
Abstract
A novel rhamnogalacturonase (RGase) acting on an acetylated substrate was detected in the commercial preparation Driselase, an enzymatic mixture derived from the basidiomycete Irpex lacteus. The activity was isolated by hydrophobic interaction chromatography, gel filtration, and preparative isoelectric focusing, resulting in the isolation of five different rhamnogalacturonan hydrolases exhibiting various isoelectric points from 6.2 to 7.7. Sodium dodecyl sulfate polyacrylamide gel electrophoresis and mass spectrometry analyses after trypsin cleavage of the five fractions revealed that the five rhamnogalacturonases have a molar mass of 55 kDa without any divergences in the identified peptides. The RGase with a pI of 7.2 exhibited a pH optimum between 4.5 and 5 and a temperature optimum between 40°C and 50°C. Its mode of action was analyzed by mass spectrometry of the oligosaccharides produced after hydrolysis of acetylated and nonacetylated rhamnogalacturonan. Oligomers esterified by an acetyl group on the reducing galacturonic acid residue or fully acetylated were detected in the hydrolysate showing that the novel enzyme is able to bind acetylated galacturonic acid in its active site.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Pectin, a major component of plant cell wall, is composed of two major regions: homogalacturonans (HG) and type I rhamnogalacturonans (RG). HG consist of a repetition of α-(1→4) linked d-galacturonic acid (GalA) which can be methylated at C-6 and/or acetylated at O-2 and/or O-3. RG are composed of alternating (1-2)-linked α-l-Rha and (1-4)-linked α-D GalA residues. Within RG regions, Rha residues often carry neutral sugars side chains, and GalA residues can be substituted by acetyl groups (Ishii 1995a; Voragen et al. 1995). The degree of acetylation (DAc) is defined as the number of acetyl groups present on 100 GalA units and depends on the plant source. Pectins isolated from apple and citrus are hardly acetylated (Axelos and Thibault 1991), whereas pectins from sugar beet are highly acetylated with a DAc around 35 (Voragen et al. 1995). In most plants, acetyl substituents are more present in RG than in HG (Ishii 1995a; Schols and Voragen 1996).
Pectin structure, composition, and substituent pattern can be studied by enzymatic tools. The enzymatic degradation of the pectic backbone involves rhamnogalacturonases and polygalacturonases. Most of the known rhamnogalacturonan-hydrolases (RG-hydrolases) have been purified from filamentous fungi (Schols et al. 1990; Suykerbuyk et al. 1997). Their activity is hampered toward pectin exhibiting a high degree of acetylation (Kofod et al. 1994; Schols et al. 1990; Schols and Voragen 1994). Therefore, an enzyme able to act on acetylated RG would be a powerful tool to study the structure of acetylated pectin, more particularly the acetylation pattern of RG regions.
Irpex lacteus is a wood-decaying basidiomycete that produces numerous cell wall polysaccharide-degrading enzymes (Hamada and Hirohashi 2000; Hamada et al. 1999b; Hoebler and Brillouet 1984; Tsumuraya et al. 1990). These proteins were purified directly from the fungus or from Driselase, a commercial preparation derived from the microorganism.
In the present study, acetylated RG isolated from sugar beet pectin has been used as substrate to demonstrate that Driselase contains, among major activities (xylanase, cellulase and polygalacturonase), a rhamnogalacturonase activity tolerant to an acetylated substrate. This novel enzyme has been purified. Its biochemical and physicochemical features have been investigated, and its mode of action has been analyzed by mass spectrometry of RG hydrolysis products.
Materials and methods
Enzymes sources
Driselase from I. lacteus was purchased from Sigma (D9515-25G; L’Isle d’Abeau, France). The RG-hydrolase from Aspergillus aculeatus was provided by Novozymes (Batch N° PPJ 4478, Bagsvaerd, Denmark).
Substrates
Different cell wall polysaccharides were used as substrates. Acetylated RG was prepared by enzymatic degradation of sugar beet pectin (Teknisk SBP, Danisco A/S, Brabrand, Denmark; Bonnin et al. 2008). Polygalacturonic acid and carboxymethylcellulose (CMC) were from commercial origin (Sigma), others substrates (nonacetylated RG (Bonnin et al. 2001), xylan (Bobin-Dubigeon et al. 1997), arabinan (Lahaye and Thibault 1990), galactan (Lahaye et al. 1991)) were obtained from the laboratory collection. To analyze the substrates, GalA content was determined by the automated m-hydroxybiphenyl method (Thibault 1979). Individual neutral sugars composition was analyzed as their alditol acetate derivatives by gas chromatography (Englyst and Cummings 1988) after hydrolysis in 2 N trifluoro acetic acid, 120°C for 2.5 h. Acetic acid released after deesterification of acetylated RG was determined using the Enzytec Acetic Acid kit (Scil diagnostics, Viernheim, Germany), and methanol was quantified by HPLC on a C18 column (Levigne et al. 2002). DM and DAc were calculated as the molar ratio of methanol and acetic acid to GalA, respectively.
Enzymatic assays
Depolymerase activity assays were performed by measuring the release of reducing ends in a reaction mixture containing substrates solubilized in 50 mM Na-acetate buffer pH 5.0 (acetylated RG at 2.5 mg/mL, other substrates at 5 mg/mL), incubated with the enzyme at 40°C. Reducing sugars were measured according to the Nelson (1944) method carried out in microplates (Sturgeon 1990) using monomer as the standard for each substrate and GalA for RG.
Acetyl esterase activity was measured by using the Enzytec Acetic Acid kit (Scil diagnostics) after incubation of acetylated RG with the enzyme in 50 mM succinate buffer pH 5.0.
RG-lyase activity was determined by measuring the absorption at 235 nm after incubation of the enzyme with nonacetylated RG (Voragen et al. 1971).
Enzymatic activities were expressed in nanokatal, 1 nkat being defined as the amount of enzyme that catalyzes the release of 1 nmol of reducing end per second in the conditions described above.
Enzyme purification
All purification steps were carried out at 4°C. Of the Driselase, 10 g were solubilized at 200 mg/mL in 50 mM acetate buffer, pH 5.0, overnight prior to centrifugation (38,000×g, 15 min, 4°C). The recovered supernatant was dialyzed against 50 mM Tris–HCl, pH 6.5, containing 1.25 M ammonium sulfate. The dialysate was loaded on a phenyl-Sepharose CL6B column (26 × 135 mm; GE Healthcare, Orsay, France) equilibrated with the dialysis buffer. The bound proteins were eluted in 50 mM Tris–HCl buffer, pH 6.5, as buffer A and 50 mM Tris–HCl and 1.25 M ammonium sulfate, pH 6.5, as buffer B according to the following gradient: 210 mL at 100% buffer B, 90 mL from 100% to 50% buffer B, 225 mL from 50% to 25% buffer B, 75 mL from 25% to 0% buffer B, and 210 mL at 0% buffer B. The elution was performed at a flow rate of 1.5 mL/min and monitored by following the absorption at 280 nm. All the fractions were tested for their activities toward nonacetylated RG, polygalacturonic acid, CMC, xylan, arabinan, and galactan. The rhamnogalacturonase rich fractions were pooled.
After dialysis and concentration, the sample was adjusted to 20 mM acetate buffer, pH 5.0, and was applied on two Superdex 75 columns (16 × 160 mm; GE Healthcare) mounted in series and equilibrated with the same buffer as the sample. The elution was carried out with the same buffer at a flow rate of 0.3 mL/min and monitored by following the adsorption at 280 nm. All the fractions were tested for their activities toward nonacetylated RG and polygalacturonic acid. The rhamnogalacturonase rich fractions were pooled.
The pool containing the rhamnogalacturonase activity was extensively dialyzed against cold deionized water to be separated in a preparative Rotofor cell (BioRad, Marnes la Coquette, France) in the presence of carrier ampholytes Bio-Lyte 6/8 (BioRad). Twenty fractions were recovered. pH, adsorption at 280 nm, and activities toward nonacetylated RG and polygalacturonic acid were measured. Fractions were pooled according to the activity profile.
Enzyme analysis
Protein content was determined using BioRad reagent and bovine serum albumin (A-3912, Sigma) as standard (Bradford 1976).
Protein homogeneity and molar mass were evaluated by polyacrylamide gel electrophoresis under denaturing conditions (sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)). It was carried out in a MiniProtean 3 apparatus (BioRad) using a continuous 10–20% polyacrylamide gel in the presence of 0.1% sodium dodecyl sulfate.
In-gel digestion
Bands of interest were excised from the SDS-PAGE gels and subjected to in-gel trypsic digestion essentially as described in Devouge et al. (2007). Briefly, gel slices were washed with 100 µL of 25 mM NH4HCO3, followed by dehydration with 100 µL of 50% (v/v) acetonitrile in 25 mM NH4HCO3. Proteins were reduced and alkylated by incubation for 1 h at 57°C in the presence of 10 mM DTT followed by 45 min at room temperature after adding 55 mM iodoacetamide. Gel slices were washed with 25 mM NH4HCO3 and dehydrated as described before. Gel slices were rehydrated with 10 µL of 12.5 ng µL–1 trypsin (sequencing grade, Promega) in 25 mM NH4HCO3. After an overnight incubation at 37°C, the supernatant was collected, and the trypsic fragments were measured by liquid chromatography–tandem mass spectrometry (LC–MS/MS).
LC–MS/MS analysis and protein identification
Mass spectrometry analyses were conducted by the platform “Biopolymers-Interaction-Structural Biology” located at the INRA Center of Angers—Nantes (INRA UR1268, F-44300 Nantes, http://www.nantes.inra.fr/plateformes_et_plateaux_techniques/plateforme_bibs).
Nanoscale capillary LC–MS/MS analysis of the digested proteins was performed using a Switchos-Ultimate II capillary LC system (Dionex, Amsterdam, the Netherlands) coupled to a hybrid quadrupole orthogonal acceleration time-of-flight mass spectrometer (Q-TOF Global, Waters, Manchester, UK). Chromatographic separation was conducted on a reverse-phase capillary column (Pepmap C18, 75-μm internal diameter, 15-cm length, LC Packings) at a flow rate of 200 nL min-1. The gradient consisted of a linear increase from 2% to 40% of acetonitrile in 50 min, followed by a rapid increase to 50% of acetonitrile within 10 min.
Mass data acquisitions were piloted by the Mass Lynx software (Waters, Milford, MA, USA) using the so-called data-dependent acquisition mode: MS data were recorded for 1 s on the mass-to-charge (m/z) range 400–1,500, after which the three most intense ions (doubly or triply charged ions) were selected and fragmented in the collision cell (MS/MS measurements).
Raw data were processed using the Protein Lynx Global Server software (PLGS, version 2.2, Waters).
Protein identification was performed by comparing the collected LC–MS/MS data with the Uniprot sequence databank (http://www.uniprot.org/), restricted to the taxonomy fungi (release 14.4). Databank searches were performed using the Mascot server 2.2 program (Matrix Science).
Mass tolerance was set at 100 ppm for parent ions (MS mode) and 0.3 Da for fragment ions (MS/MS mode); one missed cut per peptide was allowed, and the oxidation of methionines was set as a variable modification.
Validation criteria were as follows: proteins were validated if they had more than two peptides with individual MASCOT ion scores above the significance threshold (p < 0.05; cutoff score 39). For single-peptide identifications, protein hits were validated after careful inspection of MS/MS spectra and when they corresponded to proteins from I. lacteus.
Nonassigned mass spectra were further subjected to de novo sequencing using the functionality implemented in PLGS. A manual inspection and validation of the resulting de novo sequences was performed in order to retrieve high-quality stretches of amino acids. These stretches were submitted to the MS-BLAST program (Shevchenko et al. 2001).
Influence of pH and temperature
To study the influence of pH on the enzymatic activity, the enzyme reaction was performed between pH 3.5 and 6 using 50 mM sodium acetate buffer for nonacetylated RG and 50 mM succinate buffer for acetylated RG at 40°C. To study the temperature optimum, the enzyme reaction was performed between 20°C and 60°C in 50 mM Na-acetate buffer, pH 5.0 for nonacetylated RG, and 50 mM succinate buffer pH 5.0 for acetylated RG.
pH stability was studied by preincubating the enzyme at 40°C for 90 min at various pHs, using 100 mM sodium acetate buffer (pH 3.5–6). Temperature stability was evaluated by measuring the residual activity after 90 min of preincubation of the enzyme at temperatures between 20°C and 70°C in 50 mM sodium acetate buffer, pH 5.0. The residual enzyme activity was assayed using nonacetylated RG (5 mg/mL) as the substrate.
Substrate hydrolysis for analysis of the degradation products
Enzymatic degradations of acetylated and nonacetylated RG were performed as follows: 3.5 mL of substrate solution (1 mg/mL in succinate buffer 50 mM, pH 5) were incubated at 40°C with 1 nkat enzyme per milligram substrate. Aliquots were withdrawn at intervals up to 48 h and inactivated by boiling 5 min. Then, samples were analyzed by high-performance anion-exchange chromatography (HPAEC) and mass spectrometry.
High-performance anion-exchange chromatography
HPAEC analyses were performed using a Waters 626 pump equipped with a Waters 600S controller and an autosampler Waters 717 plus. Oligomers were monitored using a pulsed amperometric detector (EC 2000; Thermo Separation Products). Of the degradation samples, 20 µL were applied on an analytical Carbopac PA1 column (4 × 250 mm; Dionex, Voisins les Brotonneux, France). The elution was carried out in a gradient of Na-acetate containing 20 mM NaOH. The gradient was as follows: 0–20 min, 200–350 mM; 20–40 min, 350–600 mM; 40–60 min, 600–700 mM; 60–65 min, 700–800 mM, and then back to the starting conditions. Standard oligomers from the laboratory collection were chromatographed in similar conditions, and response factors were calculated. Chromeleon software (Dionex) was used for data acquisition and processing.
Mass spectrometry on oligosaccharides
Electrospray ionization–ion trap mass spectrometry experiments were achieved on a LCQ Advantage ion trap mass spectrometer (ThermoFinnigan, USA) using negative electrospray as ionization process. First, the samples were desalted using a Carbohydrate Membrane Desalter (Dionex). Pure methanol was added to sample solutions (2:1; v/v) to favor the spray formation into the electrospray source. Samples were then placed at 4°C for 24 h before being centrifuged. Infusion was performed at a flow rate of 2.5 µL/min. Nitrogen was used as a sheath gas (20 arbitrary units). The MS analysis was carried out under automatic gain control conditions, using a typical needle voltage of 4.0 kV and a heated capillary temperature of 200°C. For MSn analyses, the various parameters were adjusted for each sample in order to optimize the signal and get maximal structural information from the ion of interest. More than 50 scans were summed for MSn spectra acquisition.
Results
Rhamnogalacturonan substrates
Acetylated and nonacetylated RG were both extracted from sugar beet pectin by enzymatic and alkaline treatments, respectively. The main sugars present in the nonacetylated substrate were GalA, galactose, and rhamnose with molar contents of 51%, 25.1%, and 20%, respectively (Table 1). Molar ratio between GalA and rhamnose was 2.5, indicating that nonacetylated RG contained more GalA than the typical alternating structure present in RG backbone. Due to its alkaline extraction, the substrate was neither acetylated nor methylated. The main sugars present in the acetylated RG were the same as in the nonacetylated RG but in different proportions: GalA, galactose, and rhamnose represented 38%, 28.5%, and 26% of the molar composition, respectively. Molar ratio between GalA and rhamnose was 1.5, so close to 1 and characteristic of an alternating GalA-rhamnose pattern. The molar ratio between rhamnose and galactose was close to 1, indicating that some galactose residues linked on rhamnose were resistant to enzymatic extraction although it included a galactanase and a galactosidase (Bonnin et al. 2008). However, this is an average ratio, and one can think that some rhamnose carry short galactose chains with two or three residues. Arabinose residues constituted only 2.5% of the acetylated RG showing that arabinans have been efficiently removed. The DAc of acetylated RG was 46 showing that almost one GalA every two was acetylated. This acetylated RG was thus suitable to investigate the action of a RG-hydrolase tolerant to acetyl substitution.
Enzyme purification
Prior to the purification, the Driselase preparation was characterized by measuring various degradation activities on plant cell wall polysaccharides (Table 2). The major activities found were xylanase, polygalacturonase, and cellulase. Lower activities were found toward galactan and arabinan. No rhamnopyranosidase activity was detected. Hydrolase activity on nonacetylated RG was 853 nkat/mL, whereas no lyase activity was detected, meaning that it corresponds only to a hydrolase activity. An RG hydrolase activity of 33 nkat/mL was detected on RG acetylated to 46%. No acetylesterase activity was detected, demonstrating that the degradation of acetylated RG by Driselase was not consecutive to a predeesterification of the substrate. The two activities toward acetylated RG and nonacetylated RG allow defining a tolerance ratio as the activity on nonacetylated RG vs the activity on acetylated RG. Depending on the Driselase batch, the tolerance ratio was found to vary between 12 and 26. For comparison, the same activities were measured on the same substrates with the RG-hydrolase from A. aculeatus, and the tolerance ratio was found to be 140.
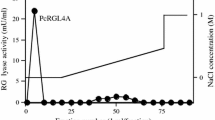
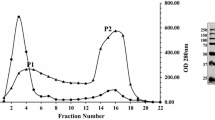
To purify this rhamnogalacturonase, a hydrophobic interaction chromatography (HIC) was first performed. Different polysaccharides degrading enzymatic activities were measured in the fractions resulting from this chromatography (Table 3). This fractionation step allowed the elimination of xylanase, cellulase, and arabinanase activities. However, the HIC fractions containing the rhamnogalacturonase still exhibited polygalacturonase and galactanase activities. A size-exclusion chromatography on Superdex 75 was then performed which resulted in two major protein peaks (Fig. 1), among which the second one corresponded to the rhamnogalacturonase activity. The size-exclusion sample was much more active on RG than the HIC sample (1.5-fold more), but the activities on polygalacturonic acid in the two samples were similar. A preparative isoelectric focusing (IEF) was performed as the last purification step. Activity profile on polygalacturonic acid showed that this activity was present only in the more acidic fractions (pH <6, Fig. 2). In the same time, the majority of the activity on nonacetylated RG was between pH 5 and 9. IEF allowed to separate RG activity in five peaks and shoulders (referred to as IEF1 to IEF5), so resulting in the isolation of different RG-hydrolases having various isoelectric points (pI): 6.2, 6.7, 7.0, 7.2, and 7.7 (Fig. 2). The purification procedure for RG-hydrolase is summarized in Table 4. Fractions IEF3, IEF4, and IEF5 exhibited the highest purification rates, each fraction containing about 1% of the initial RG activity present in Driselase.

Elution profile after Superdex chromatography. Dashed line protein; Filled triangle PG; Filled circle naRG

Elution profile after isoelectrofocalization. Dashed line protein; Filled triangle PG; Filled circle naRG
Enzyme identification
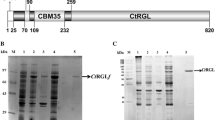
For each fraction issued from IEF fractionation, the activity was measured on acetylated and nonacetylated RG. The tolerance ratio was comprised between 11 and 14, indicating no difference between these fractions with respect to acetyl tolerance. SDS-PAGE analysis (Fig. 3) showed three bands in fractions IEF 1 and IEF 2, with Mw of 31, 43, and 55 kDa. IEF 3 exhibited the two bands of MW 31 and 55 kDa. Thus, IEF fractionation did not fully purify the different proteins. IEF 4 and IEF5 contained a single band at 55 kDa.

SDS-PAGE and Coomassie brilliant blue staining showing kaleidoscope prestained molecular weight markers (lane 1), Driselase mixture (lane 2), HIC pool (lane 3), Superdex pool (lane 4), fraction IEF1 (lane 5), fraction IEF2 (lane 6), fraction IEF3 (lane 7), fraction IEF4 (lane 8), fraction IEF5 (lane 9)
All the gel bands were excised and digested with trypsin. The resulting peptides were submitted to LC–MS/MS analysis. Data were confronted to databanks with the MASCOT program. This enabled to identify the 43-kDa band as an aspartic proteinase from I. lacteus (UniProt accession number P17576). Remaining nonassigned spectra were de novo sequenced and submitted to the sequence homology program MS-BLAST. Eight peptides were obtained from the 31-kDa protein. Four of them were found to be homologous (with more than 60% identity) to peptides from Aspergillus oryzae protease (UniProt accession number Q876Z9), whereas three of them were found highly homologous with a protease from A. oryzae (UniProt accession number Q70DX9), suggesting that the 31-kDa protein could be a protease. Fifteen peptides were de novo sequenced from the 55-kDa protein band. Eleven peptides showed high homology (more than 60% identity) with known RG-hydrolases from A. aculeatus (RGase A: Q00001), Aspergillus niger (RGase A: P87160 and RGase B: P87161), and Botryotinia fuckeliana (P87247), some of them being contiguous in the sequence. It allowed identifying the 55-kDa protein as a RG-hydrolase. This peptide analysis was carried out on the five 55-kDa proteins in the five IEF fractions, and the identified amino acids were similar. This suggests that the five 55-kDa proteins could be five isoforms of a sole enzyme.
IEF 4 fraction was chosen for further investigations as it was pure to homogeneity, and it contained the highest amount of protein.
Influence of pH and temperature
Influence of pH and temperature was examined toward nonacetylated RG and acetylated RG (Fig. 4). The optimum activity occurred between pH 4.5 and 5.0 and in a range of temperature from 40°C to 50°C. These results were similar for the two substrates. The enzyme stability was tested after 90 min preincubation in different pH or temperatures. The enzyme activity was stable from pH 3.5 to pH 6, where more than 60% of the activity remained (data not shown). After preincubation of the enzyme at different temperatures, the activity was stable up to 40°C. By increasing the temperature, the activity decreased dramatically, and less than 10% of the initial activity remained after preincubation at 60°C and beyond (data not shown).

Influence of pH (a) and temperature (b) on the activity of I. lacteus RG-hydrolase activity on nonacetylated RG (filled markers) and acetylated RG (open markers). Activity is expressed as a percentage of the maximum
Mode of action of I. lacteus RG-hydrolase
Oligomers obtained from the degradation of nonacetylated RG and acetylated RG by the RG-hydrolase from I. lacteus were examined by HPAEC and mass spectrometry. Oligomers were referred to as a combination of R for rhamnose, U for uronic acid, and G for galactose, each letter being followed by a figure indicating the number of each monomer. They constitute a homogenous series with a GalA at the reducing end. As an example, the developed formula of R2U2 is Rha-GalA-Rha-GalA.
Identification of the produced oligomers
Time course of hydrolysis of the nonacetylated and acetylated RG by the purified RG-hydrolase was followed by HPAEC pH 13 (Fig. 5), and response factors determined from standard oligomers were used to quantify the products.

Kinetics of degradation of nonacetylated RG (a) and acetylated RG (b) by I. lacteus RG-hydrolase
When nonacetylated RG was submitted to hydrolysis by the RG-hydrolase (Fig. 5a), R2U2 and R3U3 appeared as the major products of degradation. Major oligomers were, thus, of even degree of polymerisation (DP). Other oligomers in decreasing order were: R2U3, R3U3G1, and R4U4. The plateau was reached after 7 h of degradation for all the products. All the products of even DP carried GalA at the reducing end and rhamnose at the nonreducing end, confirming that the enzyme cleaves the linkage GalA-Rha. Oligomers having the highest DP (R5U5 and R4U4) reached their maximum after 30 min and 4 h, respectively, showing that they were first products and then substrates for the enzyme. The release of these linear oligomers of large DP confirmed that the nondegraded polymer contained long unsubstituted area, with contiguous Rha not carrying galactose. The presence of R2U3 suggested that there might be trace of RG-rhamnohydrolase in the RG-hydrolase fraction.
The products obtained from the hydrolysis of acetylated RG were also followed in HPAEC pH 13 (Fig. 5b). Due to the high pH of the eluent, all the ester groups were removed and separation of the oligomers took place according to a combination of DP and charge density. Thus, the same standard oligomers could be used for the quantification as for nonacetylated RG. When the acetylated substrate was degraded with the same amount of enzyme, the plateau was reached after 4 h for most of the products. The major final product formed was R3U3, followed by R4U4 and R2U2 (Fig. 5b). R3U3G1 was present as an intermediate product with a maximum at 7 h.
When comparing the two graphs (Fig. 5a and b), it can be seen that R4U4 and R5U5 were produced and further degraded in the case of nonacetylated RG, while they were produced in lower amounts in the case of acetylated RG and not further degraded. This shows that they were probably substituted and that the enzyme action was slowed down on these oligomers.
After 48 h hydrolysis, the sum of identified products was 52.6 µg/mL on acetylated RG and 158 µg/mL on nonacetylated RG, when 1 mg/mL of substrate was subjected to degradation. However, those sums were calculated only from identified products and some unknown peaks with increasing area along the time course of degradation were observed on the different chromatograms.
Location of the acetyl substituents on the hydrolysis products
The final oligosaccharides obtained from the hydrolysis of acetylated RG were analyzed by mass spectrometry. Oligosaccharides of high DP (R4U4 and R5U5) could not be detected, most probably because the highest the DP, the lowest the ionization capacity (Ralet et al. 2008). The full MS spectrum of the total hydrolysate showed a very high number of different products. It showed also that oligosaccharides came as partially or totally acetylated, whatever their DP. As an example, R3U3 was present without acetyl (m/z = 983), with 1 acetyl (m/z = 1025), 2 acetyls (m/z = 1067), and 3 acetyls (m/z = 1109). On the other hand, some of the oligomers carried galactose, as it is the case for R3U3G1. We previously explained how MSn experiments are used to assign the oligomer structure and the acetyl location, based on both the glycosidic and the cross-ring fragmentations (Quémener et al. 2003; Ralet et al. 2008). To determine the possible positions of the acetyl groups, R2U2 + 1Ac and R2U2 + 2Ac were explored in more details (Fig. 6). After isolation and collision-induced dissociation (CID) of the parent ion R2U2 + 1Ac at m/z 703 (Fig. 6a), the glycosidic cleavage ion at m/z 485 is compatible with the presence of the acetyl group on the reducing GalA. On the other hand, the glycosidic cleavage ion at m/z 527 indicates that the acetyl group can also be carried on the other GalA residue. The loss of 60, leading to an ion at m/z 643, can be attributed to a cross-ring cleavage compatible with a nonacetylated reducing GalA or a reducing GalA acetylated on C3. The loss of 102, leading to an ion at m/z 601, is representative of a cross-ring cleavage of a reducing GalA acetylated on C2 (Quémener et al. 2003). Thus, the fragmentation pattern agrees with three different structures:
-
Rha-GalA-Rha-Ac2GalA
-
Rha-GalA-Rha-Ac3GalA
-
Rha-AcGalA-Rha-GalA,
without knowing the exact location of the acetyl group in the last structure.

Chemical structure and negative CID mass spectra of R2U2, 1Ac (a), and R2U2, 2Ac (b). a MS2 experiment on R2U2, 1Ac (m/z 703 > products). b MS2 experiment on R2U2, 2Ac (m/z 745 > products). Fragments are labeled according to the Domon and Costello’s nomenclature
R2U2 + 2Ac is supposed to be fully acetylated as it contained as many acetyls as GalA. From the parent ion at m/z 745, we observed a sole glycosidic cleavage ion at m/z 527 indicating one acetyl on the reducing GalA and one acetyl on the rest of the molecule (Fig. 6b). The two cross-ring cleavages at m/z 685 (−60) and m/z 643 (−102) showed, as on R2U2 + 1Ac, that the acetyl group on the reducing GalA can be either on C3 or on C2.
The detailed analysis of the produced oligomers structures allowed us to conclude that the RGase from I. lacteus is able to cleave the linkage GalA-Rha, in a RG backbone, even if the GalA is acetylated and whatever the position of the acetyl group. This demonstrates the high tolerance of the enzyme for the presence of acetyl groups. It can be matched not only in the active site of the enzyme but even in the catalytic clamp.
As another member of the R2U2 family, R2U2G1 + 1Ac (m/z 865) was further fragmented to determine the exact position of the galactose residue (Fig. 7). As already seen with the two previous examples, the glycosidic cleavage ions at m/z 689 and 647 showed that the acetyl group can be indifferently on one or the other of the two GalA residues (Fig. 7a). The MS3 analysis of the C3 ion at m/z 647 (Fig. 7b) produced a glycosidic cleavage ion at 463 corresponding to galactose + rhamnose + GalA and a second glycosidic cleavage ion at m/z 321, corresponding to GalA + rhamnose. It was not possible to discriminate the reducing or nonreducing position of the rhamnose residue in those glycosidic cleavage ions. However, the presence of two cross-ring cleavage ions at m/z 543 and m/z 381 unambiguously shows that both rhamnose units in R2U2G1 can be galactosylated (Fig. 7c). The enzyme appears, thereby, highly tolerant to galactose moieties in the subsites −1 or −3 of its catalytic site.

Chemical structure and negative CID mass spectra of R2U2G1, 1Ac. a MS2 experiment (m/z 865 > products), b MS3 experiment (m/z 865 > 647 > products), c chemical structure of R2U2G1, 1Ac, and observed cleavages at each MS step. Fragments are labeled according to the Domon and Costello’s nomenclature (Domon and Costello 1988)
Therefore, we demonstrate here that the RG-hydrolase from I. lacteus is able to degrade not only acetylated but also galactosylated RG.
Discussion
I. lacteus is known to produce various polysaccharide degrading enzymes, as it is most often used to produce protoplasts (Duquenne et al. 2007). It has been used also for the screening of Arabidopsis thaliana stems for variation in cell wall polysaccharides (Gardner et al. 2002) and as source of several cellulose- and xylan-degrading enzymes (Hamada et al. 1999a, b; Kanda et al. 1985, 1976; Toda et al. 2005). Due to its large panel of polysaccharide degrading enzymes, Driselase has been used as a tool in structural studies of the cell walls (Brown and Fry 1993; Ishii 1995b; Needs et al. 1998).
In the present study, a new enzyme was purified from Driselase that depolymerizes acetylated rhamnogalacturonan. The use of chemical or enzymatic treatments of beet pectin allowed to obtain substrates from a unique source varying in the presence of acetyl groups and, thus, to investigate the effect of substrate substitution toward enzymatic activity. We followed the same strategy as Schols et al. (1990) who applied various enzymatic treatments on apple-modified hairy regions to obtain different polymeric substrates when reporting the first rhamnogalacturonase. Thus, we show that the I. lacteus enzyme is a RG-hydrolase able to tolerate the acetylation of its substrate. To quantify this tolerance, we used a substrate with DAc 46, and we defined a tolerance ratio as the ratio of the activities toward nonacetylated and acetylated RG. This ratio varied from 12 to 26 according to the Driselase batch, whereas A. niger RG-hydrolase exhibited a ratio of 140.
The I. lacteus enzyme had a molar mass of 55 kDa and was identified as an RG-hydrolase not only through its activity but also through its primary sequence. Indeed, 15 peptides have been elucidated after mass spectrometry analysis of the trypsin digest and de novo sequencing. Eleven of them presented high homology with RG-hydrolases from ascomycetes. The molar mass of 55 kDa is close to the 59 kDa found for the RG-hydrolase A from A. aculeatus (Kofod et al. 1994) but is less than the molar mass of RG-hydrolases A and B from A. niger which are about 70 kDa (Suykerbuyk et al. 1997). The RG-hydrolases A from the two Aspergilli are very similar (446 and 440 amino acids in A. niger and A. aculeatus RG-hydrolases A, sharing 76% identity), and their difference in molar mass may be due to difference in their glycosylation pattern. On the opposite, RG-hydrolase B from A. niger carries a C-terminal extension.
From our results, I. lacteus exhibited at least five different RG-hydrolases having different pI. As a wood-decaying fungus, I. lacteus needs to be as efficient as possible in a range of conditions. This is possible, thanks to these various proteins having the same activity but different pI. The enzyme purified from I. lacteus had a pI of 7.2. This value is higher than the one from A. aculeatus RG-hydrolase, which is of 4.5 (Kofod et al. 1994) and the values of 3.7 and 3.9 calculated for both A. niger RG-hydrolases (Suykerbuyk et al. 1997). The I. lacteus enzyme exhibited a pH optimum between 4.5 and 5 and a temperature optimum between 40°C and 50°C. These values are close to those observed for the RG-hydrolases from A. aculeatus, A. niger, and B. fuckeliana (Fu et al. 2001; Kofod et al. 1994; Suykerbuyk et al. 1997).
From mass spectroscopy analysis of the reaction products, it was confirmed that the enzyme cleaves the linkage GalA-Rha and released a reducing GalA unit, as already described for other RG-hydrolases (Mutter et al. 1994). Mass spectrometry showed also that the product could be galactosylated on the first rhamnose near the reducing GalA. The same particular product structure was previously demonstrated using 2D NMR spectroscopy on hydrolysates of apple pectin with A. aculeatus RG-hydrolase (Colquhoun et al. 1990).
Moreover, mass spectroscopy demonstrated that the hydrolysis products could be acetylated on the reducing GalA, showing the enzyme ability to bind substituted GalA in its active site, even near the catalytic site. To our knowledge, this is the first time that this behavior is shown for a RG-hydrolase. However, the data given by the protein analysis are not significantly different from those of the ascomycetes RG-hydrolases. None of these data allows explaining the tolerance of I. lacteus RG-hydrolase to acetyl groups. The amino acids involved in the active site could have a role in binding more or less substituted substrates. This was shown by comparing A. niger polygalacturonase II and Fusarium moniliforme polygalacturonase that differ in their tolerance to methyl groups (André-Leroux et al. 2005).
In the future, such an enzyme would be a powerful new tool to be used with other pectinolytic enzymes to study the acetylation pattern and structure of RG. It would be also used to produce acetylated pectic oligomers and study their biological properties, immunological, as well as prebiotic activities.
References
André-Leroux G, Tessier D, Bonnin E (2005) Endopolygalacturonases reveal molecular features for processivity pattern and tolerance towards acetylated pectin. Biochim Biophys Acta Proteins Proteomics 1749:53–64
Axelos MAV, Thibault J-F (1991) Influence of the substituents carboxyl and of the rhamnose content on the solution properties and flexibility of pectins. Int J Biol Macromol 13:77–82
Bobin-Dubigeon C, Hoebler C, Lognoné V, Dagorn-Scaviner C, Mabeau S, Barry J-L, Lahaye M (1997) Chemical composition, physico-chemical properties, enzymatic inhibition and fermentative characteristics of dietary fibres from edible seaweeds. Sci Aliment 17:619–639
Bonnin E, Brunel M, Gouy Y, Lesage-Meessen L, Asther M, Thibault J-F (2001) Aspergillus niger I-1472 and Pycnoporus cinnabarinus MUCL39533, selected for the biotransformation of ferulic acid to vanillin, are also able to produce cell wall polysaccharide-degrading enzymes and feruloyl esterases. Enza Microb Technol 28:70–80
Bonnin E, Clavurier K, Daniel S, Kauppinen S, Mikkelsen JDM, Thibault J-F (2008) Pectin acetylesterases from Aspergillus are able to deacetylate homogalacturonan as well as rhamnogalacturonan. Carbohydr Polym 74:411–418
Bradford MMA (1976) A rapid and sensitive method for the quantitation of protein utilizing principle of protein-dye binding. Anal Biochem 72:248–255
Brown JA, Fry SC (1993) Novel O-D-galacturonoyl esters in the pectic polysaccharides of suspension-cultured plant cells. Plant Physiol 103:993–999
Colquhoun IJ, de Ruiter GA, Schols HA, Voragen AGJ (1990) Identification by n.m.r. spectroscopy of oligosaccharides obtained by treatment of the hairy regions of apple pectin with rhamnogalacturonase. Carbohydr Res 206:131–144
Devouge V, Rogniaux H, Nesi N, Tessier D, Gueguen J, Larre C (2007) Differential proteomic analysis of four near-isogenic Brassica napus varieties bred for their erucic acid and glucosinolate contents. J Proteome Res 6:1342–1353
Domon B, Costello CE (1988) A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate 5:397–409
Duquenne B, Eeckhaut T, Werbrouck S, Van Huylenbroeck J (2007) Effect of enzyme concentrations on protoplast isolation and protoplast culture of Spathiphyllum and Anthurium. Plant Cell Tissue Organ Cult 91:165–173
Englyst HN, Cummings JH (1988) Improved method of measurement of dietary fiber as non-starch polysaccharides in plant foods. J Ass Off Anal Chem 71:808–814
Fu J, Prade R, Mort A (2001) Expression and action pattern of Botryotinia fuckeliana (Botrytis cinerea) rhamnogalacturonan-hydrolase in Pichia pastoris. Carbohydr Res 330:73–81
Gardner SL, Burrell MM, Fry SC (2002) Screening of Arabidopsis thaliana stems for variation in cell wall polysaccharides. Phytochemical 60:241–254
Hamada N, Fuse N, Shimosaka M, Kodaira R, Amano Y, Kanda T, Okazaki M (1999a) Cloning and characterization of a new exo-cellulase gene, cel3, in Irpex lacteus. FEMS Microbiol Lett 172:231–237
Hamada N, Hirohashi K (2000) Cloning and transcriptional analysis of exocellulase I gene from Irpex lacteus. J Tokyo Univ Fish 87:39–44
Hamada N, Okumura R, Fuse N, Kodaira R, Shimosaka M, Kanda T, Okazaki M (1999b) Isolation and transcriptional analysis of a cellulase gene (cell) from the basidiomycete Irpex lacteus. J Biosci Bioeng 87:97–102
Hoebler C, Brillouet J-M (1984) Purification and properties of an endo (1→4)-β-d-xylanase from Irpex lacteus (Polyporus tulipiferae). Carbohydr Res 128:141–155
Ishii T (1995a) Isolation and charaterization of acetylated rhamnogalacturonan oligomers liberated from bamboo shoot cell-walls by Driselase. Mokusai Gakkaishi 41:561–572
Ishii T (1995b) Pectic polysaccharides from bamboo shoot cell-walls. Mokuzai Gakkaishi 41:669–676
Kanda T, Amano Y, Nisizawa K (1985) Purification and properties of two endo-1, 4-ß-xylanases from Irpex lacteus (Polyporus tulipiferae). J Biochem 98:1545–1554
Kanda T, Wakabayashi K, Nisizawa K (1976) Xylanase activity of an endo-cellulase of carboxymethylcellulase type from Irpex lacteus (Polyporus tulipiferae). J Biochem 79:989–995
Kofod LV, Kauppinen S, Christgau S, Andersen LN, Heldt-Hansen HP, Dörreich K, Dalbøge H (1994) Cloning and characterization of two structurally and functionally divergent rhamnogalacturonases from Aspergillus aculeatus. J Biol Chem 269:29182–29189
Lahaye M, Thibault JF (1990) Purification of arabinanases and galactanases from Aspergillus niger. In: Proceedings of the 3rd International Workshop on Plant Polysaccharides, Structure, and Function, Le Croisic, France, 19–21 September
Lahaye M, Vigouroux J, Thibault JF (1991) Endo-β-1,4-D-galactanases from Aspergillus niger var. aculeatus. Purification and some properties. Carbohydr Polym 15:431–444
Levigne S, Thomas M, Ralet M-C, Quéméner B, Thibault J-F (2002) Determination of the degrees of methylation and acetylation of pectins using a C18 column and internal standards. Food Hydrocoll 16:547–550
Mutter M, Beldman G, Schols HA, Voragen AGJ (1994) Rhamnogalacturonan α-L-rhamnopyranohydrolase: a novel enzyme specific for the terminal non reducing rhamnosyl unit in rhamnogalacturonan region of pectins. Plant Physiol 106:241–250
Needs PW, Rigby NM, Colquhoun IJ, Ring SG (1998) Conflicting evidence for non-methyl galacturonoyl esters in Daucus carota. Phytochemical 48:71–77
Nelson N (1944) A photometric adaptation of the Somogyi method for determination of glucose. J Biol Chem 153:375–380
Quémener B, Cabrera-Pino JC, Ralet M-C, Bonnin E, Thibault J-F (2003) Assignment of acetyl groups to O-2 and/or O-3 of pectic oligogalacturonides using negative electrospray ionization ion trap mass spectrometry. J Mass Spectrom 38:641–648
Ralet M-C, Crépeau M-J, Bonnin E (2008) Evidence for a blockwise distribution of acetyl groups onto homogalacturonans from a commercial sugar beet (Beta vulgaris) pectin. Phytochemical 69:1903–1909
Schols HA, Geraeds CCJM, Searle-van Leeuwen MF, Kormelink FJM, Voragen AGJ (1990) Rhamnogalacturonase: a novel enzyme that degrades the hairy regions of pectins. Carbohydr Res 206:105–115
Schols HA, Voragen AGJ (1994) Occurrence of pectic hairy regions in various plant cell wall materials and their degradability by rhamnogalacturonase. Carbohydr Res 256:83–95
Schols HA, Voragen AGJ (1996) Complex pectins: structure elucidation using enzymes. In: Visser J, Voragen AGJ (eds) Pectins and pectinases, progress in biotechnology, vol 14. Elsevier, Amsterdam, pp 3–19
Shevchenko A, Sunyaev S, Loboda A, Shevchenko A, Bork P, Ens W, Standing KG (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal Chem 73:1917–1926
Sturgeon RJ (1990) Monosaccharides. In: Dey PM, Harbone JB (eds) Methods in plant biochemistry, vol 2. Academic, London, pp 1–37
Suykerbuyk MEG, Kester HCM, Schaap PJ, Stam H, Musters W, Visser J (1997) Cloning and characterization of two rhamnogalacturonases from Aspergillus niger. Appl Environ Microbiol 63:2507–2515
Thibault J-F (1979) Automatisation du dosage des substances pectiques par la méthode au méta-hydroxydiphenyl. Lebensm Wiss Technol 12:247–251
Toda H, Takada S, Oda M, Amano Y, Kanda T, Okazaki M, Shimosaka M (2005) Gene cloning of an endoglucanase from the Basidiomycete Irpex lacteus and its cDNA expression in Saccharomyces cerevisiae. Biosci Biotech Biochem 69:1262–1269
Tsumuraya Y, Mochizuki N, Hashimoto Y, Kovac P (1990) Purification of an exo-β-(1, 3)-D-galactanase of Irpex lacteus (Polyporus tulipiferae) and its action on arabino-galactan-proteins. J Biol Chem 265:7207–7215
Voragen AGJ, Pilnik W, Thibault JF, Axelos MAV, Renard CMGC (1995) Pectins. In: Stephen AM (ed) Food polysaccharides and their applications. Marcel Dekker, New York, pp 287–339
Voragen AGJ, Rombouts FM, Pilnik W (1971) The influence of the degree of esterification on the activity of pectin- and pectate-lyases. Lebensm Wiss Technol 4:126–128
Acknowledgments
We gratefully thank Audrey Geairon and David Ropartz from the ‘‘Biopolymers-Interaction-Structural Biology” platform located at the INRA Center of Nantes (http://www.nantes.inra.fr/plateformes_et_plateaux_techniques/plateforme_bibs) for the mass spectrometry analyses. Sylviane Daniel is acknowledged for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Normand, J., Ralet, MC., Thibault, JF. et al. Purification, characterization, and mode of action of a rhamnogalacturonan hydrolase from Irpex lacteus, tolerant to an acetylated substrate. Appl Microbiol Biotechnol 86, 577–588 (2010). https://doi.org/10.1007/s00253-009-2310-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-009-2310-3