 (20.6 %). Other abundant genera, such as Rhodococcus, Pseudomonas, Mycobacterium, Opitutus, and Sphingomonas, were occasionally dominant in one or more soil samples. The relative abundances of the genera in all of the samples are summarized in Table S3. Highly abundant genera were selected from each sample (a total of 38 genera for the 24 samples) and grouped based on their abundances using Bray-Curtis cluster analysis (Fig. 4). At the genus level, grouping patterns were not related to the geographical location, indicating that different microbial communities may have evolved in adjacent sampling sites. Another observation is that microbial communities of the uncontaminated control samples were only distantly related to their contaminated counterparts. Briefly, five groups were observed based on the cluster analysis.

Cluster analyses of the dominant genera in the different samples based on Bray-Curtis analysis. Different colors indicate groups that are significantly different (p < 0.05)
Further, we also applied a profile clustering network analysis (Fig. 5) to obtain a deeper insight into the differences of the microbial community in the oil-contaminated soils at the genus level. Representative microbial communities were selected from each group based on Bray-Curtis cluster analysis (Fig. 4). These five groups included oil-contaminated soils from all six oilfields. The network analysis generated by a Cytoscape network showed the most abundant 38 genera and highlighted the relative distribution and abundances. Arthrobacter was ubiquitous and was the most abundant genus in many groups, including group 1 (CQ1 and DQ3), group 2 (DQ1, JL1, and DY3), and group 3 (JS4 and JL3). It was also abundant in group 4 (DY1 and XJ7) and group 5 (JS1, CQ4, and XJ12). Dietzia was the most abundant genus in group 1, but it was also abundant in group 3 and group 5. Several genera, including Halalkalicoccus, Natronomonas, Marinobacter, Halosarcina, and Alcanivorax, were more abundant in group 5 but were less abundant in other groups, suggesting that group 5 may harbor unique microbial community.

Profile clustering Cytoscape network visualizing the 38 most abundant genera among the five groups. A comparative node (blue) indicates the size of a node that would represent the relative abundance in a group. For example, standard node 1 shows that this size would represent 1 % of the corresponding genus in a group. Standard node 15 shows that this size would represent 15 % of the corresponding genus in a group
Relationship between the microbial community and the environment
The results of CCA (Fig. 6) showed the relationship between environmental variables and microbial communities. CCA ordination showed that specific geochemical conditions shaped the variations in microbial community composition. The CCA axis 1 and soil moisture had a strong positive correlation, while sulfate had a negative correlation with CCA axis 1. The relatively small magnitude of the TOC, TN, nitrate, and pH vectors indicates that these parameters were not as strongly correlated to community composition as were moisture and sulfate. Some bacteria, notably clustered within the genera Pseudomonas, Rhodococcus, Arthrobacter, Acinetobacter, Alkanindiges, and Opitutus, were positively correlated with TOC. Remarkably, many of these genera contain species that are able to degrade petroleum components (for details, please see “Discussion”). This indicated that TOC may favor the growth of petroleum-degrading bacteria. Many archaea including Halalkalicoccus, Natronomonas, Halosarcina, and Natronococcus were positively correlated with soil moisture, suggesting that these archaea may favor environments with more soil moisture.

Ordination diagrams from canonical correspondence analysis (CCA) of dominant genera abundances and geochemical parameters. Response variables of overall microbial community (a) and specific phylotypes (b). Red arrows indicate the direction and magnitude of geochemical parameters associated with bacterial community structures. Each sample was represented by colored circles according to oilfields. Environmental abbreviations: TN total nitrogen, TOC total organic carbon. Bacteria abbreviations: Polaromo Polaromonas, Rhodo Rhodococcus, Natronoc Natronococcus, Acineto Acinetobacter, Alkan Alkanindiges, Sphin Sphingomonas, Arthro Arthrobacter, Haloterr Haloterrigena, Alcan Alcanivorax, Pseudo Pseudomonas, Natronom Natronomonas, Halakal Halalkalicoccus, Halomona Halomonas, Halosarc Halosarcina, Marinoba Marinobacter, Halobac Halobacter
Discussion
Due to oxygen penetration and the presence of sulfate and nitrate, the top soil and subsoil may harbor dynamic redox conditions. This environment may thus be a good model for investigating the response of microbial communities to oil contamination under different terminal electron-accepting conditions. The microbial communities varied significantly between different samples, indicating that the microbial communities may evolve in response to oil contamination and other in situ geochemical profiles. Consistently, the physicochemical conditions across different sites varied significantly, displaying a wide range of ionic concentrations and TOC contents. Such variations inevitably shaped the indigenous microbial communities, leading to the enrichment of those microorganisms best adapted to the geochemical conditions.
Microbial community composition
Our current knowledge of oil-degrading bacteria is still scarce because the vast majority of oil-degrading bacteria remain uncultured. Therefore, we applied high-throughput sequencing as a broad coverage approach to acquire knowledge of these uncultured microbial phylotypes. Relatively high proportions of the sequences in our samples, ranging from 5.1 to 47 % with an average of 20.6 %, could not be classified within any known phylum. In a recent study, a similar proportion (average 21 %) of sequences from diesel-contaminated soils, as derived from pyrosequencing (Sutton et al. 2013), were identified as unclassified bacteria and archaea. This high proportion of unclassified sequences suggested that a vast number of microorganisms in these environments belonged to unrecognized or novel bacterial and archaeal species. The evidence is not yet sufficient to say whether their presence is due to the biogeographical distribution or functional selection by oil contamination.
On the phylum level, a broad variety of different phyla were dominant across the 24 samples. Actinobacteria and Acidobacteria were identified as the most dominant phyla in 16 samples while Proteobacteria was identified as the most dominant phylum in only five soil samples. This observation contradicts many previous investigations in which Proteobacteria were frequently identified as the most abundant phylum in oil-contaminated environments. For instance, Proteobacteria was identified as the most dominant phylum in Algerian oilfield injection waters (Lenchi et al. 2013), an oil-contaminated mangrove (Dos Santos et al. 2011), and permafrost soils along the China-Russia crude oil pipeline (Yang et al. 2012). In this study, Proteobacteria exhibited a relatively low relative abundance in some samples, including sites CQ5, JS1, JS2, JS4, XJ6, and XJ7. In these samples, Proteobacteria accounted for less than 12 % of the total reads. Actinobacteria and Acidobacteria have been detected in high abundance in long-term diesel-contaminated soil (Sutton et al. 2013) and other soils contaminated with aliphatic or aromatic compounds (Allen et al. 2007; Kasai et al. 2005; MacNaughton et al. 1999), suggesting that these phyla contain members important for in situ oil bioremediation.
Occurrence of known oil-degrading bacteria
Analyses based on the genus level classification of the sequences allow for the further verification of functional evolution and identification of potential oil-degrading microorganisms in a community. Most of the dominant genera identified here could be further identified as aerobes, suggesting that oxygen was prevailed in these open microbial systems and facilitated the proliferation of aerobes. Although no microorganisms were isolated in this study, it may be possible to predict their metabolic capabilities from closely related cultivated isolates or uncultured clones.
A wide spectrum of genera was found at relatively high abundances in these oil-contaminated samples. It is fair to hypothesize that the dominance is correlated with oil biodegradation. Specifically, long-term oil contamination may impose a selective pressure on the indigenous microbial communities, favoring the growth of certain oil-degrading microorganisms. Consistent with this hypothesis, many of the highly abundant genera contain known oil-degrading species. Among these enriched genera, members of genus Arthrobacter were detected at high levels (>1 % relative abundance) in eight samples. As demonstrated from the Cytoscape network (Fig. 5), Arthrobacter were ubiquitously distributed in all of the groups. Arthrobacter species were highly enriched (>5 % relative abundance) in three samples taken from three different oilfields (Table S3). The ubiquity of Arthrobacter species across all of the oilfields suggested that these bacteria could be very important for oil degradation in contaminated soils. Arthrobacter spp. have been reported to degrade various hydrocarbons, including aromatic hydrocarbons (Stevenson 1967) and PAHs (Puškárová et al. 2013). Additionally, Arthrobacter spp. were noted for their capability to produce emulsifying agents (Rosenberg et al. 1979) and biosurfactants (Morikawa et al. 1993), which could be used as oil removers. For example, Arthrobacter RAG-1 produced an extracellular nondialyzable emulsifying agent when grown on hexadecane, ethanol, or acetate media (Rosenberg et al. 1979), and Arthrobacter sp. strain MIS38 produces a biosurfactant, termed arthrofactin (Morikawa et al. 1993). Two other phylotypes belonging to the genera Pseudomonas and Rhodococcus showed high relative abundances in many samples. These two genera also contain a number of species that are capable of producing biosurfactants (Bicca et al. 1999; Iqbal et al. 1995; Ivshina et al. 1998; Patel and Desai 1997). The presence of emulsifiers has been shown to enhance hydrocarbon degradation in the environment (Atlas 1993; Atlas and Bartha 1992). These emulsifier-producing bacteria hold great potential for oil remediation: During petroleum bioremediation, microorganisms utilize long and short chain hydrocarbons and numerous aromatic compounds as energy and carbon sources. However, all of these compounds have low solubility in water, making them less accessible to microorganisms. The emulsifying agents produced by these bacteria can improve the solubility of crude oil, making the substrates more accessible and enhancing the biodegradation potential of the crude oil.
Dietzia was at relatively high abundances in many soil samples including two samples from the CQ, one sample from the JS, and all of the samples from the XJ oilfield. Members of Dietzia isolated from other oil-contaminated areas have been shown to degrade hydrocarbons (Alonso-Gutiérrez et al. 2011; Bødtker et al. 2009; Plakunov et al. 2008; von der Weid et al. 2007; Wang et al. 2011a). Interestingly, Dietzia psychralcaliphila sp. were isolated from cold water (6 °C) and identified as psychrophilic hydrocarbon-degrading bacteria (Yumoto et al. 2002), while Dietzia cinnamea was isolated from tropical soil contaminated with crude oil (von der Weid et al. 2007), suggesting that Dietzia can survive under a wide range of temperatures. This could explain why Dietzia spp. were dominant across different geoclimatic regions in this study. Sequences related to Marinobacter accounted for more than 20 % in two soil samples (JS3 and XJ12) but less than 1 % in all other samples. Marinobacter is a widely distributed bacterium that is frequently isolated from costal marine sediments and seawater (Bowman et al. 1997; Pinhassi et al. 1997). Marinobacter species are important hydrocarbon-degrading bacteria that display versatility in their metabolic capabilities. They are capable of using a wide range of carbon sources including petroleum components, especially in marine environments (Al-Awadhi et al. 2007; Harwati et al. 2007; Yakimov et al. 2007). The presence of Marinobacter in the JS oilfield might be attributed to the location of the sampling site, which is close to a coastal area. However, the enrichment of Marinobacter in the XJ oilfield in inland China suggested that Marinobacter may not be limited to marine environments but may also inhabit terrestrial environments. Opitutus species demonstrated high abundances in five samples from three different oilfields. The enrichment of Opitutus is noteworthy because Opitutus spp. were the only obligate anaerobic bacteria widely enriched in this study. Unlike the oil-degrading bacteria mentioned above, Opitutus were seldom correlated with crude oil bioremediation. The only potential link between Opitutus and biodegradation is from a study where Opitutus was identified in a microbial system contaminated by the artificial release of ethanol, benzene, and toluene (Ma et al. 2013). In that study, Opitutus exhibited a relatively high abundance after 10 months of exposure to the spill. The presence and dominance of Opitutus in the current study are ambiguous and need further investigation.
Enrichment of halophilic archaea
An important observation from this study is that a number of archaeal phylotypes were found at high levels in some contaminated soils. Archaeal phylotypes were especially enriched in soil samples taken from the JS oilfield. For instance, 67.9 and 29.3 % of the total reads from JS1 and JS2, respectively, were associated with the phylum Euryarchaeota. However, only 0.4 % of the total reads were associated with Euryarchaeota in uncontaminated samples in this region (JS4), suggesting that oil contamination may stimulate the enrichment of such Euryarchaeota-related phylotypes. In addition, Euryarchaeota has been detected in some other oil-contaminated environments. For instance, Euryarchaeota have been identified as dominant culturable members in low-temperature biodegraded oil reservoir (Grabowski et al. 2005). Euryarchaeota have been detected as the most predominant phylum in oil sands tailing ponds (An et al. 2013). All these observations indicated a potential link between members of Euryarchaeota and oil biodegradation. In this study, Euryarchaeota-related sequences were associated with the genera Halalkalicoccus, Natronomonas, Haloterrigena, and Natrinema. These halophilic archaea occurred at high levels in all contaminated samples of the JS oilfield (JS1, JS2, and JS3) and site CQ4. Methanogenic archaea have been frequently detected in oil-contaminated aquifers and soils (Nilsen and Torsvik 1996; Ren et al. 2011). In this study, however, methanogens were only found in very low abundance in each sample. The most dominant archaeal phylotype was Halalkalicoccus, which are alkaliphilic halophilic archaea, dominating in sites JS1 (15.7 %) and JS2 (3.4 %), and were also found at high levels in one sample from the CQ oilfield (CQ4 (9.5 %)). The dominance of Halalkalicoccus was concomitant with the dominance of other halophilic archaea, such as Natrinema, Natronomonas, and Haloterrigena, suggesting that certain in situ geochemical conditions, probably the alkaline soil in the JS oilfield, may stimulate the enrichment of such halophiles. Halophilic archaea have been linked to hydrocarbon degradation in many studies (Al-Mailem et al. 2010; Bertrand et al. 1990; Tapilatu et al. 2010). For instance, five extreme halophilic archaeal strains related to Haloarcula and Haloferax have been reported to degrade hydrocarbons in an uncontaminated hypersaline pond (Tapilatu et al. 2010). In another study, the halophilic genera Haloferax and Natronomonas were reported to play a role in the natural attenuation of a petroleum-contaminated saline-alkali soil (Wang et al. 2011b). Our observation of the high abundance of halophilic archaea in some of the oilfields provides additional evidence that halophilic archaea may play a central role in the natural attenuation of oxygen-rich petroleum-contaminated soil. Further investigations to explore their metabolic capabilities are underway, including the isolation of these halophilic archaea and stable isotope probing.
Correlation between the environmental parameters and bacterial community
Phylogenetic analyses suggest that microbial community composition is largely affected by environmental parameters but provide little information about the physicochemical conditions that might be responsible. Therefore, CCA was used to explore the relationship between microbial communities and environmental factors. Soil moisture appeared to be one of the most important environmental parameters and varied significantly in samples taken from different oilfields. This variation may be attributed to the different geoclimatic zones in which the oilfields are located. For example, soil samples taken from the XJ and CQ oilfields have comparatively lower soil moisture than other sites because these two oilfields are located in a continental arid and monsoon area, which sees low rainfall and high evaporation rate. Higher soil moisture may increase the bioavailability of certain oil components and other nutrients, which may then favor the enrichment of oil-degrading microorganisms. Notably, a number of archaeal phylotypes were positively correlated with soil moisture, indicating that these phylotypes also prefer more moist environments. Pearson correlation analysis confirmed the correlation between soil moisture and microbial diversity, as shown in Fig. S1. Alpha diversity, measured as number of phylotypes (OTUs at 97 % similarity), was positively correlated with soil moisture (r = 0.6094, P < 0.001). One environmental implication of these results is that the increasing soil moisture in oil-contaminated sites in arid regions could potentially facilitate the bioremediation of oil contaminants.
Sulfate was also strongly linked to microbial community variance. Sulfate can be utilized as an electron acceptor by a number of diversity of anaerobic bacteria such as sulfate reducing bacteria (SRB). In this study, we detected a wide diversity of SRB in sites from different oil-contaminated soils. For example, Desulfosporosinus, Geobacter, and Desulfitobacterium were detected in all of the samples. Although aerobic environments prevailed in the surface soil, some microenvironments may exist in the soil where oxygen is depleted due to consumption by aerobes and other physicochemical processes. This would create suitable habitats for these strictly anaerobic SRBs. In many oil-contaminated environments, biological sulfate reduction contributes to the utilization of approximately 70 % BTEX compounds (Kniemeyer et al. 2007). The sulfate reducing bacteria found in oilfields could use these hydrocarbons and organic acids as electron donors for sulfate reduction. Specifically, Desulfosporosinus, Geobacter, and Desulfitobacterium detected in this study have frequently been linked to the degradation of petroleum components (Fowler et al. 2014; Kunapuli et al. 2010; Sun and Cupples 2012; Sun et al. 2014a, b; Winderl et al. 2008, 2010). Given that a wide diversity of SRB were detected in these oilfields, it follows that sulfate could be used to stimulate the growth of hydrocarbon-degrading microorganisms and increase biodegradation rates. In addition, the relatively small magnitude of the pH, nitrate, TOC, and total nitrogen vectors indicates that these environmental parameters are not as strongly correlated to community composition as sulfate and soil moisture.
This study illustrates how the application of emerging molecular techniques can contribute substantially toward the advancement of our knowledge of indigenous microbial communities in oil-contaminated areas. Many different phylotypes exhibited high abundance in all six oilfields in contrast to uncontaminated soils, indicating that they may be functionally important in the bioremediation of hydrocarbons and the production of biosurfactants in raw oil-contaminated sites. The determination of the correlations between physicochemical conditions and microbial communities indicated that soil moisture and sulfate were the most significant factors that influenced the soils.
References
Al-Awadhi H, Sulaiman RH, Mahmoud HM, Radwan S (2007) Alkaliphilic and halophilic hydrocarbon-utilizing bacteria from Kuwaiti coasts of the Arabian Gulf. Appl Microbiol Biot 77(1):183–186
Allen JP, Atekwana EA, Atekwana EA, Duris JW, Werkema DD, Rossbach S (2007) The microbial community structure in petroleum-contaminated sediments corresponds to geophysical signatures. Appl Environ Microb 73(9):2860–2870
Al-Mailem D, Sorkhoh N, Al-Awadhi H, Eliyas M, Radwan S (2010) Biodegradation of crude oil and pure hydrocarbons by extreme halophilic archaea from hypersaline coasts of the Arabian Gulf. Extremophiles 14(3):321–328
Alonso-Gutiérrez J, Teramoto M, Yamazoe A, Harayama S, Figueras A, Novoa B (2011) Alkane‐degrading properties of Dietzia sp. H0B, a key player in the Prestige oil spill biodegradation (NW Spain). J Appl Microbiol 111(4):800–810
An D, Brown D, Chatterjee I, Dong X, Ramos-Padron E, Wilson S, Bordenave S, Caffrey SM, Gieg LM, Sensen CW (2013) Microbial community and potential functional gene diversity involved in anaerobic hydrocarbon degradation and methanogenesis in an oil sands tailings pond 1. Genome 56(10):612–618
Atlas R (1993) Bacteria and bioremediation of marine oil spills. Oceanus;(United States) 36(2)
Atlas RM (1995) Petroleum biodegradation and oil spill bioremediation. Mar Pollut Bull 31(4):178–182
Atlas RM, Bartha R (1992) Hydrocarbon biodegradation and oil spill bioremediation Advances in microbial ecology. Springer, pp 287-338
Bell TH, Yergeau E, Martineau C, Juck D, Whyte LG, Greer CW (2011) Identification of nitrogen-incorporating bacteria in petroleum-contaminated Arctic soils by using 15N DNA-based stable isotope probing and pyrosequencing. Appl Environ Microb 77(12):4163–4171
Bertrand J, Almallah M, Acquaviva M, Mille G (1990) Biodegradation of hydrocarbons by an extremely halophilic archaebacterium. Lett Appl Microbiol 11(5):260–263
Bicca FC, Fleck LC, Ayub MAZ (1999) Production of biosurfactant by hydrocarbon degrading Rhodococcus ruber and Rhodococcus erythropolis. Rev Microbiol 30(3):231–236
Bødtker G, Hvidsten IV, Barth T, Torsvik T (2009) Hydrocarbon degradation by Dietzia sp. A14101 isolated from an oil reservoir model column. Anton Leeuw Int J G 96(4):459–469
Bowman JP, McCammon SA, Brown MV, Nichols DS, McMeekin TA (1997) Diversity and association of psychrophilic bacteria in Antarctic sea ice. Appl Environ Microb 63(8):3068–3078
Brandt CA, Becker JM, Porta A (2002) Distribution of polycyclic aromatic hydrocarbons in soils and terrestrial biota after a spill of crude oil in Trecate, Italy. Environ Toxicol Chem 21(8):1638–1643
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Dos Santos HF, Cury JC, Do Carmo FL, Dos Santos AL, Tiedje J, Van Elsas JD, Rosado AS, Peixoto RS (2011) Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: bacterial proxies for oil pollution. PLoS One 6(3), e16943
Fowler SJ, Gutierrez‐Zamora ML, Manefield M, Gieg LM (2014) Identification of toluene degraders in a methanogenic enrichment culture. FEMS Microbiol Ecol
Grabowski A, Nercessian O, Fayolle F, Blanchet D, Jeanthon C (2005) Microbial diversity in production waters of a low-temperature biodegraded oil reservoir. FEMS Microbiol Ecol 54(3):427–443
Grassia GS, McLean KM, Glénat P, Bauld J, Sheehy AJ (1996) A systematic survey for thermophilic fermentative bacteria and archaea in high temperature petroleum reservoirs. FEMS Microbiol Ecol 21(1):47–58
Gray N, Sherry A, Hubert C, Dolfing J, Head I (2010) Methanogenic degradation of petroleum hydrocarbons in subsurface environments: remediation, heavy oil formation, and energy recovery. Adv Appl Microbiol 72:137–161
Gray N, Sherry A, Grant R, Rowan A, Hubert C, Callbeck C, Aitken C, Jones D, Adams J, Larter S (2011) The quantitative significance of Syntrophaceae and syntrophic partnerships in methanogenic degradation of crude oil alkanes. Environ Microb 13(11):2957–2975
Harwati TU, Kasai Y, Kodama Y, Susilaningsih D, Watanabe K (2007) Characterization of diverse hydrocarbon-degrading bacteria isolated from Indonesian seawater. Microbes Environ 22(4):412
Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL (2010) Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science 330(6001):204–208
Iqbal S, Khalid Z, Malik K (1995) Enhanced biodegradation and emulsification of crude oil and hyperproduction of biosurfactants by a gamma ray-induced mutant of Pseudomonas aeruginosa. Lett Appl Microbiol 21(3):176–179
Ivshina I, Kuyukina M, Philp J, Christofi N (1998) Oil desorption from mineral and organic materials using biosurfactant complexes produced by Rhodococcus species. World J Microb Biot 14(5):711–717
Kasai Y, Takahata Y, Hoaki T, Watanabe K (2005) Physiological and molecular characterization of a microbial community established in unsaturated, petroleum-contaminated soil. Environ Microb 7(6):806–818
Kharaka YK, Thordsen JJ, Kakouros E, Herkelrath WN (2005) Impacts of petroleum production on ground and surface waters: results from the Osage–Skiatook petroleum environmental research a site, Osage County, Oklahoma. Environ Geosci 12(2):127–138
Kniemeyer O, Musat F, Sievert SM, Knittel K, Wilkes H, Blumenberg M, Michaelis W, Classen A, Bolm C, Joye SB (2007) Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449(7164):898–901
Kostka JE, Prakash O, Overholt WA, Green SJ, Freyer G, Canion A, Delgardio J, Norton N, Hazen TC, Huettel M (2011) Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl Environ Microb 77(22):7962–7974
Kunapuli U, Jahn MK, Lueders T, Geyer R, Heipieper HJ, Meckenstock RU (2010) Desulfitobacterium aromaticivorans sp. nov. and Geobacter toluenoxydans sp. nov., iron-reducing bacteria capable of anaerobic degradation of monoaromatic hydrocarbons. Int J Syst Evol Micr 60(3):686–695
Lenchi N, İnceoğlu Ö, Kebbouche-Gana S, Gana ML, Llirós M, Servais P, García-Armisen T (2013) Diversity of microbial communities in production and injection waters of algerian oilfields revealed by 16S rRNA gene amplicon 454 pyrosequencing. PLoS One 8(6), e66588
Liang Y, Van Nostrand JD, Deng Y, He Z, Wu L, Zhang X, Li G, Zhou J (2010) Functional gene diversity of soil microbial communities from five oil-contaminated fields in China. ISME J 5(3):403–413
Ma J, Nossa CW, Xiu Z, Rixey WG, Alvarez PJ (2013) Adaptive microbial population shifts in response to a continuous ethanol blend release increases biodegradation potential. Environ Pollut 178:419–425
MacNaughton SJ, Stephen JR, Venosa AD, Davis GA, Chang Y-J, White DC (1999) Microbial population changes during bioremediation of an experimental oil spill. Appl Environ Microb 65(8):3566–3574
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437(7057):376–380
Morikawa M, Daido H, Takao T, Murata S, Shimonishi Y, Imanaka T (1993) A new lipopeptide biosurfactant produced by Arthrobacter sp. strain MIS38. J Bacteriol 175(20):6459–6466
Nilsen RK, Torsvik T (1996) Methanococcus thermolithotrophicus isolated from North Sea oil field reservoir Water. Appl Environ Microb 62(2):728–731
Olsgard F, Gray J (1995) A comprehensive analysis of the effects of offshore oil and gas exploration and production on the benthic communities of the Norwegian continental shelf. Marine Ecol Prog ies Oldendorf 122(1):277–306
O'Rourke D, Connolly S (2003) Just oil? The distribution of environmental and social impacts of oil production and consumption. Annu Rev Env Resour 28(1):587–617
Orphan V, Taylor L, Hafenbradl D, Delong E (2000) Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl Environ Microb 66(2):700–711
Osborn SG, Vengosh A, Warner NR, Jackson RB (2011) Methane contamination of drinking water accompanying gas-well drilling and hydraulic fracturing. proceedings of the. Natl Acad Sci 108(20):8172–8176
Patel R, Desai A (1997) Biosurfactant production by Pseudomonas aeruginosa GS3 from molasses. Lett Appl Microbiol 25(2):91–94
Pinhassi J, Zweifel UL, Hagstroëm A (1997) Dominant marine bacterioplankton species found among colony-forming bacteria. Appl Environ Microb 63(9):3359–3366
Plakunov V, Zhurina M, Belyaev S (2008) Resistance of the oil-oxidizing microorganism Dietzia sp. to hyperosmotic shock in reconstituted biofilms. Microbiology 77(5):515–522
Pritchard PH, Costa CF (1991) EPA's Alaska oil spill bioremediation project. Part 5. Environ Sci Technol 25(3):372–379
Puškárová A, Bučková M, Chovanová K, Harichová J, Karelová E, Godočíková J, Polek B, Ferianc P, Pangallo D (2013) Diversity and PAH growth abilities of bacterial strains isolated from a contaminated soil in Slovakia. Biologia 68(4):587–591
Ren H, Zhang X, Song Z, Rupert W, Gao G, Guo S, Zhao L (2011) Comparison of microbial community compositions of injection and production well samples in a long-term water-flooded petroleum reservoir. PLoS One 6(8), e23258
Rosenberg E, Zuckerberg A, Rubinovitz C, Gutnick D (1979) Emulsifier of Arthrobacter RAG-1: isolation and emulsifying properties. Appl Environ Microb 37(3):402–408
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75(23):7537–7541
Song H, Wang X, Bartha R (1990) Bioremediation potential of terrestrial fuel spills. Appl Environ Microb 56(3):652–656
Stevenson I (1967) Utilization of aromatic hydrocarbons by Arthrobacter spp. Can J Microbiol 13(2):205–211
Sun W, Cupples AM (2012) Diversity of five anaerobic toluene-degrading microbial communities investigated using stable isotope probing. Appl Environ Microb 78(4):972–980
Sun W, Xie S, Luo C, Cupples AM (2010) Direct link between toluene degradation in contaminated-site microcosms and a Polaromonas strain. Appl Environ Microb 76(3):956–959
Sun W, Sun X, Cupples AM (2014a) Identification of Desulfosporosinus as toluene-assimilating microorganisms from a methanogenic consortium. Int Biodeter Biodegr 88:13–19
Sun W, Sun X, Cupples AM (2014b) Presence, diversity and enumeration of functional genes (bssA and bamA) relating to toluene degradation across a range of redox conditions and inoculum sources. Biodegradation 25(2):189–203
Sutton NB, Maphosa F, Morillo JA, Al-Soud WA, Langenhoff AA, Grotenhuis T, Rijnaarts HH, Smidt H (2013) Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microb 79(2):619–630
Swannell R, Lee K, McDonagh M (1996) Field evaluations of marine oil spill bioremediation. Microbiol R 60(2):342–365
Tapilatu YH, Grossi V, Acquaviva M, Militon C, Bertrand J-C, Cuny P (2010) Isolation of hydrocarbon-degrading extremely halophilic archaea from an uncontaminated hypersaline pond (Camargue, France). Extremophiles 14(2):225–231
Viñas M, Sabaté J, Guasp C, Lalucat J, Solanas AM (2005) Culture-dependent and-independent approaches establish the complexity of a PAH-degrading microbial consortium. Can J Microbiol 51(11):897–909
von der Weid I, Marques JM, Cunha CD, Lippi RK, Dos Santos SC, Rosado AS, Lins U, Seldin L (2007) Identification and biodegradation potential of a novel strain of Dietzia cinnamea isolated from a petroleum-contaminated tropical soil. Adv Appl Microbiol 30(4):331–339
Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, Knight R (2011) PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27(8):1159–1161
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73(16):5261–5267
Wang X, Chi C, Nie Y, Tang Y, Tan Y, Wu G, Wu X (2011a) Degradation of petroleum hydrocarbons (C6–C40) and crude oil by a novel Dietzia strain. Bioresource Technol 102(17):7755–7761
Wang X, Han Z, Bai Z, Tang J, Ma A, He J, Zhuang G (2011b) Archaeal community structure along a gradient of petroleum contamination in saline-alkali soil. J Environ Sci 23(11):1858–1864
Winderl C, Anneser B, Griebler C, Meckenstock RU, Lueders T (2008) Depth-resolved quantification of anaerobic toluene degraders and aquifer microbial community patterns in distinct redox zones of a tar oil contaminant plume. Appl Environ Microb 74(3):792–801
Winderl C, Penning H, von Netzer F, Meckenstock RU, Lueders T (2010) DNA-SIP identifies sulfate-reducing Clostridia as important toluene degraders in tar-oil-contaminated aquifer sediment. ISME J 4(10):1314–1325
Yakimov MM, Timmis KN, Golyshin PN (2007) Obligate oil-degrading marine bacteria. Curr Opin Biotech 18(3):257–266
Yang S, Wen X, Jin H, Wu Q (2012) Pyrosequencing investigation into the bacterial community in permafrost soils along the China-Russia Crude Oil Pipeline (CRCOP). PLoS One 7(12), e52730
Yumoto I, Nakamura A, Iwata H, Kojima K, Kusumoto K, Nodasaka Y, Matsuyama H (2002) Dietzia psychralcaliphila sp. nov., a novel, facultatively psychrophilic alkaliphile that grows on hydrocarbons. Int J Syst Evol Micr 52(1):85–90
Acknowledgments
We thank Dr. Ying Huang for her help and suggestions on the statistical analysis. We thank Dr. Amanda Luther for thoughtful reviews of the manuscript and discussion of the topics therein.
Funding
This study was funded by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB06020200), the National Natural Science Foundation of China (Grant No. 41376077 and No. 41406067), the Knowledge Innovation Program of the Chinese Academy of Sciences (Grant No. SIDSSE-QN-201303), and the Open Fund of Key Laboratory of Marine Spill Oil Identification and Damage Assessment Technology (Grant No. 201307).
Conflict of interest
Weimin Sun declares that he has no conflict of interest. Jiwei Li declares that he has no conflict of interest. Lei Jiang declares that he has no conflict of interest. Zhilei Sun declares that he has no conflict of interest. Meiyan Fu declares that she has no conflict of interest. Xiaotong Peng declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 240 kb)
Rights and permissions
About this article

Cite this article
Sun, W., Li, J., Jiang, L. et al. Profiling microbial community structures across six large oilfields in China and the potential role of dominant microorganisms in bioremediation. Appl Microbiol Biotechnol 99, 8751–8764 (2015). https://doi.org/10.1007/s00253-015-6748-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-6748-1