
Abstract
Carnitine transporter defect is an autosomal recessive disorder caused by mutations in the SLC22A5 gene that encodes the high-affinity carnitine transporter OCTN2. Affected patients can present with predominant metabolic or cardiac manifestations. Early recognition of this disorder in a context of life-threatening cardiac failure and treatment with carnitine can be lifesaving in this inborn error of fatty acid oxidation. Here we describe a boy with a severe cardiomyopathy and severe anemia who improved with carnitine therapy. Physiopathology of anemia, a probably less recognized symptom of carnitine deficiency, is also discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
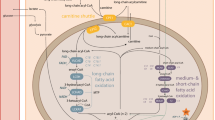
Carnitine plays an essential role in the transfer of long-chain fatty acids inside mitochondria for β-oxidation. Carnitine accumulates inside the cells by the high-affinity OCTN2 carnitine transporter present in the heart, muscle, and kidney. During periods of fasting, fatty acids are the predominant substrate for energy production. If fatty acid oxidation is defective, fat cannot be used, glucose is consumed without regeneration via neoglucogenesis and hypoglycemia may be present. Fat accumulates in the heart and skeletal muscle and impairs organ function (cardiomyopathy/myopathy) in addition to the failure in energy production [4].
Primary carnitine deficiency (OMIM 212140) is an autosomal recessive disorder of fatty acid oxidation due to lack of functional OCTN2 carnitine transporter. In the United States and Europe, its frequency has not been reported but, from the reported cases, it seems similar to that in Japan (1/40,000 newborn [3]). Although most of the fatty acid oxidation disorders affect heart and skeletal muscles and liver, cardiac failure is seen as the major presenting manifestation only in carnitine transporter deficit. Over half of the reported cases of this disease first presented between 12 months to 7 years of age with progressive heart failure and generalized muscle weakness [7]. Although the outcome is usually very good with carnitine therapy, the cardiac failure can rapidly progress to death without this specific treatment. Affected patients can also have a “metabolic” presentation: extended fasting stress may induce hypoketotic hypoglycemic coma (with or without cardiomyopathy). The hepatic presentation occurs less frequently than the myopathic presentation because the liver has a separate transporter for carnitine that can usually maintain sufficient levels of carnitine to support ketogenesis. The key to the diagnosis is the measurement of plasma carnitine levels that are extremely decreased (free carnitine <5 μM, controls 25–50 μM). Diagnosis is confirmed by demonstrating reduced carnitine transport in skin fibroblasts from the patient. Numerous mutations have been described in the organic cation transporter OCTN2, encoded by the SLC22A5 gene [5]. We describe here a boy with a severe anemia and cardiomyopathy who rapidly improved with carnitine therapy.
Case Report
A 7-month-old boy from Guyana was transferred to our unit after diagnosis in his native region of cardiomyopathy. He was born at term, after a normal gestation, from nonconsanguineous parents. He had 2 healthy brothers and a third brother who died at 5 years of age in a context of pulmonary disease. In our patient, a systolic murmur found during neonatal physical examination first led to the diagnosis of an asymptomatic ventricular septal defect, which subsequently closed spontaneously. At 6 months of age, because of the recurrence of a systolic murmur, he underwent, in Guyana, a cardiac ultrasound examination that confirmed a closed ventricular septal defect but revealed left ventricular failure. The patient was secondarily transferred to La Timone Enfants Hospital in Marseille (France). On physical examination, the patient presented with well-tolerated tachycardia (160 beats per minute) and with a moderate hepatomegaly (2 cm below the costal margin). Growth and psychomotor development were normal. Cardiac ultrasound showed normal anatomy but a dilated, hypokinetic, and hypertrophic left ventricle: ejection fraction 18% (normal >60%), shortening fraction 8% (normal >30%), end-diastolic left ventricle diameter 43 mm (z-score +7.9), and end-diastolic intraventricular septal diameter 7.3 mm (z-score +3.9). Laboratory testings revealed a normocytic hypochromic nonregenerative anemia (hemoglobin levels: 69 g/L, mean globular volume: 75 fl, mean corpuscular hemoglobin: 24 pg, reticulocytes: 54 Giga/L) and cytolysis (aspartate aminotransferase: 170 IU/L, alanine aminotransferase: 98 IU/L) without cholestasis. Blood electrolytes, urea, and creatinine were normal. Plasma amino acid chromatography was normal and urinary organic acid profile revealed a moderate and transient dicarboxylic aciduria. Plasma carnitine was severely reduced (in various samples: free carnitine: 1–3 μmol/L, total carnitine: 2–4.5 μmol/L). A carnitine transporter defect was suspected and confirmed by the study of l-[methyl-3H] carnitine uptake by fibroblasts (according to Treem et al. [11]). Treatment with carnitine was started as soon as samples for laboratory testings were taken (and thus before confirmation of diagnosis), intravenously at 185 mg/kg per day for 6 days and then orally at 145 mg/kg per day. The child tolerated PO intake. Oral carnitine supplementation was then progressively increased because of the persistence of low plasma carnitine levels. It was increased up to 215 mg/kg per day divided in 3 doses, with the same dose in the morning and in the afternoon and a higher dose at night so as to maintain stable levels. The patient was concomitantly started on diuretics (spironolactone, furosemide), β-blockers (carvedilol) and an inhibitor of angiotensin I-converting enzyme (captopril). He underwent blood transfusion because of the severity of the anemia in the context of cardiomyopathy. Iron supplementation was also started. Cardiac function clearly and rapidly improved while on treatment (Table 1). Four months after starting the treatment, the patient was allowed to go back to Guyana with his family, where local doctors will continue treatment.
Discussion
Carnitine transporter defect is a rare but treatable etiology of metabolic cardiomyopathy. Over half of the known cases of carnitine transporter deficiency first presented with progressive heart failure and generalized muscle weakness [7]. Tein et al. described the features of 11 cases of primary carnitine deficiency [10]. Cardiomyopathy was present in all cases. A prompt response to carnitine therapy occurred in 8 cases within the first month of therapy. Of the remaining 3 children, 2 died during an acute episode before diagnosis was made and therapy started. In the last child, the diagnosis was made at 3.5 months when cardiac features were limited to mild echocardiography abnormalities; thus, the clinical response to treatment was difficult to evaluate in this asymptomatic child. Those data as well as our case report highlight the need to look for this disease in children with cardiomyopathy and the necessity to start treatment very early, even before receiving the results of blood sampling if suspicion is high. As shown by Tein et al. [10] and in our patient, rapid improvement of ventricular function can be obtained with appropriate treatment. The therapeutic effect of high-dose oral or intravenous carnitine supplementation may be the result of a passive diffusion of carnitine or an uptake by alternative lower affinity organic cation/carnitine transporters on the plasma membrane [9]. Our patient, after 6 days of high-dose intravenous carnitine therapy, required high oral doses unequally distributed over the day with higher doses at night, in order to achieve satisfactory plasma levels. This is consistent with literature data recommending intravenous therapy of 100–400 mg/kg per day of carnitine during life-threatening events, and for chronic treatment 100–300 mg/kg per day of oral carnitine [6]. In addition to cardiomyopathy, our patient presented with normocytic hypochromic nonregenerative anemia. In the literature, anemia was noted in 6 cases of primary carnitine transporter deficiency [10]. The red blood cell morphology on smear was variable: hypochromic, normochromic, normocytic, or macrocytic. The investigations made to search for an underlying etiology were negative in these 6 described patients. Carnitine is known to have a role in red blood cell metabolism: it stabilizes the cellular membrane and raises the red blood cell osmotic resistance [2]. Iron metabolism is also linked to carnitine because various authors showed low serum carnitine concentrations in healthy children with iron deficiency anemia [1, 8]. Thus, anemia may be present in carnitine transporter deficiency and an iron deficiency may worsen anemia in this context. Moreover, iron deficiency may be a cause of secondary carnitine deficiency (with moderately low plasma carnitine levels). In our patient, the hemoglobin level remained stable after transfusion, while he was receiving iron and carnitine therapy. Although it was not clear, iron deficiency was probably associated with his primary carnitine deficit (normal ferritinemia but low siderophilin saturation were present).
Even though our patient did not present these symptoms, subjects with primary carnitine transporter deficiency may also suffer from myopathy, central nervous system abnormalities (mostly secondary to hypoglycemic episodes), and gastrointestinal symptoms (such as recurrent episodes of abdominal pain and diarrhea).
Conclusion
Carnitine membrane transporter deficiency is one of the rare treatable etiologies of metabolic cardiomyopathies. It should be suspected and searched for by measuring the levels of free and total carnitine in plasma and urine from such patients. Then, in a context of cardiomyopathy, a potentially life-saving treatment with carnitine at 100–400 mg/kg per day has to be introduced as soon as samples for metabolic investigations have been collected, and even before a carnitine deficiency is proven. Moreover, anemia should be considered as a symptom of carnitine transporter deficiency when other causes have been searched for and eliminated.
References
Cemeroglu AP, Kocabas CN, Coskun T, Gurgey A (2001) Low serum carnitine concentrations in healthy children with iron deficiency anemia. Pediatr Hematol Oncol 18:491–495
Evangeliou A, Vlassopoulos D (2003) Carnitine metabolism and deficit—when supplementation is necessary? Curr Pharm Biotechnol 4:211–219
Koizumi A, Nozaki J, Ohura T, Kayo T, Wada Y, Nezu J, Ohashi R, Tamai I, Shoji Y, Takada G, Kibira S, Matsuishi T, Tsuji A (1999) Genetic epidemiology of the carnitine transporter OCTN2 gene in a Japanese population and phenotypic characterization in Japanese pedigrees with primary systemic carnitine deficiency. Hum Mol Genet 8:2247–2254
Longo N, Amat di San Filippo C, Pasquali M (2006) Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet 142:77–85
Nezu J, Tamai I, Oku A, Ohashi R, Yabuuchi H, Hashimoto N, Nikaido H, Sai Y, Koizumi A, Shoji Y, Takada G, Matsuishi T, Yoshino M, Kato H, Ohura T, Tsujimoto G, Hayakawa J, Shimane M, Tsuji A (1999) Primary systemic carnitine deficiency is caused by mutations in a gene encoding sodium ion-dependent carnitine transporter. Nat Genet 21:91–94
Ogier de Baulny H, Superti-Furga A (2006) Disorders of mitochondrial fatty acid oxidation and ketone body metabolism. In: Blau N, Hoffman GF, Leonard J, Clarke JTR (eds) Physician’s guide to the treatment and follow-up of metabolic disease 2006. Springer, Berlin, Heidelberg, New York, pp 147–160
Stanley CA, Bennett MJ, Mayatepek E (2006) Disorders of fatty acid oxidation and related metabolic pathways. In: Fernandes J, Saudubray JM, van Den Berghe G, Walter JH (eds) Inborn metabolic disease diagnostic and treatment 2006, 4th ed. Springer, Heidelberg, pp 177–188
Tanzer F, Hizel S, Cetinkaya O, Sekreter E (2001) Serum free carnitine and total triglyceride levels in children with iron deficiency anemia. Int J Vitam Nutr Res 71:66–69
Tein I (2003) Carnitine transport: pathophysiology and metabolism of known molecular defects. J Inherit Metab Dis 26:147–169
Tein I, Di Mauro S (1992) Primary systemic carnitine deficiency manifested by carnitine-responsive cardiomyopathy. In: Ferrari R, Di Mauro S, Sherwood G (eds) L-carnitine and its role in medicine: from function to therapy 1992. Academic Press, San Diego, pp 155–183
Treem WR, Stanley CA, Finegold DN, Hale DE, Coates PM (1988) Primary carnitine deficiency due to a failure of carnitine transport in kidney, muscle, and fibroblasts. N Engl J Med 319:1331–1336
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cano, A., Ovaert, C., Vianey-Saban, C. et al. Carnitine Membrane Transporter Deficiency: A Rare Treatable Cause of Cardiomyopathy and Anemia. Pediatr Cardiol 29, 163–165 (2008). https://doi.org/10.1007/s00246-007-9051-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-007-9051-9