
Abstract
The Great Salt Plains of Oklahoma is a natural inland terrestrial hypersaline environment that forms evaporite crusts of mainly NaCl. Previous work described the bacterial community through the characterization of 105 isolates from 46 phylotypes. The current report describes the archaeal community through both microbial isolation and culture-independent techniques. Nineteen distinct archaea were isolated, and ten were characterized phenetically. Included were isolates phylogenetically related to Haloarcula, Haloferax, Halorubrum, Haloterrigena, and Natrinema. The isolates were aerobic, non-motile, Gram-negative organisms and exhibited little capacity for fermentation. All of the isolates were halophilic, with most requiring at least 15% salinity for growth, and all grew at 30% salinity. The isolates were mainly mesothermic and could grow at alkaline pH (8.5). A 16S rRNA gene library was generated by polymerase chain reaction amplification of direct soil DNA extracts, and 200 clones were sequenced and analyzed. At 99% and 94% sequence identity, 36 and 19 operational taxonomic units (OTUs) were detected, respectively, while 53 and 22 OTUs were estimated by Chao1, respectively. Coverage was relatively high (100% and 59% at 89% and 99% sequence identity, respectively), and the Shannon Index was 3.01 at 99% sequence identity, comparable to or somewhat lower than hypersaline habitats previously studied. Only sequences from Euryarchaeota in the Halobacteriales were detected, and the strength of matches to known sequences was generally low, most near 90% sequence identity. Large clusters were observed that are related to Haloarcula and Halorubrum. More than two-thirds of the sequences were in clusters that did not have close relatives reported in public databases.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Great Salt Plains (GSP) of Oklahoma are part of the Salt Plains National Wildlife Refuge in north-central Oklahoma. The National Science Foundation-funded Salt Plains Microbial Observatory was established there to isolate and characterize the diverse halotolerant microbes in hypersaline soils at the site. The GSP are barren sandy mud flats covering 65 km2 that are crusted with evaporite salt deposits derived from briny aquifers [24, 41]. Areas of the surface have small ponds of saturated brine, mainly NaCl, while others support only transient pools. The environment is extreme not only in salinity, as the surface is exposed to unobstructed UV irradiance, desiccation, alkaline pH, and high temperature (50°C or more). In addition, the environment is extremely dynamic, with rain events creating transient freshwater pools on the salt-saturated soils. These pools dry in a matter of days, increasing in salinity over time, and associated with this are shifts in the microbial community [27].
The GSP is an environment of extremes, particularly in salinity and pH. For organisms adapted to these extremes, this is a normative environment. What makes this a poikiloenvironment in sensu Gorbushina & Krumbein [18] are the unpredictable and sporadic rainfall events that cause unexpected and drastic changes in salinity. Organisms successful in this environment need to overcome unfavorable times and may be expected to exhibit wide environmental tolerances. Many of the GSP bacterial isolates exhibit such characteristics [8, 26]. Competition is expected to be central to controlling diversity in such a restricted environment, but spatial separation in soils may lower competition [20]. Ecosystem disturbance also can drive changes in diversity. The intermediate disturbance hypothesis suggests that very stable or very unstable environments will have less diversity than moderately unstable environments [35]. Bacterial community structure has been shown to change due to environmental disturbance [1, 16, 32, 49]. The implication is that poikiloenvironments would have relatively little diversity. However, since the disturbance is episodic, although severe, the GSP could be considered an environment with an intermediate level of disturbance and hence be expected to be more diverse.
Previous work at the Salt Plains Microbial Observatory has focused on halotolerant aerobic bacteria [8, 26, 51]. The collection of bacterial isolates obtained by enrichment culture was dominated by Halomonas and Bacillus species, although 46 phylotypes were represented. In some cases, the isolates were halotolerant strains of bacteria common in oligohaline soils. A wide salt tolerance range was observed for most of the GSP bacterial isolates, with growth in media from 0.1% salinity to over 20% salinity. Some showed tolerance to a wide range of pHs and temperatures, and high resistance to UV irradiation. In a changing environment, microbes need to be flexible to survive. The current report describes halophilic archaeal isolates obtained by enrichment culture of GSP soils. The isolates have been characterized biochemically, physiologically, and phylogenetically. Culture-independent 16S rRNA gene clone libraries were used to estimate archaeal diversity at the GSP. This is the first large molecular analysis of archaeal communities from natural inland terrestrial hypersaline soils. Preliminary accounts of this work have been presented previously [7, 9, 44].
Materials and Methods
Sample Collection
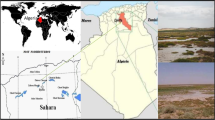
Soil samples were collected from three sites at the GSP between August 2001 and August 2002 (Fig. 1): WP3, N 36°43.044′, W 98°15.702′; WP5, N 36°43.661′, W 98°15.526′; and WP6, N 36°43.851′, W 98°15.561′. These grab samples of surface crusts included the top centimeter of soil on the unvegetated salt-crusted areas of the salt plains with a pH of approximately 7.8 and a salinity of 7.5% as described previously [8]. Each bulk sample was mixed by hand and aliquots taken for inoculation of enrichment cultures and storage for molecular analyses. Samples for molecular analysis (approximately 100 g) in sterile Whirl-Pak bags were frozen in the field on dry ice, transported to the laboratory on dry ice, and stored at −80°C for later use. Samples for establishing enrichment cultures and dilution-plating experiments were placed in sterile Whirl-Pak bags or directly into tubes of media, transported to an on-site laboratory at 25°C using a cold pack, and processed within 1 to 2 h of collection. One-gram aliquots of soil samples were added to 30 mL of sterile media in polypropylene tubes. The tubes were capped with sterile foam plugs to allow for aeration and transported at 25°C to the laboratory at Wichita State University.

Map of the Salt Plains National Wildlife Refuge showing the salt flats and sampling sites used for the current study
Enrichment and Isolation
Direct plating, liquid enrichment, and dilution plating were used to isolate halophiles from GSP soils. Halophiles moderate (HM) medium has high salinity (~25%) and high magnesium and was used at 37°C, with plates being kept in a moist chamber to prevent rapid drying of the surface. The composition of HM medium was based on earlier reports [38, 42] and previously described in detail [8]. Although this medium was directed at the isolation of halophilic archaea, several bacterial isolates were obtained as well, including GSP 64, 65, and 66 [8].
In some cases, soil aliquots (approximately 0.5 g) were spread directly onto the surface of plates. The plates were maintained at the appropriate temperature, and colonies were collected after several days. The plates were kept for several weeks, and representatives of new colony types were harvested to capture slower growing organisms. The 30-mL enrichment tubes inoculated at the field laboratory were aseptically transferred to 250-mL Erlenmeyer flasks containing 70 mL of HM medium. Liquid enrichment cultures were incubated on a rotary shaking platform (2.5-cm stroke diameter) at 150 rpm. Aliquots (100 µl) were plated after 24 and 48 h. One milliliter of the liquid enrichment cultures was transferred to fresh liquid medium after 1 week, and 100-µl aliquots were again plated after 24 and 48 h. For dilution plating, soil samples (approximately 1 g) were added to tubes containing 10 ml of 20% brine and shaken for 15 min. The liquid was then serially diluted in 20% brine and plated on HM medium.
Colonies arising on the plates were selected for isolation based on gross morphological and physiological features, differing in pigmentation, size, margin, or rate of growth. Colonies were transferred to fresh HM agar plates and isolated using the streak plate method. Each isolate was subjected to at least five successive streak platings to ensure clonal purity. Isolates were subjected to phenetic and phylogenetic analyses as described below. The isolates were maintained on agar slants and at −80°C as 50% glycerol stocks.
Morphological, Physiological, and Biochemical Tests
Differential media were supplemented with 25% (w/v) NaCl unless otherwise noted. All tests were incubated in duplicate at 37°C and scored at 24, 48, and 72 h unless otherwise noted, and all tests were accompanied by positive and negative controls using known laboratory strains and oligohaline media.
Isolates were Gram-stained using the PROTOCOL Gram-staining kit (Fisher Diagnostics) following the manufacturer's instructions. Motility was assessed by examining wet mounts of 24-h cultures at 1,000× and by stab inoculation of sulfur–indole–motility medium deeps (SIM, BBL Fisher Scientific). The addition of 3% hydrogen peroxide solution to confluent plates or smears of confluent culture on slides was used to detect catalase. Oxidase testing was performed using the BBL DrySlide system according to the manufacturer's instructions. Hydrolysis of gelatin was determined using nutrient gelatin agar deeps (Difco) and lipid hydrolysis with Spirit Blue agar (Difco) containing homogenized olive oil and Tween 80 solution. Hydrogen sulfide production was assayed using SIM media (BBL). Urease activity was determined at 5 days using urea broth with phenol red indicator. Production of acid and gas from carbohydrates was tested with 0.5% (w/v) glucose, lactose, or sucrose in culture tubes containing inverted Durham tubes, using a 25% NaCl solution supplemented with Bacto tryptone (10 g L−1) and phenol red (18 mg L−1; pH 7.3). HM medium composition was modified for different salt concentrations, pHs, and magnesium concentrations. Temperature tolerance was measured in liquid shake tubes. Results are reported as the maximum and minimum conditions for growth, with optima representing those cultures that first showed growth.
PCR, Cloning, and DNA Sequencing
Crude DNA extracts from each isolate were prepared using a freeze–thaw technique as described in Caton et al. [8]. Genomic DNA in the supernatant was the target for polymerase chain reaction (PCR) amplification of 16S rRNA genes using the universal archaeal primers Arch69F (5′ TAA GCC ATG CRA GTC GAA YG; [25]) and Arch958R (5′ YCC GGC GTT GAM TCC AAT; [10]). PCR was performed in a thermal cycler (Eppendorf Mastercycler) as 100-µL reactions containing 1× PCR buffer (ABI), 20 µM of each dNTP, 0.2 μM of each primer, 1 U of Taq DNA polymerase (ABI), and 5 µL of cell extract. DNA was denatured at 95°C for 2 min, followed by 40 cycles of 95°C for 1 min, 50°C for 1 min, and 72°C for 1 min, with a final 5-min extension at 72°C.
For cultivated isolates, two or more amplicons were pooled for each set of 16S rRNA gene sequencing reactions in order to maximize sequencing template concentration. Amplicons were prepared for sequencing reactions using Millipore filtration units that remove excess dNTPs and the oligonucleotides used in the amplification. Sequence data were obtained with the protocols and reagents that accompany the four-color PRISM BigDye kit designed for use with the ABI 373 automated DNA sequencing system. Base calls were completed using the ABI PRISM DNA Sequencing Analysis Software package (v. 3.3). All sequence fragments generated from a given template were edited against electropherograms and then assembled into contigs using Sequencher 4.1 (Genecodes, Ann Arbor, MI, USA). For most sequences, two to four overlapping fragments (from both the coding and non-coding strand) were used to assemble the contigs.
Genomic DNA extracts were made directly from GSP soils using the protocol of Bürgmann et al. [5], with some modifications as follows. Soil samples (0.5 g) were weighed in 2-ml sterile microcentrifuge tubes, and 0.75 g of glass silica/zirconia beads (0.2 mm) were added with 1.25 ml of extraction buffer (0.2% (w/v) CTAB, 1 mM dithiothreitol, 0.2 M sodium phosphate [pH 8.0], 0.1 M NaCl, and 50 mM EDTA). The samples were processed with a bead-beater (Genie Cell Disruptor; Scientific Industries, Inc.) at 4°C, at maximum speed, for 3 min. The homogenates were clarified by centrifugation at 16,000 × g for 5 min, and the supernatant was transferred to a fresh sterile 2-ml microcentrifuge tube. Buffer-equilibrated phenol (350 µL; pH 8.0) was added to the supernatant, well mixed by vortexing, and centrifuged for 5 min at 16,000×g. The supernatant was transferred to a fresh sterile 2-ml microcentrifuge tube, and 350 µL of water-equilibrated chloroform-isoamyl alcohol (24:1, v/v) was added; the solution was mixed by vortexing and then centrifuged at 16,000×g for 5 min. An aliquot of the aqueous phase (700 μL) was incubated for 1 h at 37°C with 750 μL of precipitation solution (20% (w/v) polyethyleneglycol 6000 in 2.5 M NaCl solution). Nucleic acids were pelleted by centrifugation at 16,000×g for 30 min at room temperature. The pellet was rinsed with ice-cold 70% ethanol, air dried for 5 min at room temperature, and resuspended in 100–300 µL of TE buffer (10 mM Tris–HCl [pH 8.0] and 1 mM EDTA). The purity of the extracted DNA was assessed spectrophotometrically by measuring A 260/A 280 (DNA to protein) and A 260/A 230 (DNA to humic acids) ratios with a Genesys 5 spectrophotometer (Fisher).
Extracts of genomic DNA were used as templates for PCR amplification of archaeal 16S rRNA gene sequences as described above. Ten separate PCR amplicon populations were purified by band excision from a 2% agarose gel after electrophoresis and pooled. Clone libraries were generated from the pooled amplicons using a TOPO-TA blue-white cloning system in Escherichia coli (Invitrogen) following the manufacturer's instructions. More than 200 clones were randomly collected and inoculated into 96-well plates, with plasmid isolation and insert sequencing by a commercial vendor (Agencourt) using the Arch69F primer. Sequences were trimmed to remove remaining vector regions, leaving sequences of approximately 650 bp for analysis. All sequences appear in GenBank with accession numbers FJ696218 to FJ696385.
Sequence Analyses
All sequences were automatically aligned using CLUSTAL W [47] and then manually aligned and trimmed in MacClade v4.08 (Sinauer Associates). Contextual 16S rRNA gene sequences were identified in GenBank using BLAST [2] or from comparison to relevant literature, including Bergey's Manual [19]. PAUP 4.0 b10 [46] generated phylogenetic trees using distance analysis with Jukes–Cantor rules and the neighbor-joining algorithm. Sequences were trimmed to equal lengths, with short sequences removed and positions with gaps and ambiguous bases ignored, giving 556 positions for analysis. Jackknife was used to assess the relative support for each branch with a total of 100 bootstrap replicates conducted heuristically using the distance-based neighbor-joining algorithm and the nearest-neighbor-interchange algorithm in PAUP 4.0 b10. The tree was rooted using the sequence from Methanospirillum hungatei (M60880) as the functional outgroup. Putative chimeras were identified through several iterative analyses using the BELLEROPHON server [22]. Each putative chimera was manually checked by building trees where the two sections of the sequence were treated separately. Those that clustered together were not considered chimeras. True chimeras were removed from the analysis. The complete tree (Fig. S1) was edited for presentation in the manuscript by grouping closely related sequences.
Distance files were further analyzed using the DOTUR statistical package [43] to determine Chao1 estimators, Simpson indices, Shannon indices, accumulation curves, rarefaction curves, and to define operational taxonomic units (OTUs) at various levels of sequence identity. Coverage was determined according to the equation C = 1 − (n1/N), where n1 is the number of OTUs with only one representative and N is the total number of clones, as described previously [17].
Results
Collection and Identification of Archaeal Isolates
Nearly 100 bacterial isolates were obtained from GSP soil samples and characterized as described in earlier reports [8, 26, 51]. Archaeal isolates were obtained at the same time, through enrichment at 37°C in HM medium containing 25% salt. A total of 19 distinct archaeal isolates were recovered, and ten were retained in culture for characterization. Phylogenetic analysis using 16S rRNA gene amplicons is shown in Figs. 2 and S1. Isolates maintained in culture are designated with ‘GSP’ numbers (GSP 100–109), while those isolates obtained from dilution plating and not retained in culture are designated with ‘MO’ numbers. All of the MO isolates were obtained from the WP6 site, along with GSP 104 and 107. The isolates GSP 100, 102, 103, 105, 106, and 109 were from WP3, while isolates GSP 101 and 108 were from WP5. These sites were all roughly equivalent salt-crusted mud flats.

Summary phylogenetic tree for GSP archaea based on 16S rRNA gene sequences. GSP sequences are included from both the clone library and cultured isolates. Solid triangles represent clusters of clones. Bootstrap values greater than 50% are shown. An expanded phylogenetic tree including all GSP clones and isolates and additional contextual sequences is presented in Fig. S1
All of the GSP archaeal isolates were related to known haloarchaeal isolates from hypersaline environments. Dilution plating indicated that the most abundant haloarchaeons culturable under the conditions used (GSP 103 and 8 other isolates) were most closely related to Haloferax mediterranei. Another abundant isolate, MO51, was within the Haloferax but part of a separate cluster not far removed from H. mediterranei. Isolates GSP 102 and 109 clustered with Natrinema pallidum, while a nearby cluster included the abundant isolates from dilution plating, MO 19, 23, and 24, that were most closely related to Haloterrigena thermotolerans. Two isolates, GSP 101 and 108, were most closely related to Haloarcula hispanica, and GSP100 was most closely related to Halorubrum sodomense. Archaeal abundance values are not reported here, as it was not possible to clearly distinguish archaeal and bacterial colonies. However, overall microbial abundance on HM plates at 25% salinity was approximately 1 × 103 cfu per gram soil, compared with approximately 5 × 105 cfu per gram soil on 10% salinity SP plates [8].
Characterization of Archaeal Isolates
The GSP archaeal isolates were characterized phenetically through biochemical and physiological tests (Table 1). All of the isolates were aerobic, nonmotile, and stained Gram-negative. All of the isolates were oxidase-negative, while nearly all were catalase-positive. Haloarchaea are typically oxidase-positive [21], so this finding is unusual and may be due to high salinity interfering with the commercially available assay kit used. Aerobic bacterial isolates from these soil samples showed a similar pattern, with >90% being catalase-positive and fewer than 30% being oxidase-positive [8]. All of the archaeal isolates were negative for gelatinase, lipase, urease, and hydrogen sulfide production, while some isolates could hydrolyze starch. None of the GSP bacterial isolates from this soil were positive for gelatinase or sulfide production; however, 75% were lipase-positive. Fermentation was limited, with glucose being the substrate most used by the archaeal isolates, as also was seen for the GSP bacterial isolates.
Environmental conditions at the GSP can be harsh, so the isolates were tested for their ability to grow under extreme conditions. All of the isolates were halophilic, requiring at least 5% salinity (Fig. 3). In general, the GSP isolates tended to have broader tolerance ranges than are typical for their genera, by extending growth to lower salinities [21, 37]. For instance, Haloferax-like isolate GSP106 grew at 5% salinity (less than 1 M), while three others in the Haloferax cluster could grow at 10% salinity. Haloferax spp. from other hypersaline soils have shown prolonged survival at 4% NaCl [14]. Halobacterium and Natrinema isolates typically will not grow below 17% (3 M) salinity, but related strains from the GSP could grow at 15% salinity or even 10% salinity in the case of Natrinema-like GSP109. All of the GSP isolates had salinity optima of 20% or more.

Salinity tolerance of GSP archaeal isolates. The ranges permissible for growth are indicated by bars, and the optimal salinities for growth are indicated by closed circles
None of the GSP isolates grew at pH 5.5, although this is within the range observed for these genera. All of the GSP isolates, except GSP 104 and 109, were able to grow at pH 8.5, not surprising given the alkalinity of most GSP soils. None of the GSP isolates could grow at pH 9.5. All GSP isolates grew at 25 and 37°C. Only GSP 105 and 106 could grow at 45°C, and none could grow at 50°C, although haloarchaea are expected to perform well at these temperatures [21]. None of the isolates grew at 4°C, which is not surprising as isolates from these genera typically do not grow below 15°C. Isolates from these genera typically have modest magnesium requirements, in the 10–50 mM range. All of the GSP isolates grew at 10 mM Mg. GSP 102 and 104 showed growth increases of 50% or more at 100 mM Mg relative to 10 mM Mg. All of the isolates exhibited growth increases of twofold or more at 0.5 M Mg relative to 10 mM Mg.
Culture-Independent Clone Libraries
Archaeal 16S rRNA gene clone libraries were prepared from direct DNA extracts from a WP6 soil sample collected in June 2002 that was used previously for the enrichment and isolation of archaea and bacteria [8] and from which bacterial 16S rRNA gene clone libraries had been constructed [7]. To reduce biases in the library, the amplicons from ten separate PCRs were combined in the cloning reaction. A total of 200 archaeal 16S rRNA gene clones were sequenced. From this group, 45 (23%) sequences were identified as potential chimeras, consistent with chimera frequencies previously reported [49, 50]. Each was checked manually, and 24 (12%) sequences were excluded because their 5′ and 3′ ends were closely related to members of different phylogenetic groups. Other sequences were rejected as being too short or too full of ambiguous bases for analysis. The overall clustering of major monophyletic groups closely resembled that of Bergey's Manual [19].
Estimation of Diversity and Coverage
The results of statistical estimates [43] of species richness, evenness, and the efficiency of species collection are given in Table 2 and Fig. 4. The data are presented at several levels of identity, reflecting commonly used thresholds for distinction of strain (99%), species (97%), genus (94%), and division (89%). The number of OTUs increases at higher levels of sequence identity as expected. With a threshold of 99% sequence identity, 36 OTUs were identified within the archaeal library. Good's coverage values were high at lower levels of identity, reaching 100% at the 89% sequence identity threshold, indicating that the library likely included all of the diversity at these higher taxonomic levels. Coverage was relatively high (59%) at 99% sequence identity but lower than at 89% sequence identity, indicating that more sampling is needed to fully describe diversity at the 99% sequence identity level.

Rarefaction curves based on 16S rRNA gene sequences obtained from the archaeal clone library as analyzed using DOTUR. The curves represent different levels of sequence identity
This is reinforced by the rarefaction curves presented in Fig. 4. Again, the data is presented at several sequence identity thresholds. The rarefaction curve based on 89% sequence identity quickly leveled off, indicating that nearly all of the diversity at this level of distinction is represented in the clone library. The rarefaction curves based on 94% and 97% sequence identity indicate that much of the diversity at the species and genus levels is represented in the clone library, although these curves did not fully level off. At 99% sequence identity, the rarefaction curve did not level off, indicating that more sampling is needed to adequately describe the archaeal assemblage at this taxonomic level. Accumulation (collector's) curves gave slopes similar to the rarefaction curves, except at 99% sequence identity, suggesting that the rarefaction curve overestimates the number of potential OTUs at this level.
Chao1 estimates were used to project the total number of OTUs at different levels of sequence identity (Table 2). At the 99% sequence identity level, 53 OTUs were predicted, while 27 OTUs were predicted at the 97% sequence identity level. By comparing the Chao1 estimates to the OTUs delineated in the sequence library, it is estimated that 68%, 85%, and 86% of the archaeal diversity was sampled at 99%, 97%, and 94% sequence identity, respectively. At 89% sequence identity, all of the estimated diversity was present in the sequence library. The Shannon and Simpson indices both indicate that archaeal diversity increases at higher levels of sequence identity. Based on the Shannon index, archaeal assemblage diversity at the GSP is comparable to, or somewhat lower than, those reported in previous studies of other hypersaline habitats [29, 49], although the GSP is the first natural inland hypersaline soil to be examined. A measure of evenness, H/H max, increases at higher levels of sequence identity but is relatively high across all levels of identity, indicating that the distribution of archaeal taxa in the library is relatively even, as was reported for the Wadi An Natrun lakes [29].
Phylogenetic Groups Recovered
Phylogenetic trees based on the 16S rRNA clone library were generated, including sequences from cultured and uncultured archaea obtained from the GenBank database (Figs. 2 and S1). Initial BLAST analyses of the GSP sequences found few published sequences with greater than 90–92% sequence identity. This level of divergence has been observed in earlier studies (e.g., [29]), and even the larger database now available did not uncover sequences of greater similarity. Within the overall phylogenetic tree generated (Fig. 2 and S1), several clusters of GSP sequences did not closely match known sequences. These clusters (GSP clusters A, B, and C) contained the most abundant archaeal clones in the GSP sequence library.
All of the GSP clones fell within the Euryarchaeota. Only Halobacteriales sequences were recovered, and no Methanosarcinales sequences were detected. No crenarchaeal or korachaeal sequences were recovered. Initial PCR amplifications directed at crenarchaeal 16S rRNA gene sequences in GSP soils have been negative (Seth A. Perkins, unpublished observation). Both Methanosarcinales and Crenarchaeota sequences were detected in the previously reported Salt Spring clone libraries [49], while the Wadi An Natrun clone libraries included Methanosarcinales [29].
Generally, the cultured GSP isolates were not closely related to sequences found in the GSP clone library. Previous studies have shown that the vast majority of species identified through culture-independent molecular analyses have not been cultivated [11, 40]. In the current study, for instance, the group of cultured isolates that includes MO55, GSP103, and several others clusters with known Haloferax spp. sequences (94–99% sequence identity), but this cluster contains no GSP clones. None of the cultured isolates clustered with the most abundant GSP clones in the major GSP clusters of Fig. 2. In contrast, the Halorubrum cluster includes GSP100 and is closely related to GSP clones arch72, arch194, and arch195.
The larger clusters of GSP clones represent the most abundant sequences recovered and, yet, have little similarity to previously reported sequences, including the UEG clusters of Walsh et al. [49]. GSP clusters A and B represent the most abundant groups in the library, with 55 clones each, more than two-thirds of all archaeal clones. None of the previously reported sequences intercalate into the main GSP clones in clusters A and B. GSP cluster A is a separate branch that appears near Haloarcula and Halorubrum; however, the deeper branches are not statistically supported, and their relationships change easily with different contextual sequences (cf. Figs. 2 and S1). The cluster only includes uncultured isolates from salterns, hypersaline marsh sediments, and mangrove forest soil (Fig. S1). GSP cluster B is a branch from the Halococcus cluster that only includes a few environmental clones from rock salt, salterns, and Salt Spring. The most deeply branching sequences, those in GSP cluster C, include GSP clones arch43 and arch158. These clustered with sequences (90–98% sequence identity) from the Wadi An Natrun included in cluster 5 of Mesbah et al. [29] and did not formally fall within the Haloarchaea cluster.
Discussion
This is the first published culture-independent study of archaea in a natural inland terrestrial hypersaline environment. Previous studies that include large rRNA clone libraries have examined solar salterns [3, 6, 33, 34, 48], hypersaline lakes [12, 15, 23, 27, 29, 30], marshes [13], streams [14, 49], and rock salt deposits [28, 39]. The current study found evidence for 36 OTUs at the 99% sequence identity level, representing some 23 species and 19 genera, in the archaeal assemblage of the GSP. Although coverage was high, estimated levels of diversity by Chao1 suggested that 50–100% more diversity is present at the site than captured in the culture-independent clone libraries. Nonetheless, archaeal diversity is not high at the GSP. It has to be acknowledged in this context that PCR is not quantitative and can be subject to biases [36, 45]. The disparate copies of rRNA genes found in single genomes among the archaea may lead to an overestimate of diversity [4, 31]. Finally, the results summarized in this paper are a snapshot of the archaeal community at one spot on the GSP and may not be wholly representative.
Of the published studies, several report on 16S rRNA clone libraries that are rich in Halorubrum spp. [3, 6, 28, 29, 34, 39]. The GSP library had some representatives from Halorubrum, but the vast majority of sequences were associated with Halococcus (GSP cluster B) and the unaffiliated GSP cluster A. The dominance of Halococcus spp. in the GSP library is rare with some studies not finding any Halococcus in their libraries [6, 34, 39, 49]. Alkaliphilic (clade 1) haloarchaea have been found often in previous studies [29, 39, 49], but these were not seen in the GSP library. Although several GSP isolates fall into this clade, a greater abundance of alkaliphilic organisms was expected, given the high pH of the soils. In some environments, such as transient Salt Spring [49], a large proportion of the archaeal sequences are not within the haloarchaea. Only haloarchaea were detected at the GSP, with no other Euryarchaeota, Crenarchaeota, or Korarchaeota. No sequences were detected from the SHOW or ADL groups, although these were numerous in a previous saltern study [6].
Culturable archaeal diversity at the GSP was very low. The species isolated were those commonly enriched in collections from hypersaline systems, namely, Haloarcula, Halobacterium, Haloferax, Halorubrum, Haloterrigena, and Natrinema [3, 6, 30, 34]. This might be expected using the single medium that many studies employ; however, very low culturable diversity also was observed in a study using more varied media [3]. No isolates were cultured from the GSP clusters A and B that dominated the clone library, and no known cultured organisms fall within those clusters.
Previous studies at the GSP characterized bacterial isolates captured from the same soil samples used in the current study to isolate archaea [7, 26, 51]. The bacterial study found that most isolates had very wide ranges of salt tolerance. It was suggested that the rapidly changing salinity conditions in terrestrial hypersaline environments enrich for organisms that can grow in a wide range of salinities. Growth at low salinity (1%) was common, even for bacterial isolates that could grow at 15% salinity or higher. Only four bacterial isolates required 5% salinity or more to grow. As expected, all of the archaeal isolates required high salinity for growth, with only one isolate (GSP106) growing below 10% salinity. All of the archaeal isolates grew at 30% salinity. The temperature tolerance of the archaeal isolates is remarkable in that the tolerance ranges are narrow relative to most of the bacterial isolates and lower than expected for haloarchaea. The narrow range of permissible growth temperatures observed for the archaea does not include the highest temperatures (greater than 50°C) common at the surface in summer. The narrow temperature tolerance ranges observed for the archaea are similar to those of GSP alkaline bacilli (GSP 67–78) previously described [7]. The pH tolerance range for the GSP archaeal isolates was also relatively narrow, with no growth below pH 7 or above pH 8.5. The average pH at the site is 8.5, with many areas having higher pHs. Only six of 37 bacterial phylotypes had similar ranges.
It is not clear how these halophiles survive at the GSP when rain events greatly reduce salinities near the surface. One suggestion is that there are microniches of higher salinity and then migration to recolonize areas impacted by low salinity. A related suggestion is that fresh water on the surface does not penetrate very deeply into the saline soil, thus preserving a suitable environment for halophiles. It has been suggested that mass mortality events may be an important factor in the assembly of haloarchaeal communities in poikilotrophic environments and that some individuals survive due to the heterogeneity of the soil environment [49]. Disturbance, in the form of rain events, may have increased microbial diversity in these terrestrial hypersaline systems. The observation of increased microdiversity in hypersaline environments [29] is consistent with the suggestion that increased diversity is driven by rapidly changing conditions. Mutation rates were relatively high in Halomonas isolates from the GSP [51], implying a mechanism for the microdiversity observed.
References
Abed RMM, Kohls K, de Beer D (2007) Effect of salinity changes on the bacterial diversity, photosynthesis and oxygen consumption of cyanobacterial mats from an intertidal flat of the Arabian Gulf. Environ Microbiol 9:1384–1392
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Benlloch S, Acinas SG, Antón J, López-López A, Luz SP, Rodriguez-Valera F (2001) Archaeal biodiversity in crystallizer ponds from a solar saltern: culture versus PCR. Microb Ecol 41:12–19
Boucher Y, Douady CJ, Sharma AK, Kamekura M, Doolittle WF (2004) Intragenomic heterogeneity and intergenomic recombination among haloarchaeal rRNA genes. J Bacteriol 186:3980–3990
Bürgmann H, Pesaro M, Widmer F, Zeyer J (2001) A strategy for optimizing quality and quantity of DNA extracted from soil. J Microbiol Methods 45:7–20
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004) Combined use of cultivation-dependent and cultivation-independent methods indicates that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol 70:5258–5265
Caton IR, Schneegurt MA (2008) Culture-independent analysis of microbial diversity at the Great Salt Plains of Oklahoma. Abstract and Program, 108th General Meeting of the American Society for Microbiology, Boston, MA, June 2008
Caton TM, Witte LR, Ngyuen HD, Buchheim JA, Buchheim MA, Schneegurt MA (2004) Halotolerant aerobic heterotrophic bacteria from the Great Salt Plains of Oklahoma. Microb Ecol 48:449–462
Caton TM, Witte LR, Buchheim JA, Buchheim MA, Schneegurt MA (2003) Phenetic and phylogenetic characterization of the prokaryotic assemblage at the Great Salt Plains of Oklahoma. Abstract and Program, 103rd General Meeting of the American Society for Microbiology, Washington, DC, May 2003
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci USA 89:5685–5689
DeLong EF, Pace NR (2001) Environmental diversity of bacteria and archaea. Syst Biol 50:470–478
Dorador C, Vila I, Witzel K-P, Imhoff JF (2007) Unique microbial communities in contrasting aquatic environments of the high altitude Andean Altiplano (northern Chile). Max Planck Institute for Evolutionary Biology, Germany
Eilmus S, Rösch C, Bothe H (2007) Prokaryotic life in a potash-polluted marsh with emphasis on N-metabolizing microorganisms. Environ Pollut 146:478–491
Elshahed MS, Najar FZ, Roe BA, Oren A, Dewers TA, Krumholz LR (2004) Survey of archaeal diversity reveals an abundance of halophilic Archaea in a low-salt, sulfide- and sulfur-rich spring. Appl Environ Microbiol 70:2230–2239
Fan H, Xue Y, Ma Y, Ventosa A, Grant WP (2004) Halorubrum tibetense sp. nov., a novel haloalkaliphilic archaeon from Lake Zabuye in Tibet, China. Int J Syst Evol Microbiol 54:1213–1216
Fierer N, Schimel JP, Holden PA (2003) Influence of drying-rewetting frequency on soil bacterial community structure. Microb Ecol 45:63–71
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Gorbushina AA, Krumbein WE (1998) The poikilotrophic micro-organism and its environment. In: Seckbach J (ed) Enigmatic microorganisms and life in extreme habitats. Kluwer, The Netherlands, pp 177–185
Grant WD, Kamekura M, McGenity TJ, Ventosa A (2001) Class III. Halobacteria class. nov. In: Boone DR, Castenholz RW (eds) Bergey's manual of systematic bacteriology. The Archaea and the deeply branching and phototrophic Bacteria, vol 1, 2nd edn. Springer, New York, pp 294–334
Hardin G (1960) The competitive exclusion principle. Science 131:1292–1297
Holt JG, Krieg NR, Sneath PHA, Staley JT, Williams ST (2000) Bergey's manual of determinative bacteriology, 9th edn. Lippincott Williams & Wilkins, Philadelphia, pp 739–755
Huber T, Faulkner G, Hugenholtz P (2004) BELLEROPHON: a program to detect chimeric sequences in multiple alignments. Bioinformatics 20:2317–2319
Jiang H, Dong H, Yu B, Liu X, Li Y, Ji S et al (2007) Microbial response to salinity change in Lake Chaka, a hypersaline lake on Tibetan plateau. Environ Microbiol 9:2603–2621
Johnson KS (1980) Guidebook for geologic field trips in Oklahoma. Book II: Northwest Oklahoma. University of Oklahoma, Norman, pp 13–15
Leadbetter JR, Breznak JA (1996) Physiological ecology of Methanobrevibacter cuticularis sp. nov. and Methanobrevibacter curvatus sp. nov., isolated from the hindgut of the termite Reticulitermes flavipes. Appl Environ Microbiol 62:3620–3631
Litzner BR, Caton TM, Schneegurt MA (2006) Carbon substrate utilization, antibiotic sensitivity, and numerical taxonomy of bacterial isolates from the Great Salt Plains of Oklahoma. Arch Microbiol 185:286–296
Major KM, Kirkwood AE, Major CS, McCreadie JW, Henley WJ (2005) In situ studies of algal biomass in relation to physicochemical characteristics of the Salt Plains National Wildlife Refuge, Oklahoma, USA. Saline Syst 1:11–20
McGenity TJ, Gemmell RT, Grant WD, Stan-Lotter H (2000) Origins of halophilic microorganisms in ancient salt deposits. Environ Microbiol 2:243–250
Mesbah NM, Abou-El-Ela SH, Wiegel J (2007) Novel and unexpected prokaryotic diversity in water and sediments of the alkaline, hypersaline lakes of the Wadi An Natrun, Egypt. Microb Ecol 54:598–617
Mutlu MB, Martínez-García M, Santos F, Peña A, Guven K, Antón J (2008) Prokaryotic diversity in Tuz Lake, a hypersaline environment in inland Turkey. FEMS Microbiol Ecol 65:474–483
Mylvaganam S, Dennis PP (1992) Sequence heterogeneity between the two genes encoding 16S rRNA from the halophilic archaebacterium Haloarcula marismortui. Genetics 130:399–410
Nusslein K, Tiedje JM (1999) Soil bacterial community shift correlated with change from forest to pasture vegetation in a tropical soil. Appl Environ Microbiol 65:3622–3626
Park S-J, Kang C-H, Rhee S-K (2006) Characterization of microbial diversity in a solar saltern of Korea based on 16S rRNA gene analysis. J Microbiol Biotechnol 16:1640–1645
Pašić L, Ulrih NP, Črnigoj M, Grabnar M, Velikonja BH (2007) Haloarchaeal communities in the crystallizers of two Adriatic solar salterns. Can J Microbiol 53:8–18
Petraitis PS, Latham RE, Niesenbaum RA (1989) The maintenance of species diversity by disturbance. Q Rev Biol 64:393–418
Polz MF, Cavanaugh CM (1998) Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol 64:3724–3730
Purdy KJ, Cresswell-Maynard TD, Nedwell DB, McGenity TJ, Grant WD, Timmis KN et al (2004) Isolation of haloarchaea that grow at low salinities. Environ Microbiol 6:591–595
Quesada E, Ventosa A, Rodriquez-Valera F, Megias L, Ramos-Cormenzana A (1983) Numerical taxonomy of moderately halophilic Gram-negative bacteria from hypersaline soils. J Gen Microbiol 129:2649–2657
Radax C, Gruber C, Stan-Lotter H (2001) Novel haloarchaeal 16S rRNA gene sequences from Alpine Permo-Triassic rock salt. Extremophiles 5:221–228
Rappé MS, Giovannoni SJ (2003) The uncultured microbial majority. Annu Rev Microbiol 57:369–394
Reed JE (1982) Preliminary projections of the effects of chloride-control structures on the quaternary aquifer at the Great Salt Plains, Oklahoma. US Geological Survey, Water-Resources Investigations 80–120, Oklahoma City, OK
Rodriguez-Valera F, Ruiz-Berraquero F, Ramos-Cormenzana A (1980) Isolation of extremely halophilic bacteria able to grow in defined organic media with single carbon sources. J Gen Microbiol 119:535–538
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schneegurt MA, Caton IR, Landon BA, Castro SL, Perkins SA (2007) Microbial diversity at the Great Salt Plains of Oklahoma. Abstract and Program, 107th General Meeting of the American Society for Microbiology, Toronto, ON, May 2007
Suzuki MT, Giovannoni SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Swofford DL (1998) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sunderland, MA: Sinauer
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Valenzuela-Encinas C, Neria-González I, Alcántara-Hernández RJ, Enríques-Aragón A, Estrada-Alvarado I, Hernández-Rodríquez C et al (2008) Phylogenetic analysis of archaeal community in an alkaline-saline soil of the former lake Texcoco (Mexico). Extremophiles 12:247–254
Walsh DA, Papke RT, Doolittle WF (2005) Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol 7:1655–1666
Wang GC, Wang Y (1997) Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl Environ Microbiol. 63:4645–4650
Wilson C, Caton TM, Buchheim JA, Buchheim MA, Schneegurt MA, Miller RV (2004) DNA-Repair potential of Halomonas spp. from the Salt Plains Microbial Observatory of Oklahoma. Microbial Ecol 48:541–549
Acknowledgments
The authors are grateful to Julie Buchheim and Mark Buchheim (University of Tulsa) for sequencing archaeal isolate genes and to Sarah Castro and Seth Perkins for preliminary data. Thanks are extended to Darren Francisco, Brooke Landon, Hieu Ngyuen, Roberta Pettriess, Holly Pinkart, Noah Schneegurt, and Russell Vreeland for their help with the project. Primary support for this work was provided by grants from the Microbial Observatories program of the National Science Foundation (MCB-0131659 and MCB-0132083). Additional support was provided by grants from the NIH National Center for Research Resources through the Kansas Biomedical Research Infrastructure Network (KBRIN; P20 RR16475).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Fig. S1
Full phylogenetic tree for GSP archaea based on 16S rRNA gene sequences. GSP sequences are included from both the clone library and cultured isolates. (JPEG 1.34 MB)
Rights and permissions
About this article
Cite this article
Caton, T.M., Caton, I.R., Witte, L.R. et al. Archaeal Diversity at the Great Salt Plains of Oklahoma Described by Cultivation and Molecular Analyses. Microb Ecol 58, 519–528 (2009). https://doi.org/10.1007/s00248-009-9507-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-009-9507-y