
Abstract
Genome sequencing of Aspergillus species including Aspergillus nidulans has revealed that there are far more secondary metabolite biosynthetic gene clusters than secondary metabolites isolated from these organisms. This implies that these organisms can produce additional secondary metabolites, which have not yet been elucidated. The A. nidulans genome contains 12 nonribosomal peptide synthetase (NRPS), one hybrid polyketide synthase/NRPS, and 14 NRPS-like genes. The only NRPS-like gene in A. nidulans with a known product is tdiA, which is involved in terrequinone A biosynthesis. To attempt to identify the products of these NRPS-like genes, we replaced the native promoters of the NRPS-like genes with the inducible alcohol dehydrogenase (alcA) promoter. Our results demonstrated that induction of the single NRPS-like gene AN3396.4 led to the enhanced production of microperfuranone. Furthermore, heterologous expression of AN3396.4 in Aspergillus niger confirmed that only one NRPS-like gene, AN3396.4, is necessary for the production of microperfuranone.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aspergillus species are known to produce medicinally important natural products, such as lovastatin as well as toxins such as aflatoxin (Kennedy et al. 1999; Minto and Townsend 1997). Genome sequencing of Aspergillus species has revealed that there are far more secondary metabolism genes than secondary metabolites that have been ever isolated from these organisms (Galagan et al. 2005; Machida et al. 2005; Nierman et al. 2005). This implies that more secondary metabolites await discovery. The availability of genome sequencing information has facilitated secondary metabolite discovery in a strategy often termed “genome mining” (Chiang et al. 2011a; Winter et al. 2011). This approach involves the use of bioinformatic analysis of genomic data for the identification of putative biosynthesis genes followed by gene deletions or heterologous expression for the verification of gene function. Genome mining of secondary metabolism genes in Aspergillus nidulans has been greatly facilitated by the continuous refinement of the genome annotation and the creation, development, and refinement of the community databases, Aspergillus Genome Database (AspGD), Broad Institute Aspergillus Comparative Database, Central Aspergillus Data REpository (CADRE), Secondary Metabolite Unique Regions Finder (SMURF) (Khaldi et al. 2010), and the Department of Energy Joint Genome Institute (JGI) Fungal Genomics Program. We and others have initiated programs to identify the products of the secondary metabolism genes in A. nidulans (Bergmann et al. 2007, 2010; Bok et al. 2006, 2009; Chiang et al. 2008, 2009, 2010; Sanchez et al. 2010; Scherlach and Hertweck 2006; Scherlach et al. 2010; Schroeckh et al. 2009; Szewczyk et al. 2008). With the rapid development of next generation sequencing, whole fungal genome sequencing is now within the budget of individual labs (Nowrousian et al. 2010), and A. nidulans is an excellent model organism for the development of strategies and tools that can be translated to many genome-sequenced fungal species.
The initial genome analysis of A. nidulans identified 29 polyketide synthases (PKSs) and 12 nonribosomal peptide synthetases (NRPSs). Several single module NRPS-related genes were not identified in this initial genome annotation effort (Galagan et al. 2005). A recent comprehensive review by von Dohren reevaluated the NRPS genes in A. nidulans and grouped them into 12 NRPS, one hybrid PKS/NRPS, and 14 NRPS-like genes (von Dohren 2009). Monomodular NRPS-like genes in A. nidulans are not well characterized either genetically or biochemically. NRPS-like genes share the catalytic domains found in NRPS but are missing the critical condensation domain necessary for peptide formation. The only NRPS-like gene in A. nidulans, the product of which is known, is tdiA (AN8513.4 using the CADRE gene designation), which is involved in terrequinone A (compound 2, Fig. 1) biosynthesis (Balibar et al. 2007; Bok et al. 2006; Schneider et al. 2007, 2008). The tdiA gene contains three domains found in a typical NRPS gene. They are an adenylation (A) domain, which loads a specific amino acid, a thiolation (T) domain, and a thioesterase (TE) domain, but the condensation domain is missing. Terrequinone A is a secondary metabolite derived from amino acids but does not have peptide bonds in its structure. Examination of the 14 NRPS-like genes in A. nidulans revealed that one additional NRPS-like gene (AN3396.4) contains the A-T-TE domain architecture found in TdiA (von Dohren 2009). The remaining 12 NRPS-like genes contain either a NAD-binding domain in place of the TE domain or are missing both domains. We cultivated A. nidulans in a variety of growth conditions and media in the hopes of identifying conditions that would enable production of a metabolite from the 13 NRPS-like genes that have not yet been characterized at a level detectable by high performance liquid chromatography–diode-array detection–mass spectrometry (HPLC-DAD-MS) (Sanchez et al. 2010). However, despite numerous attempts, we were unable to detect new metabolites that correspond to biosynthetic pathways that include the 13 NRPS-like genes. This suggests that these genes are silent or expressed in very low amounts under the conditions we examined. Since it was difficult to obtain conditions to activate the native promoters, we initiated a strategy to replace the native promoters with inducible promoters to turn on expression of these genes. We first replaced the native promoters of the 13 NRPS-like genes with the alcohol dehydrogenase promoter [alcA(p)] that can be induced to very high levels of expression using cyclopentanone. We observed that induction of the single NRPS-like gene AN3396.4 led to enhanced production of microperfuranone (compound 1, Fig. 1). Microperfuranone was first isolated from the fungus Anixiella micropertusa and also isolated from a marine strain of Emericella nidulans (Fujimoto et al. 1998; Kralj et al. 2006). We named the gene AN3396.4 micA for microperfuranone synthase. To verify that indeed only one NRPS-like protein is necessary to produce microperfuranone, we heterologously expressed micA in Aspergillus niger, which has a well-characterized secondary metabolome and is known to not produce microperfuranone or similar compounds (Nielsen et al. 2009).

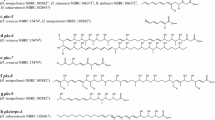
Chemical structures of microperfuranone and terrequinone A
Materials and methods
Generation of fusion PCR fragments, A. nidulans protoplasting, and transformation
We generated fusion PCR fragments to replace the native promoters of the 13 NRPS-like genes with an alcA promoter. For example, to obtain strains overexpressing micA, a 100-bp fragment immediately upstream of the micA start codon was replaced with a fragment containing the A. fumigatus pyroA gene (AfpyroA) followed by a 404-bp fragment containing the A. nidulans alcA promoter such that the coding sequence of micA was placed under the control of the alcA promoter. Construction of fusion PCR products, protoplast production, and transformation were carried out as described (Szewczyk et al. 2006). For construction of fusion PCR fragments, two ~1,000-bp fragments, one upstream and one downstream of the targeted endogenous promoter, were amplified from genomic A. nidulans DNA by PCR. Using two nested primers, a fusion PCR reaction attached the two 1,000-bp fragments to flank the A. fumigatus pyroA selective marker. The primers for fusion PCR are listed in Table 1. An A. nidulans strain, LO2026, carrying a deletion of the stcJΔ that prevents sterigmatocystin production was used as a recipient strain for transformation (Bok et al. 2009). The transformants with correct promoter replacements were further verified with diagnostic PCR using the external primers used in the first round of PCR (Table 1 and Fig. S1). In each case, at least two transformants carrying the correct promoter replacement were used for further study. A. nidulans strains used in this study are listed in Table 2. Deletions of ten genes, designated AN3391.4–AN3395.4 and AN3397.4–AN3401.4, were generated by replacing each gene with the A. fumigatus pyrG gene in the A. nidulans strain CW3023 (stcJΔ, alcA(p)-micA). The primers for gene deletion and complete genotypes are listed in Table S1 and Table S2, respectively.
Reverse transcription polymerase chain reaction analysis
Total RNA of A. nidulans parental and mutant strains was extracted using the Qiagen RNeasy Plant Mini Kit according to the manufacturer’s instructions. First-strand complementary DNA (cDNA) was synthesized using TaqMan Reverse Transcription Reagents (Applied Biosystems) following the supplied protocols. The cDNA was then used as template for PCR amplification with following specific primer sets that flanking the intron except for AN3397.4, which does not have a predicted intron: AN3391.4, 5'-ACCTATACCAGTGCGGAAC-3' and 5'-GAGCCACGCACTCAATATTC-3'; AN3392.4, 5'-GAGGCACGGTTAGCTCTAC-3' and 5'-CCCAAACGCAATAGGCATG-3'; AN3393.4, 5'-ATCAGGACCAGCACCACTG-3' and 5'-CAGCTCGTTGGAGGTGTAG-3'; AN3394.4, 5'-CTGCGTCACAATTCAGTGC-3' and 5'-GCTTGTAAGTCAAGGCTGC-3'; AN3395.4, 5'-GTCTTCGCCTTGTCAACAC-3' and 5'-GCGATACTAGTATGGCCAC-3'; AN3396.4, 5'-GACCACGTTGCTAGTTTGAC-3' and 5'-AATCACTTCGGCTTGGACAC-3'; AN3397.4, 5'-CCACGTCGAGGTGATCAAG-3' and 5'-GGCAGTGAAGTCGACGTTC-3'; AN3398.4, 5'-GACTCGCAAAGACCTATGC-3' and 5'-GCATTCTAAGCTGGCGCTG-3'; AN3399.4, 5'-CTGCACTGTGACGAGAGTC-3' and 5'-GAACCACTCCTCGATTGCAC-3'; AN3400.4, 5'-TGCAATTGCTGTAGAGGC-3' and 5'-CCATACTTGGGAGGAAGCT-3'; AN3401.4, 5'-GACTCCAAAGATCGCTC-3' and 5'-CTTTGCAGTGGCCACAAC-3'; β-tubulin, 5'-CATGATGACAGCTGCCAAC-3' and 5'-GAGCAGTTTGGACGTTGTTG-3'. Amplification products were analyzed by electrophoresis in 1.5 % agarose gels stained with ethidium bromide.
Heterologous expression of micA
Expression of micA was achieved in A. niger by fusing the coding sequence of the gene to a promoter sequence taken from the A. oryzae amyB gene. The amyB gene from A. oryzae (locus ID, AO090120000196) has been used for heterologous expression in other systems and is known to be responsive to growth on different carbon sources (Kanemori et al. 1999). The construct was built using the yeast gap repair method and plasmid described in a previous study for creating fungal gene fusions (Bourett et al. 2002). In brief, the amyB promoter was amplified from A. oryzae strain RIB40 genomic DNA using primers AN3396.amyF and AN3396.amyR. The micA coding region was amplified using primers AN3396.ATG and AN3396.TAA. The five prime ends of AN3396.amyF and AN3396.TAA have homology to regions within pSM565 (GenBank, AY142483.1) that can repair an XhoI digestion of this plasmid when transformed into Saccharomyces cerevisiae. Sequences on primers AN3396.amyR and AN3396.ATG directly fuse the start codon from the amylase promoter to the micA coding sequence. The plasmid created was isolated from yeast colonies, amplified in Escherichia coli cultures and subsequently transformed into A. niger strain KB1001 (Chiang et al. 2011b). Positive transformants are resistant to hygromycin due to the presence of the hph gene on the plasmid. Spores were collected from the OE:micA strains by cultivating 1.0 × 107 spores per 10-cm plate for 5 days at 30 °C on YAG medium (5 g of yeast extract/l, 15 g of agar/l, and 20 g of d-glucose/l supplemented with a 1 ml/l trace element solution) containing 100 μg/ml hygromycin B. To test for expression of the micA gene and production of microperfuranone, a 30-ml liquid YG culture supplemented with 100 μg/ml hygromycin B was inoculated with 3.0 × 107 spores and grown at 30 °C with shaking at 170 rpm for 18 h. The hyphae were collected with miracloth and put into medium to induce the amylase promoter of OE:micA using GMM medium with 2 % (w/v) maltose as the carbon source. After 2 days of induction, the medium were collected by filtration and then extracted as described below.
Fermentation and purification
For fermentation, 3.0 × 107 spores of A. nidulans were grown in 125-ml flasks containing 30 ml liquid LMM medium (15 g/l lactose, 6 g/l NaNO3, 0.52 g/l KCl, 0.52 g/l MgSO4⋅7H2O, 1.52 g/l KH2PO4, and 1 ml/l trace elements) supplemented when necessary with uracil (1 mg/ml) and uridine (10 mM) at 37 °C with shaking at 200 rpm (Chiang et al. 2008). For alcA promoter induction, cyclopentanone at a final concentration of 10 mM was added to the medium after 18 h of incubation. Culture medium was collected 48 h after cyclopentanone induction by filtration and extracted with the same volume of EtOAc two times. The combined EtOAc layers were evaporated in vacuo, redissolved in 0.75 ml of 1:4 dimethyl sulfoxide/MeOH, and 10 μl was injected for HPLC-DAD-MS analysis. Conditions for MS included a capillary voltage 5.0 kV, a sheath gas flow rate at 60 arbitrary units, an auxiliary gas flow rate at 10 arbitrary units, and the ion transfer capillary temperature at 350 °C. HPLC-MS was carried out in positive mode using a ThermoFinnigan LCQ Advantage ion trap mass spectrometer with an RP C18 column (Alltech Prevail C18; particle size, 3 μm; column, 2.1 × 100 mm) at a flow rate of 125 μl/min. Microperfuranone was eluted at 26.5 min.
For structure elucidation, a strain carrying alcA(p)-micA was cultivated in 2 liter LMM medium. After 2 days of induction, the medium was collected by filtration and then extracted with equal amount of EtOAc twice. The combined EtOAc extracts were evaporated in vacuo. The crude extract in EtOAc layer (448 mg) was coated on 6,720 mg of C18 reverse phase gel (Cosmosil 75C18-OPN, Nacalai USA), which was then suspended in 10 % of MeOH/ddH2O and applied to a C18 reverse phase column (30 × 60 mm). This column was then eluted with MeOH/ddH2O mixtures of decreasing polarity (fraction A, 10 % MeOH, 300 ml; fraction B, 30 % MeOH, 300 ml; fraction C, 70 % MeOH, 300 ml; and fraction D, 100 % MeOH, 300 ml). All fractions were analyzed by HPLC-DAD-MS. Fraction C containing microperfuranone was further subjected to semi-preparative reverse phase HPLC (Phenomenex Luna 5 μm C18, 250 × 10 mm) with a flow rate of 5.0 ml/min and measured by a UV detector at 254 nm. The solvent gradient for HPLC was 100 % MeCN (solvent B) in 5 % MeCN/H2O (solvent A), 20 % B from 0 to 20 min, 60 to 100 % B from 20 to 22 min, maintained at 100 % B from 22 to 25 min, 100 to 20 % B from 25 to 26 min, and re-equilibration with 20 % B from 26 to 30 min. Microperfuranone (20 mg) was eluted at 20.0 min.
Compound identification
1H and 13C nuclear magnetic resonance (NMR) spectra were collected on a Varian Mercury Plus 400 spectrometer, whereas HRESIMS spectra were obtained on an Agilent Technologies 1200 series high-resolution mass spectrometer. Microperfuranone was isolated as colorless plates, and its molecular formula was deduced to be C17H14O3 ([M+H]+ m/z found 267.1022; calcd. for C17H15O3: 267.1021, Fig. S2). The 1H (Fig. S3 and S4) and 13C (Fig. S5 and S6) NMR data in CDCl3 or acetone-d 6 were in good agreement with the published data (Fujimoto et al. 2006, 1998).
Results
Induction of the micA gene using an inducible promoter stimulates the production of microperfuranone
Previous data have shown that A. nidulans secondary metabolite production is heavily dependent on culture conditions (Sanchez et al. 2010; Scherlach and Hertweck 2006; Scherlach et al. 2010). Attempts to grow A. nidulans under 20 different conditions failed to produce any additional metabolites that could be produced by NRPS-like genes. Analysis by von Dohren identified 14 NRPS-like genes in A. nidulans. Since one of the NRPS-like is tdiA (AN8513.4), we focused on the other 13 genes for promoter replacements (AN1680.4, AN2064.4, AN2924.4, AN3396.4, AN3495.4, AN4827.4, AN5318.4, AN6444.4, AN8105.4, AN8504.4, AN9291.4, AN10297.4, and AN10486.4) (von Dohren 2009). We replaced the native promoters of the 13 genes with the inducible alcohol dehydrogenase promoter. The replacement was accomplished using our previously reported strategy involving an nkuAΔ A. nidulans strain and fusion PCR (Chiang et al. 2008; Nayak et al. 2006; Szewczyk et al. 2006). The promoter replacements were carried out in an A. nidulans strain carrying a deletion of the stcJΔ gene (stcJΔ), which prevents the production of the major polyketide sterigmatocystin (Chiang et al. 2009). We cultivated three separate promoter exchanged strains per gene and a control strain LO2026 in lactose minimal media and used cyclopentanone as an inducer. All strains were verified by diagnostic PCR (Fig. S1). Metabolite profiles from each induced strain were analyzed by HPLC-DAD-MS, and only the alcA(p)-micA strains were able to produce a new metabolite with the molecular weight of 266 (m/z = 267 [M+H]+) (Fig. 2 and S2). The compound was isolated from large-scale cultivation of the alcA(p)-micA strain by purification initially from flash chromatography followed by preparative HPLC. Both 1H and 13C NMR analysis of the compound (Fig. S3–S6) identified the product as microperfuranone, which has been isolated from A. micropertusa and E. nidulans var. acristata (Fujimoto et al. 1998, 2006; Kralj et al. 2006). The 12 other strains failed to produce new metabolites, suggesting that co-overexpression of additional genes in their pathways might be necessary to elicit detectable and isolatable products.

HPLC-DAD-MS analysis of wild-type A. nidulans and alcA(p)-micA mutant metabolites. a HPLC profiles of extracts as detected by UV absorption. b UV-vis and ESIMS spectra (positive mode) of microperfuranone
Heterologous expression of micA in the heterologous host A. niger demonstrates that only one gene is necessary for microperfuranone biosynthesis
Our data so far suggested that overexpression of a single gene, micA, was sufficient to elicit microperfuranone production. Since genes that involved in secondary metabolite biosynthesis are generally clustered in A. nidulans, we wondered if genes proximal to micA also participated in the biosynthesis of microperfuranone. Reverse transcription polymerase chain reaction (RT-PCR) analysis of the genes near micA confirmed that micA was actively transcribed in the induction condition (Fig. S7). Transcription of two genes near micA, AN3394.4 and AN3395.4, was also detected by RT-PCR, although their levels of transcription resembled the wild-type control. To determine, definitively, if genes nearby micA were necessary for the biosynthesis microperfuranone, we created deletions of ten additional genes (AN3391.4–AN3395.4 and AN3397.4–AN3401.4) surrounding micA. The deletions were accomplished using our previously reported strategy involving a nkuAΔ strains and fusion PCR (Chiang et al. 2008). The targeted genes were replaced with the A. fumigatus pyrG gene (Nayak et al. 2006). Metabolite analysis of the extracts from the ten deletion mutant strains revealed that all ten mutant strains continue to produce microperfuranone (data not shown). To confirm that only one single gene micA without any other accessory genes is sufficient for microperfuranone biosynthesis, we expressed micA in a heterologous host, A. niger. We selected A. niger as a heterologous host because the A. niger secondary metabolome has been extensively studied and is not known to produce microperfuranone (Chiang et al. 2011b; Nielsen et al. 2009). We fused micA with the amylase promoter and transformed into A. niger (Kanemori et al. 1999). The amylase promoter can be induced using maltose as a carbon source in the media and is inhibited using fructose. We cultivated the OE:micA A. niger using maltose as a carbon source, and as a control, we also cultivated parental strain A. niger. Using HPLC-DAD-MS, we detected microperfuranone in OE:micA A. niger and not in the A. niger parental strain control strain, demonstrating that only micA is necessary for microperfuranone biosynthesis (Fig. 3).

HPLC-DAD-MS analysis of parental strain (Control) A. niger and amylase(p)-micA (OE:micA) mutant metabolites. Asterisk A related compound that has similar MS/MS fragmentation with microperfuranone
Discussion
There are 14 NRPS-like genes in the A. nidulans genome, but only the product of tdiA from the terrequinone A pathway has been characterized genetically and biochemically. By replacing each of the promoters of the 13 NRPS-like genes in A. nidulans with an inducible promoter we show that one of the NRPS-like genes micA (AN3396.4) is apparently sufficient for the biosynthesis of the metabolite microperfuranone. Replacement of the promoter of the other 12 NRPS-like genes in A. nidulans with the inducible promoter failed to produce metabolites, suggesting that activation of additional genes in the genome are necessary for their production. Creation of double and other multiple promoter replacements strains in A. nidulans are currently underway and their results will be reported in due course.
From our genetic analysis and heterologous overexpression experiments, we propose a speculative but plausible mechanism for microperfuranone biosynthesis (Fig. 4). These steps include the activation of phenylpyruvic acid (PPA), a precursor available in both A. nidulans and A. niger, by the MicA A domain to AMP-phenylpyruvic acid followed by loading of the PPA unit to the T domain and eventually transferring to the TE domain. After loading another PPA unit onto the T domain, aldol condensation establishes the carbon–carbon bond between the α- and β-carbon of the two PPA units. The carbon–carbon bond formation by the TE domain is not unprecedented and has been demonstrated biochemically in terrequinone A biosynthesis by the Walsh group (Balibar et al. 2007). Sulfur-assisted furan ring formation, TE domain mediated hydrolysis, decarboxylation, and keto-enol tautomerization would generate microperfuranone attached to the T domain of MicA. Finally, microperfuranone is released by the TE domain, and the catalytic cycle continues. Recently, the biosynthesis pathway for furanone from the Gram-negative bacterium Ralstonia solanacearum, a secondary metabolite sharing structural similarities to microperfuranone, was characterized genetically and biochemically (Wackler et al. 2011). Our data suggested that microperfuranone is biosynthesized in A. nidulans using a similar pathway.

Proposed model of microperfuranone biosynthesis
In summary, our studies demonstrate that induction of the expression of micA, a NRPS-like gene, stimulates the production of microperfuranone. To verify that indeed micA is sufficient, we have demonstrated that overexpression of micA in a heterologous host, A. niger, enables the production of microperfuranone. Our results confirm that only micA is necessary for microperfuranone biosynthesis in A. nidulans.
References
Balibar CJ, Howard-Jones AR, Walsh CT (2007) Terrequinone a biosynthesis through L-tryptophan oxidation, dimerization and bisprenylation. Nat Chem Biol 3:584–592
Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, Hertweck C (2007) Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat Chem Biol 3:213–217
Bergmann S, Funk AN, Scherlach K, Schroeckh V, Shelest E, Horn U, Hertweck C, Brakhage AA (2010) Activation of a silent fungal polyketide biosynthesis pathway through regulatory cross talk with a cryptic nonribosomal peptide synthetase gene cluster. Appl Environ Microbiol 76:8143–8149
Bok JW, Hoffmeister D, Maggio-Hall LA, Murillo R, Glasner JD, Keller NP (2006) Genomic mining for Aspergillus natural products. Chem Biol 13:31–37
Bok JW, Chiang YM, Szewczyk E, Reyes-Dominguez Y, Davidson AD, Sanchez JF, Lo HC, Watanabe K, Strauss J, Oakley BR, Wang CC, Keller NP (2009) Chromatin-level regulation of biosynthetic gene clusters. Nat Chem Biol 5:462–464
Bourett TM, Sweigard JA, Czymmek KJ, Carroll A, Howard RJ (2002) Reef coral fluorescent proteins for visualizing fungal pathogens. Fungal Genet Biol 37:211–220
Chiang YM, Szewczyk E, Nayak T, Davidson AD, Sanchez JF, Lo HC, Ho WY, Simityan H, Kuo E, Praseuth A, Watanabe K, Oakley BR, Wang CC (2008) Molecular genetic mining of the Aspergillus secondary metabolome: discovery of the emericellamide biosynthetic pathway. Chem Biol 15:527–532
Chiang YM, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CC (2009) A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J Am Chem Soc 131:2965–2970
Chiang YM, Szewczyk E, Davidson AD, Entwistle R, Keller NP, Wang CC, Oakley BR (2010) Characterization of the Aspergillus nidulans monodictyphenone gene cluster. Appl Environ Microbiol 76:2067–2074
Chiang YM, Chang SL, Oakley BR, Wang CC (2011a) Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol 15:137–143
Chiang YM, Meyer KM, Praseuth M, Baker SE, Bruno KS, Wang CC (2011b) Characterization of a polyketide synthase in Aspergillus niger whose product is a precursor for both dihydroxynaphthalene (DHN) melanin and naphtho-gamma-pyrone. Fungal Genet Biol 48:430–437
Fujimoto H, Satoh Y, Yamaguchi K, Yamazaki M (1998) Monoamine oxidase inhibitory constituents from Anixiella micropertusa. Chem Pharm Bull 46:1506–1510
Fujimoto H, Asai T, Kim YP, Ishibashi M (2006) Nine constituents including six xanthone-related compounds isolated from two ascomycetes, Gelasinospora santi-florii and Emericella quadrilineata, found in a screening study focused on immunomodulatory activity. Chem Pharm Bull 54:550–553
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, Selker EU, Archer DB, Penalva MA, Oakley BR, Momany M, Tanaka T, Kumagai T, Asai K, Machida M, Nierman WC, Denning DW, Caddick M, Hynes M, Paoletti M, Fischer R, Miller B, Dyer P, Sachs MS, Osmani SA, Birren BW (2005) Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 438:1105–1115
Kanemori Y, Gomi K, Kitamoto K, Kumagai C, Tamura G (1999) Insertion analysis of putative functional elements in the promoter region of the Aspergillus oryzae Taka-amylase A gene (amyB) using a heterologous Aspergillus nidulans amdS-lacZ fusion gene system. Biosci Biotechnol Biochem 63:180–183
Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284:1368–1372
Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND (2010) SMURF: genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol 47:736–741
Kralj A, Kehraus S, Krick A, Eguereva E, Kelter G, Maurer M, Wortmann A, Fiebig HH, Konig GM (2006) Arugosins G and H: prenylated polyketides from the marine-derived fungus Emericella nidulans var. acristata. J Nat Prod 69:995–1000
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438:1157–1161
Minto RE, Townsend CA (1997) Enzymology and molecular biology of aflatoxin biosynthesis. Chem Rev 97:2537–2556
Nayak T, Szewczyk E, Oakley CE, Osmani A, Ukil L, Murray SL, Hynes MJ, Osmani SA, Oakley BR (2006) A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172:1557–1566
Nielsen KF, Mogensen JM, Johansen M, Larsen TO, Frisvad JC (2009) Review of secondary metabolites and mycotoxins from the Aspergillus niger group. Anal Bioanal Chem 395:1225–1242
Nierman WC, Pain A, Anderson MJ, Wortman JR, Kim HS, Arroyo J, Berriman M, Abe K, Archer DB, Bermejo C, Bennett J, Bowyer P, Chen D, Collins M, Coulsen R, Davies R, Dyer PS, Farman M, Fedorova N, Feldblyum TV, Fischer R, Fosker N, Fraser A, Garcia JL, Garcia MJ, Goble A, Goldman GH, Gomi K, Griffith-Jones S, Gwilliam R, Haas B, Haas H, Harris D, Horiuchi H, Huang J, Humphray S, Jimenez J, Keller N, Khouri H, Kitamoto K, Kobayashi T, Konzack S, Kulkarni R, Kumagai T, Lafon A, Latge JP, Li W, Lord A, Lu C, Majoros WH, May GS, Miller BL, Mohamoud Y, Molina M, Monod M, Mouyna I, Mulligan S, Murphy L, O'Neil S, Paulsen I, Penalva MA, Pertea M, Price C, Pritchard BL, Quail MA, Rabbinowitsch E, Rawlins N, Rajandream MA, Reichard U, Renauld H, Robson GD, Rodriguez de Cordoba S, Rodriguez-Pena JM, Ronning CM, Rutter S, Salzberg SL, Sanchez M, Sanchez-Ferrero JC, Saunders D, Seeger K, Squares R, Squares S, Takeuchi M, Tekaia F, Turner G, Vazquez de Aldana CR, Weidman J, White O, Woodward J, Yu JH, Fraser C, Galagan JE, Asai K, Machida M, Hall N, Barrell B, Denning DW (2005) Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438:1151–1156
Nowrousian M, Stajich JE, Chu M, Engh I, Espagne E, Halliday K, Kamerewerd J, Kempken F, Knab B, Kuo HC, Osiewacz HD, Poggeler S, Read ND, Seiler S, Smith KM, Zickler D, Kuck U, Freitag M (2010) De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet 6:e1000891
Sanchez JF, Chiang YM, Szewczyk E, Davidson AD, Ahuja M, Elizabeth Oakley C, Woo Bok J, Keller N, Oakley BR, Wang CC (2010) Molecular genetic analysis of the orsellinic acid/F9775 gene cluster of Aspergillus nidulans. Mol Biosyst 6:587–593
Scherlach K, Hertweck C (2006) Discovery of aspoquinolones A-D, prenylated quinoline-2-one alkaloids from Aspergillus nidulans, motivated by genome mining. Org Biomol Chem 4:3517–3520
Scherlach K, Schuemann J, Dahse HM, Hertweck C (2010) Aspernidine A and B, prenylated isoindolinone alkaloids from the model fungus Aspergillus nidulans. J Antibiot 63:375–377
Schneider P, Weber M, Rosenberger K, Hoffmeister D (2007) A one-pot chemoenzymatic synthesis for the universal precursor of antidiabetes and antiviral bis-indolylquinones. Chem Biol 14:635–644
Schneider P, Weber M, Hoffmeister D (2008) The Aspergillus nidulans enzyme TdiB catalyzes prenyltransfer to the precursor of bioactive asterriquinones. Fungal Genet Biol 45:302–309
Schroeckh V, Scherlach K, Nutzmann HW, Shelest E, Schmidt-Heck W, Schuemann J, Martin K, Hertweck C, Brakhage AA (2009) Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA 106:14558–14563
Szewczyk E, Nayak T, Oakley CE, Edgerton H, Xiong Y, Taheri-Talesh N, Osmani SA, Oakley BR (2006) Fusion PCR and gene targeting in Aspergillus nidulans. Nat Protoc 1:3111–3120
Szewczyk E, Chiang YM, Oakley CE, Davidson AD, Wang CC, Oakley BR (2008) Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl Environ Microbiol 74:7607–7612
von Dohren H (2009) A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans. Fungal Genet Biol 46:S45–S52
Wackler B, Schneider P, Jacobs JM, Pauly J, Allen C, Nett M, Hoffmeister D (2011) Ralfuranone biosynthesis in Ralstonia solanacearum suggests functional divergence in the quinone synthetase family of enzymes. Chem Biol 18:354–360
Winter JM, Behnken S, Hertweck C (2011) Genomics-inspired discovery of natural products. Curr Opin Chem Biol 15:22–31
Acknowledgments
This project was supported by grant PO1GM084077 from the National Institute of General Medical Sciences. Research conducted at the Pacific Northwest National Lab was supported by the PNNL Energy and Environment Directorate’s Laboratory Directed Research and Development office. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.17 MB)
Rights and permissions
About this article
Cite this article
Yeh, HH., Chiang, YM., Entwistle, R. et al. Molecular genetic analysis reveals that a nonribosomal peptide synthetase-like (NRPS-like) gene in Aspergillus nidulans is responsible for microperfuranone biosynthesis. Appl Microbiol Biotechnol 96, 739–748 (2012). https://doi.org/10.1007/s00253-012-4098-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-012-4098-9