
Abstract
Twin and genealogy studies suggest a strong genetic component of nephrolithiasis. Likewise, urinary traits associated with renal stone formation were found to be highly heritable, even after adjustment for demographic, anthropometric and dietary covariates. Recent high-throughput sequencing projects of phenotypically well-defined cohorts of stone formers and large genome-wide association studies led to the discovery of many new genes associated with kidney stones. The spectrum ranges from infrequent but highly penetrant variants (mutations) causing mendelian forms of nephrolithiasis (monogenic traits) to common but phenotypically mild variants associated with nephrolithiasis (polygenic traits). About two-thirds of the genes currently known to be associated with nephrolithiasis code for membrane proteins or enzymes involved in renal tubular transport. The thick ascending limb of Henle and connecting tubules are of paramount importance for renal water and electrolyte handling, urinary concentration and maintenance of acid–base homeostasis. In most instances, pathogenic variants in genes involved in thick ascending limb of Henle and connecting tubule function result in phenotypically severe disease, frequently accompanied by nephrocalcinosis with progressive CKD and to a variable degree by nephrolithiasis. The aim of this article is to review the current knowledge on kidney stone disease associated with inherited defects in the thick ascending loop of Henle and the connecting tubules. We also highlight recent advances in the field of kidney stone genetics that have implications beyond rare disease, offering new insights into the most common type of kidney stone disease, i.e., idiopathic calcium stone disease.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Kidney stone formation depends on dietary, environmental and genetic factors. Familial aggregation of kidney stone disease was already recognized in 1874 [1]. A positive family history is present in 20–50% of renal stone formers (SF) [2,3,4] and both twin [5] and genealogy [6] studies revealed a strong heritability of nephrolithiasis. The genetics of nephrolithiasis is heterogeneous and complex. Driven by technical progress in genomic sequence analysis and bioinformatics, there has been an enormous increase of knowledge in the field of kidney stone genetics in the last two decades. The spectrum ranges from infrequent but highly penetrant variants (mutations) causing mendelian forms of nephrolithiasis (monogenic traits) to common but phenotypically mild variants associated with nephrolithiasis (polygenic traits). Genome-wide association studies (GWAS) with large sample sizes up to 300,000 individuals revealed several variants that are associated with an increased or decreased risk of kidney stone disease [7,8,9]. The clinical relevance and contribution of these common variants to the overall burden of kidney stone disease, however, remain unclear. The situation is different at the other end of spectrum, the mendelian forms of nephrolithiasis. About 30 monogenetic forms of nephrolithiasis have been identified thus far and the list is continuously growing [10, 11]. A decade ago, less than 2% of all SF were estimated to have an underlying monogenetic cause [12]. In a recent study with a selected group of individuals suffering from recurrent nephrolithiasis or isolated nephrocalcinosis, high-throughput sequencing revealed a monogenetic cause in 11.4% of adult cases and 20.8% of childhood-onset cases [11]. Recessive mutations were more frequent among children, dominant disease occurred more abundantly in adults. About two-thirds of the genes (~ 20 out of 30) currently known to be associated with monogenetic stone disease code for membrane proteins or enzymes involved in renal tubular transport.
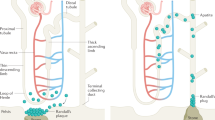
The thick ascending limb of Henle (TALH) and connecting tubules (CNT) have a central role in renal water and electrolyte handling as well as in the maintenance of acid–base homeostasis. As such it is not surprising that mutations in genes involved in TALH and CT function can result in phenotypically severe disease. Mutations in at least ten genes expressed in these two tubular segments are known to be associated with recurrent nephrolithiasis, including Bartter’s syndrome (BS), familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) and distal renal tubular acidosis (dRTA) [13]. Apart from rare genetic forms of stone disease, the medullary TALH is also involved in the formation of interstitial apatite deposits, known as Randall’s plaques, which are thought to represent the starting point for Ca oxalate stone formation [14, 15].
Anatomical and physiological aspects
We will briefly review in this section several anatomical and physiological aspects that are necessary to understand the inherited defects in the TALH and CNT leading to secondary nephrolithiasis.
Loop of henle
The loop of Henle comprises the descending thin limb, the ascending thin limb, and the thick ascending limb. Superficial and midcortical nephrons have a short thin descending limb or may even lack a thin descending limb, so that the TALH begins at the hairpin turn. In contrast, juxtamedullary nephrons contain a long loop extending deep into the medulla with the TALH beginning at the boundary between the inner and outer medulla. Unlike the thin descending limb, the thin ascending limb and the TALH do not express aquaporin-1 and hence are water impermeable [16]. Physiological functions of descending and ascending thin limbs have been reviewed in detail elsewhere [17]. For the sake of this review, we will place special emphasis on the TALH, which is critically involved in fluid and electrolytes homeostasis, urine concentration/dilution, acid–base homeostasis, as well as urinary protein composition [18].
The TALH is responsible for the reabsorption of 25–40% of filtered NaCl and thus plays a crucial role in the maintenance of extracellular volume [19]. Due to the impermeability to water, the TALH contributes to the urinary concentrating mechanism by diluting the luminal fluid, establishing a gradient of increasing osmolarity along the medulla via a countercurrent multiplication process [20]. NaCl reabsorption occurs through the co-transport of 1 Na, 1 K and 2 Cl ions mediated by NKCC2, a co-transporter expressed at the apical membrane along the entire TALH (Fig. 1) [21]. This electroneutral co-transport is driven by the Na-gradient generated by the Na/K-ATPase on the basolateral membrane and can specifically be inhibited by “loop” diuretics such as furosemide or bumetanide [22, 23]. The proper functioning of NKCC2 also requires apical K channels, mainly renal outer medullary K channels (ROMK) and to a lesser extent big K (BK) channels, whose function is to “recycle” K ions back into the lumen [24, 25]. Without this recycling, the low luminal K content of the TALH would indeed result in a severely impaired NKCC2-mediated NaCl transport [26]. On the basolateral side of TALH cells, the chloride channels CLC-Ka and CLC-Kb with their associated Barttin subunit mediate electrogenic efflux of Cl ions [27], whereas the electroneutral K–Cl cotransporter KCC4 is responsible for K-dependent Cl exit (Fig. 1) [28].

Schematic representation of a TALH cell. NKCC2 Na–K–2Cl cotransporter, ROMK renal outer medullary K channel, ClC-Kb voltage-gated Cl channel Kb isoform, KCC4 K–Cl cotransporter, CaSR calcium-sensing receptor
The apical co-transport of 1 Na+, 1 K+ and 2 Cl− ions through NKCC2 coupled to the apical recycling of K generates a lumen-positive transepithelial voltage that drives paracellular reabsorption of cations such as Na, Mg, and Ca [29]. The TALH reabsorbs 50–70% of filtered Mg and up to 25% of filtered Ca [30, 31]. Charge and size selective tight junctions composed of different types of claudins facilitate and regulate this paracellular transport [32]. In particular, claudin-16 and claudin-19 critically contribute to this task, genetic disruption of claudin-16 or -19 causes urinary Mg and Ca wasting in both mice and men [33,34,35,36]. Claudin-16 and claudin-19 form a complex with claudin-3 to build a pore for divalent cations in the TALH [37]. Claudin-14 interacts with claudin-16 and thereby attenuates Ca permeability through this tight junction pores (Fig. 1) [38]. Claudin-14 transcript and protein are upregulated by high Ca diet and downregulated by low Ca diet. A second TALH tight junction pore mainly permeable for Na is formed by claudin-10b. Conditional TALH claudin-10b KO mice exhibit increased paracellular permeabilities of Mg and Ca but greatly decreased Na permeability, resulting in hypermagnesemia, hypocalciuria, nephrocalcinosis and polyuria [39]. Together these findings indicate that there are two different paracellular permeability pathways in the TALH, a Na-permeable pathway formed by claudin-10b and a Ca- and Mg-permeable pathway formed by claudin-16 and claudin-19. If one type of claudin complex is perturbed or eliminated, the other predominates conferring only its functional permeability properties. In support of this notion, deletion of claudin-10b rescues claudin-16 deficient mice from hypomagnesemia and hypercalciuria [40]. Double mutant mice display a complete loss of paracellular cation selectivity at the level of the TALH with recruitment of downstream compensatory mechanisms.
The calcium-sensing receptor (CaSR) is strongly expressed at the basolateral membrane of TALH cells, and regulates NaCl and Ca transport in this segment [41]. Activation of the CaSR by elevated extracellular Ca has been shown to reduce apical NaCl transport via the inhibition of ROMK [42]. This mitigates the lumen-positive transepithelial potential necessary for the paracellular transport of Ca, resulting in hypercalciuria. However, a recent study reported that inhibition of the CaSR causes hypocalciuria without concomitant alteration in NaCl transport, suggesting that the CaSR may primarily influence the paracellular transport pathway [43]. Indeed, results from several recent studies demonstrate that the CaSR directly regulates the expression of claudin-14 and claudin-16 [38, 43,44,45,46].
In addition to their resorptive tasks, TALH cells express and secrete uromodulin (also known as Tamm–Horsfall protein) into the urine [47]. Uromodulin is the most abundant protein physiologically present in urine and regulates blood pressure via modulation of ROMK and NKCC2 activity [48,49,50], prevents tubular crystallization of Ca oxalate [51, 52] and also has a protective role against urinary tract infections [53].
Connecting and collecting tubules
After the TALH, the distal nephron encompasses the distal convoluted tubule (DCT), the connecting tubule (CNT) and the collecting duct (CD). With the exception of the rabbit kidney, in which the boundaries between these segments are clearly defined, the delineation is gradual in most species [54]. The CNT appears thus as a transition segment between the DCT and the CD. The CNT contains CNT-specific cells, which resemble principal cells of the CD, and intercalated cells. The CD is classically divided into three parts: the cortical collecting duct, the outer medullary collecting duct and inner medullary collecting duct. As for the CNT, the CD contains principal cells and intercalated cells, which are further subdivided into three subtypes including type A, type B, and non-A/non-B intercalated cells. The CD is a “salt and pepper” type epithelium, with principal cells being the main cell type with intercalated cells “sprinkled” throughout [55]. Hereditary defects in type A intercalated cells but not in other CNT and CD cells cause kidney stone disease in humans. Type A intercalated cells are found from the late DCT to the inner medullary collecting duct and are critical for urinary net acid excretion [56]. They exhibit basolateral expression of the Cl /bicarbonate exchanger AE1 and V-ATPase activity at the apical membrane (Fig. 2) [54, 57]. The V-ATPase consists of two multi-subunit complexes, the V1 (head) and V0 (membrane anchored) subunits [58]. The 640 kDa V1 subunit is composed of subunits A, B, C, D, E, F, G and H. Mammals have two B subunits, the ubiquitous B2 isoform and the B1 isoform, which is restricted to specialized epithelia of the inner ear, epididymis and intercalated cells [59, 60]. Protons furnished by the cytosolic carbonic anhydrase type II present in type A intercalated cells are translocated against a concentration gradient by the ATP-driven V-ATPase into the urine. Contemporaneous with apical proton secretion, carbonic anhydrase liberates bicarbonate ions, which exit basolaterally via AE1.

Schematic representation of a type A intercalated cell. AE1 anion exchanger 1, CAII carbonic anhydrase type II, V membrane-anchored subunit of the V-ATPase, V head subunit of the V-ATPase
Genetic disorders of the thick ascending loop of henle
Monogenetic disease
Bartter syndrome
Antenatal and classical Bartter syndromes constitute a congenital heterogeneous family of salt reabsorption disorders, which share common features such as hypokalemic metabolic alkalosis, elevated renin and aldosterone levels without hypertension, hypercalciuria, nephrolithiasis and nephrocalcinosis. The antenatal form of Bartter syndrome presents with additional severe symptoms including maternal polyhydramnios, premature birth, dehydration due to polyuria and early-onset nephrocalcinosis [61]. Five genes are currently known to cause Bartter syndrome: Mutations in the SLCA12A1 gene coding for the Na, K, 2 Cl cotransporter NKCC2 cause Bartter syndrome type I, mutations in the KCNJ1 gene coding for the K channel ROMK cause Bartter syndrome type II, mutations in the CLCNKB gene coding for the voltage-gated Cl channel ClC-Kb cause Bartter syndrome type III, mutations in the BSND gene coding for barttin cause Bartter syndrome type IV, and mutations in the CASR gene coding for the CaSR causes Bartter syndrome type V (Table 1) [62,63,64,65,66,67]. With the exception of Bartter syndrome type V, which is due to a dominant activating mutation of the CaSR, the other types of Bartter syndrome result from recessive loss-of-function mutations.
Mutations in the CLCN5 gene coding for the voltage-gated chloride channel ClC-5 in the proximal tubule cause Dent disease and nephrolithiasis and may also be associated with a Bartter-like syndrome [68,69,70]. In addition, a severe but transient form of antenatal Bartter syndrome due to mutations in the melanoma-associated antigen D2 (encoded by the X-chromosomal MAGED2 gene) has recently been reported [71]. MAGED2 gene mutations are associated with polyhydramnios, prematurity and increased perinatal mortality but an association with kidney stones has not been reported.
In 2002, the first patient with Bartter syndrome and kidney stones was described [72]. The reported patient was a 28-year-old male with genetically confirmed Bartter syndrome type III. He had hypercalciuria but no evidence of nephrocalcinosis, stone analysis results were not reported. In the last 20 years, several large case-series with Bartter patients were published. In an Italian long-term follow-up study of 15 patients with Bartter syndrome type I and II, an increased Ca/creatinine ratio was found in 10 of 11 investigated children and all but one child had signs of nephrocalcinosis at diagnosis. Nephrolithiasis was not observed at baseline or during a median follow-up of 11 years [73]. In another series with 40 patients, all patients with Bartter syndrome type II had evidence of hypercalciuria and medullary nephrocalcinosis within the first weeks of life, whereas only two of 20 patients with Bartter syndrome type III displayed hypercalciuria and nephrocalcinosis [74]. The prevalence of nephrolithiasis was not reported. Similar findings were reported in another study with 52 Bartter patients: nephrocalcinosis was present in 31 of 32 cases with Bartter syndromes type I and II but only in two of 20 patients with Bartter syndrome type III [75]. The prevalence of nephrolithiasis was also not described in this study. In a nationwide Korean cohort, Bartter syndrome type III was found to be the most common form of Bartter syndrome, affecting 23 of 26 genotyped patients [76]. Hypercalciuria and nephrocalcinosis were present in ~ 60% and ~ 20% of patients, respectively, but the prevalence of nephrolithiasis was not described. In another long-time follow-up study (median follow-up 8.3 years) with 42 patients with Bartter syndrome type I–IV, all patients with Bartter syndrome type I and II had hypercalciuria and nephrocalcinosis. Out of four children with Bartter syndrome type III for which biochemical data were available, three children had hypercalciuria but only one child also displayed nephrocalcinosis. Of the four children with Bartter syndrome type IV reported, two were also found to have hypercalciuria and nephrocalcinosis [61]. Again, prevalence of nephrolithiasis was not reported.
The high phenotypic variability of Bartter syndrome type III is well illustrated by a recent large retrospective study with 115 patients [77]. Clinical features of the antenatal/neonatal form were found in ~ 30% of patients, features of the classical form in ~ 45% of patients and features of a Gitelman-like form in ~ 25% of patients. Urinary Ca/creatinine ratio was highest in the antenatal form and lowest in the Gitelman-like form. Nephrocalcinosis followed the same distribution pattern with prevalence rates of 29.4% in the antenatal form, 14% in the classical form and only 3% in the Gitelman-like form. Follow-up data (median follow-up was 8 years) were available in 77 of the 115 patients. Overall, seven patients developed nephrolithiasis (two of 26 patients with the antenatal form, five of 35 patients with the classical form but none of the 16 patients with the Gitelman-like presentation). The exact distribution between symptomatic stone events and asymptomatic stones discovered during imaging, stone composition results and urinary risk factors of stone formation other than calciuria were not reported.
It is remarkable that in the published case-series on Bartter syndrome, prevalence of nephrolithiasis is often not reported and if reported, seems to be considerably lower than the prevalence of nephrocalcinosis. Strikingly, we have not found a single published case of Bartter syndrome with the results of a stone analysis reported. In our own stone clinic, we follow one patient with Bartter syndrome and recurrent nephrolithiasis. This currently 27-year-old male with a compound heterozygous SLC12A1 mutation (c.799G>A, p.A267T and c.2752G>A, p.D918N) developed recurrent calcium nephrolithiasis at age 24. Two infrared spectroscopy-based stone analyses were performed on two consecutive stones, both yielding the same composition: 60% octa-Ca-dihydrogen phosphate and 40% carbonate apatite. Although the patient had mild compensated metabolic alkalosis (venous plasma bicarbonate 33.4 mmol/l), he was found to be profoundly hypocitraturic (0.03–0.06 mmol/24 h), likely due to medullary nephrocalcinosis with mild CKD (plasma creatinine 125–148 µmol/l) and a urinary acidification deficit (24 h urinary pH values between 6.6 and 6.8). Urinary Ca was significantly elevated (8.6–8.8 mmol/24 h), urinary volumes were between 3.1 and 3.4 l/24 h. In addition, the patient also had a constellation compatible with normocalcemic primary hyperparathyroidism with elevated PTH (129 pg/ml), high normal ionized plasma Ca (1.21–1.29 mmol/l) and low normal plasma P (0.78–0.94 mmol/l). The association of Bartter syndrome type I and primary hyperparathyroidism has only recently been recognized [78, 79]. None of the reported six pediatric cases suffered from symptomatic kidney stones but one child had radiologic evidence of asymptomatic nephrolithiasis. Cinacalcet and potassium citrate improved biochemical abnormalities in these children. As in the reported cases, sestamibi scintigraphy and neck MRI failed to reveal a parathyroid adenoma in our patient. The patient was started on cinacalcet 30 mg twice daily and potassium citrate 30 mmol twice daily, which resulted in a significant rise of urinary citrate to 0.87 mmol/24 h and a large drop in urinary Ca excretion to 2.8 mmol/24 h. The patient had a last symptomatic stone event 20 months after treatment initiation (22 months after the first stone event) and has remained asymptomatic now since 12 months.
As outlined above, large series suggest a correlation between degree of hypercalciuria and prevalence of nephrocalcinosis/nephrolithiasis in Bartter syndrome patients. Factors favoring the development of kidney stones in Bartter syndrome remain elusive so far. Indeed, high urinary volume and high urinary citrate due to metabolic alkalosis should prevent stone formation. Similarly, reduced Ca reabsorption at the level of the TALH should prevent Randall plaque formation and thus CaOx stone formation. These findings raise several questions. Do Bartter patients only form stones in the setting of high urinary Ca, low urinary citrate and increased urinary pH as in our case ? Are all Bartter patients forming CaP stones? What is the site of stone formation (ductal plugging versus growth on plaques)? Clearly, carefully conducted prospective studies with detailed clinical, genetic, biochemical and high-resolution imaging data are needed to better understand the pathophysiological mechanisms leading to kidney stone formation in Bartter patients.
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis
The autosomal-recessive disorder familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is caused by homozygous mutations in the claudin-16 (alias paracellin-1) and claudin-19 genes CLDN16 and CLDN19, respectively (Table 1) [33, 34]. Symptoms include seizures, tetany, failure to thrive, polyuria, recurrent urinary tract infections, nephrolithiasis and CKD with a substantial portion of patients eventually progressing to ESRD [80]. The phenotype of CLDN19 mutation is similar to the CLDN16 mutation syndrome but affected individuals additionally display ocular involvement (macular coloboma, myopia, retinitis pigmentosa, nystagmus and visual loss) [34]. Similar to Bartter syndrome, nephrocalcinosis seems to be much more prevalent than nephrolithiasis in FHHNC. While nephrocalcinosis is a universal finding in FHHNC patients, nephrolithiasis was reported in 25% and 42% of patients with claudin-16 and claudin-19 mutations, respectively [34, 81].
CKD progression was found to be unaffected by magnesium salts or thiazide diuretics but early treatment with vitamin D and Ca was essential to maintain growth in children [82]. In a more recent small cross-over study with four male and four female patients with FHHNC due to homozygous CLDN16 mutations, short-time administration of hydrochlorothiazide effectively reduced urinary Ca to a variable degree without significant alteration of urinary Mg excretion, but long-term treatment data are lacking [83]. Interestingly, in larger pedigree analyses, hypercalciuria and nephrolithiasis but not nephrocalcinosis or hypomagnesemia were observed in otherwise healthy family members, suggesting that pathogenic CLDN16 variants may be associated with “idiopathic” Ca stones [80, 81, 84].
Variants
Claudin-14
A GWAS in 3773 SF and 42,510 healthy controls from Iceland and the Netherlands found common synonymous variants in the CLDN14 gene encoding claudin-14 to be associated with an increased risk for kidney stones and reduced bone mineral density (Table 1) [8]. The synonymous variant rs219780(C) was present in a homozygous state in 62% of the population and conferred an estimated 1.64-fold increased risk for the development of kidney stones compared to noncarriers. The rs219780(C) variant was also significantly associated with increased urinary Ca and serum parathyroid hormone and reduced serum total CO2, but was not associated with serum Ca, P or vitamin D.
Another study investigated the association of rare variants (allele frequencies < 2%) in 40 candidate genes in 960 individuals with low and high 24-h urinary Ca excretion from the Nurses’ Health Studies I and II and the Health Professionals Follow-up Study [85]. None of the rare gene variants assessed were found with increased frequency in the low vs. high urinary Ca groups. The analysis of variants with allele frequencies ≥ 2% suggested an association of the non-synonymous CLDN14 variant rs11383113 (c.11C>T, p.Thr4Met) with lower urinary Ca excretion. The CLDN14 gene variant rs219780, which had been reported to increase the risk for kidney stones in the Icelandic and Dutch population, did not segregate between the groups with low and high 24-h urinary Ca excretion [8, 85]. In another study with 200 kidney SF and 200 healthy controls from the eastern part of India, the two variants rs219777 and rs219778 in the CLDN14 gene were found to be strongly associated with kidney stone disease, but no significant association was found with urinary or serum Ca [86]. A more recent study described the association of the CLDN14 gene variant rs78250838:C>T in an intronic cis-regulatory element with early-onset hypercalciuria and kidney stones in children [87]. In vitro studies revealed that this variant enhances claudin-14 expression, thereby increasing urinary Ca excretion and promoting calcium stone formation.
Calcium-sensing receptor
Vezzoli et al. were the first to report a positive association of the non-synonymous CaSR gene variant rs1042636 (p.R990G) in exon 7 with urinary Ca excretion in a cohort of kidney SF [88]. The association of this CASR variant with urinary Ca excretion and calcium nephrolithiasis has been reproduced in other ethnic cohorts [86, 89]. Several other non-synonymous (rs1801725 and rs1801726), intronic (rs17251221 and rs1501899) and 5′ UTR (rs7652589) CASR gene variants have also been associated with kidney stone disease in recent years (Table 1) [86, 89,90,91,92,93,94].
In a large follow-up GWAS with 5419 kidney stone cases and 279,870 controls in Iceland, additional genomic sequence variants associated with kidney stone disease were identified [7]. Relevant for this review is the observed association of a novel variant in a regulatory region in the first intron of the CASR gene rs7627468(A) that was found to be positively associated with kidney stone disease (odds ratio 1.16), increased serum total and ionized Ca and decreased 25-hydroxy vitamin D (Table 1).
Uromodulin
Rare mutations in the UMOD gene cause autosomal-dominant tubulointerstitial kidney disease (ADTIK), which is associated with progressive worsening of kidney function, hyperuricemia, gout and arterial hypertension but not kidney stones [47]. In a GWAS for CKD in an Icelandic population, a common variant rs4293393(T) at the UMOD gene locus was found to be associated with an increased risk for the development of CKD and gout but to confer protection against kidney stones (Table 1) [95].
Genetic disorders of the connecting tubules
Monogenetic disease
Distal renal tubular acidosis
Lightwood provided the first description of congenital distal renal tubular acidosis (dRTA) in an autopsy series of six children in 1935 [96]. Five monogenetic forms of dRTA are currently known: autosomal-recessive and autosomal-dominant mutations in the anion exchanger 1 (AE1, encoded by SLC4A1 gene), autosomal-recessive mutations in the B1 and a4 subunits of the V-ATPase (encoded by ATP6V1B1 and ATP6V0A4 genes, respectively), autosomal-recessive mutations in the transcription factor Foxi1 (encoded by the FOXI1 gene) and autosomal-recessive mutations in carbonic anhydrase type II (encoded by the CA2 gene) (Table 2) [97,98,99,100,101,102,103]. Sensu stricto, CA2 mutations cause a combined proximal–distal RTA, also known as type III RTA, but the renal phenotype is very similar to isolated cases of inherited dRTA.
Clinically, diagnosis of dRTA (also known as RTA type I) is straightforward and provocative tests are usually not needed. Typical laboratory features include hypokalemia, systemic normal anion gap acidosis (in the absence of extrarenal alkali losses), hypocitraturia, hypercalciuria and urinary pH > 6.5.
DRTA can be acquired or inherited, and separation between the two is critical as it will have diagnostic and therapeutic consequences (e.g., immunosuppression for Sjögren’s syndrome). Apart from obtaining a detailed family history, extrarenal manifestations can be helpful in identifying and differentiating inherited cases. In general, phenotype is milder and age of diagnosis significantly later with dominant SLC4A1 mutations compared to recessive forms of dRTA [104, 105]. Systemic metabolic acidosis may even be absent in dominant SLC4A1 mutations, a constellation known as incomplete dRTA [106,107,108]. B1 and a4 subunits of the V-ATPase are expressed in the stria vascularis of the inner ear and almost all patients with recessive ATP6V1B1 mutations and about half of patients with recessive ATP6V0A4 mutations develop progressive, bilateral sensorineural hearing loss [104]. For unknown reasons, onset of hearing loss occurs at a younger age and the phenotype is more severe in patients with ATP6V1B1 mutations [104, 105, 109]. Sensorineural hearing loss can be accompanied by enlargement of the vestibular aqueduct and endolymphatic sac and may contribute to the onset or the progression of hearing impairment [110,111,112]. SLC4A1 mutations can cause isolated dRTA, isolated hemolytic anemia or both [113, 114]. DRTA due to dominant SLC4A1 mutations is not associated with hemolytic anemia, unless dominant mutations are present in a homozygous or compound heterozygous state [113, 114]. Recessive SLC4A1 mutations with combined dRTA and hemolytic anemia have been reported in Southeast Asian patients, but recessive SLC4A1 mutations can also cause isolated dRTA [114, 115]. Patients with recessive CA2 mutations can be separated from other forms of inherited dRTA by a wealth of characteristic extrarenal features including intracerebral calcifications, psychomotor retardation, abnormal teeth with enamel hypoplasia and osteopetrosis, which can be severe, leading to optic nerve entrapment and blindness [102, 116].
The sequelae of dRTA are recurrent nephrolithiasis, nephrocalcinosis and low bone mass. While nephrocalcinosis is almost a universal finding in inherited dRTA, the prevalence of nephrolithiasis is unknown and has not been reported in large cohort studies [104, 105]. Hypercalciuria (due to increased release of Ca from bone and decreased renal Ca reabsorption), hypocitraturia (due to augmented proximal tubular citrate reabsorption) and alkaline urinary pH are the three principal prolithogenic factors in dRTA and favor CaP precipitation [117,118,119,120]. The typical renal calculus in dRTA consists of carbonate apatite and has a characteristic morphology with a smooth aspect and a glazed brown-yellow appearance with tiny cracks [121, 122]. Nephrocalcinosis is almost a universal finding in inherited dRTA and frequently associated with reduced GFR [104]. The pathogenesis of nephrocalcinosis in dRTA is unknown but possibly involves the same lithogenic factors that also foster the development of stones. Patients with dRTA typically present with low bone mass (exception are patients with CA2 mutations), primarily due to reduced bone formation and turnover rates, and to some extent defective mineralization and reduced non-collagenous proteins [123,124,125]. Alkali therapy is the cornerstone of dRTA treatment. In children, alkali improves bone turnover and somatic growth [126, 127]. In adults with dRTA, alkali therapy decreases calciuria, increases citraturia, reduces stone formation, normalizes bone formation and increases bone density [118, 125, 128, 129]. K-citrate is preferable over Na-citrate because K-citrate tends to reduce calciuria in addition to increasing citraturia, whereas Na-citrate at equimolar doses can increase calciuria [128].
Variants
V-ATPase B1 subunit
Impaired urinary acidification capacity in the absence of systemic acidosis is a frequent finding in recurrent SF and has been designated “incomplete dRTA” [108, 130,131,132,133]. There is a positive association of kidney stone CaP content and the prevalence of a urinary acidification deficit in SF [134, 135]. In most SF, the cause of the urinary acidification deficit remains obscure and maybe due to acquired or inherited conditions. Recent studies revealed that heterozygous carriers of a recessive ATP6V1B1 truncation mutation c.1401_1402dupGT; p.F468fsX487 or SF heterozygous for the non-synonymous ATP6V1B1 polymorphism c.481G>A; p.E161K exhibit a urinary acidification deficit in the absence of systemic acidosis, reduced urinary citrate and an increased prevalence of CaP containing kidney stones (Table 2) [136, 137]. In an unselected cohort of SF referred for metabolic work-up to a tertiary care stone clinic, the prevalence of the ATP6V1B1 c.481G>A;p.E161K polymorphism was 5.8% [137]. Compound heterozygosity or homozygosity for the ATP6V1B1 c.481G>A; p.E161K polymorphism was reported to be associated with overt dRTA [11, 138]. In vitro studies demonstrated intact integration of p.E161K B1 subunits into the V-ATPase complex but greatly reduced pump function [136, 139]. No evidence for a dominant-negative effect of p.E161K B1 subunits was observed in these studies, suggesting haploinsufficiency in heterozygous carriers as underlying mechanism. If other known V-ATPase B1 or a4 subunit mutations also cause a detectable deficit in urinary acidification in a heterozygous state is currently unknown. Thus, it is conceivable but yet unproven that additional allelic variants in ATP6V1B1 or in other genes associated with urinary net acid excretion are associated with a urinary acidification deficit, reduced urinary citrate excretion and an increased risk of CaP stone formation. In mice genetic deletion of the type A intercalated cell apical ammonia transporter Rhcg, the basolateral K–Cl− cotransporter KCC4 or the basolateral chloride/bicarbonate exchanger SLC26A7 cause dRTA [63,64,65]. To date, however, pathogenic variants in these transporters have not been reported in patients with dRTA and nephrolithiasis.
Conclusions
Recognition of monogenetic forms of nephrolithiasis in the TALH and CNT is of prime clinical importance. The phenotype is well defined and traits are highly penetrant, making genetic testing a powerful diagnostic tool with prognostic and therapeutic relevance. In contrast, the impact of common, phenotypically mild variants in TALH or CNT genes on stone recurrence, disease progression and treatment response remains unclear. Thus, genetic testing for these common variants therefore has currently no role in the clinical care of patients suffering from kidney stones.
The identification of patients with inherited forms of nephrolithiasis remains a challenge, the main obstacle being differentiation from idiopathic stone disease. A detailed history and thorough clinical examination of the patient as well as the alertness of the treating physician are key. Red flags pointing to inherited kidney stone disease are early onset of disease, positive family history, consanguinity of parents, progressive renal failure, nephrocalcinosis, tubular dysfunction or extrarenal manifestations. Diagnosis is greatly facilitated if an expert environment with established collaborations between nephrologists, urologists and geneticists is available. With novel and specific treatment options at the horizon and a continued high rate of discoveries in the field, the relevance of recognition and diagnosis of inherited forms of nephrolithiasis will increase considerably in the next years.
About half of all inherited disorders of the kidney currently known to be associated with nephrolithiasis or nephrocalcinosis localize to the TALH or the CT. Molecular and phenotypic investigations of these rare disorders have greatly expanded our knowledge in renal physiology. Nevertheless, our understanding of the basic mechanisms leading to stone formation and/or interstitial calcifications in these patients remains poor. A large knowledge gap also exists on how to best treat these patients to prevent stone recurrence and progression of nephrocalcinosis, and the associated CKD. Clearly, there is a dire need for more basic and clinical research efforts in this neglected area. Studying these rare monogenetic forms of stone disease offers a unique opportunity to unravel general mechanisms of calcium stone formation and may lead to the development of novel diagnostic and therapeutic strategies for “idiopathic” SF.
References
Clubbe WH (1874) Family disposition to urinary concretions. Lancet 1:823
Resnick M, Pridgen DB, Goodman HO (1968) Genetic predisposition to formation of calcium oxalate renal calculi. N Engl J Med 278(24):1313–1318
Curhan GC, Willett WC, Rimm EB, Stampfer MJ (1997) Family history and risk of kidney stones. J Am Soc Nephrol 8(10):1568–1573
Guerra A, Ticinesi A, Allegri F, Nouvenne A, Pinelli S, Lauretani F, Maggio M, Cervellin G, Borghi L, Meschi T (2017) Calcium urolithiasis course in young stone formers is influenced by the strength of family history: results from a retrospective study. Urolithiasis 45(6):525–533
Goldfarb DS, Fischer ME, Keich Y, Goldberg J (2005) A twin study of genetic and dietary influences on nephrolithiasis: a report from the Vietnam Era Twin (VET) Registry. Kidney Int 67(3):1053–1061
Edvardsson VO, Palsson R, Indridason OS, Thorvaldsson S, Stefansson K (2009) Familiality of kidney stone disease in Iceland. Scand J Urol Nephrol 43(5):420–424
Oddsson A, Sulem P, Helgason H, Edvardsson VO, Thorleifsson G, Sveinbjornsson G, Haraldsdottir E, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Masson G, Holm H, Gudbjartsson DF, Thorsteinsdottir U, Indridason OS, Palsson R, Stefansson K (2015) Common and rare variants associated with kidney stones and biochemical traits. Nat Commun 6:7975
Thorleifsson G, Holm H, Edvardsson V, Walters GB, Styrkarsdottir U, Gudbjartsson DF, Sulem P, Halldorsson BV, de Vegt F, d’Ancona FC, den Heijer M, Franzson L, Christiansen C, Alexandersen P, Rafnar T, Kristjansson K, Sigurdsson G, Kiemeney LA, Bodvarsson M, Indridason OS, Palsson R, Kong A, Thorsteinsdottir U, Stefansson K (2009) Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet 41(8):926–930
Urabe Y, Tanikawa C, Takahashi A, Okada Y, Morizono T, Tsunoda T, Kamatani N, Kohri K, Chayama K, Kubo M, Nakamura Y, Matsuda K (2012) A genome-wide association study of nephrolithiasis in the Japanese population identifies novel susceptible Loci at 5q35.3, 7p14.3, and 13q14.1. PLoS Genet 8(3):e1002541
Gee HY, Jun I, Braun DA, Lawson JA, Halbritter J, Shril S, Nelson CP, Tan W, Stein D, Wassner AJ, Ferguson MA, Gucev Z, Sayer JA, Milosevic D, Baum M, Tasic V, Lee MG, Hildebrandt F (2016) Mutations in SLC26A1 cause nephrolithiasis. Am J Hum Genet 98(6):1228–1234
Halbritter J, Baum M, Hynes AM, Rice SJ, Thwaites DT, Gucev ZS, Fisher B, Spaneas L, Porath JD, Braun DA, Wassner AJ, Nelson CP, Tasic V, Sayer JA, Hildebrandt F (2015) Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol 26(3):543–551
Griffin DG (2004) A review of the heritability of idiopathic nephrolithiasis. J Clin Pathol 57(8):793–796
Mohebbi N, Ferraro PM, Gambaro G, Unwin R (2017) Tubular and genetic disorders associated with kidney stones. Urolithiasis 45(1):127–137
Randall A (1936) An hypothesis for the origin of renal calculus. N Engl J Med 214(6):234–242
Evan AP, Lingeman JE, Coe FL, Parks JH, Bledsoe SB, Shao Y, Sommer AJ, Paterson RF, Kuo RL, Grynpas M (2003) Randall’s plaque of patients with nephrolithiasis begins in basement membranes of thin loops of Henle. J Clin Investig 111(5):607–616
Nielsen S, Pallone T, Smith BL, Christensen EI, Agre P, Maunsbach AB (1995) Aquaporin-1 water channels in short and long loop descending thin limbs and in descending vasa recta in rat kidney. Am J Physiol 268(6 Pt 2):F1023–F1037. https://doi.org/10.1152/ajprenal.1995.268.6.F1023
Pannabecker TL (2012) Structure and function of the thin limbs of the loop of Henle. Compr Physiol 2(3):2063–2086. https://doi.org/10.1002/cphy.c110019
Mount DB (2014) Thick ascending limb of the loop of Henle. Clin J Am Soc Nephrol 9(11):1974–1986. https://doi.org/10.2215/CJN.04480413
Greger R (1985) Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol Rev 65(3):760–797. https://doi.org/10.1152/physrev.1985.65.3.760
Dantzler WH, Layton AT, Layton HE, Pannabecker TL (2014) Urine-concentrating mechanism in the inner medulla: function of the thin limbs of the loops of Henle. Clin J Am Soc Nephrol 9(10):1781–1789. https://doi.org/10.2215/CJN.08750812
Castrop H, Schiessl IM (2014) Physiology and pathophysiology of the renal Na–K–2Cl cotransporter (NKCC2). Am J Physiol Ren Physiol 307(9):F991–F1002. https://doi.org/10.1152/ajprenal.00432.2014
Hebert SC, Andreoli TE (1984) Control of NaCl transport in the thick ascending limb. Am J Physiol 246(6 Pt 2):F745–F756. https://doi.org/10.1152/ajprenal.1984.246.6.F745
Burg M, Stoner L, Cardinal J, Green N (1973) Furosemide effect on isolated perfused tubules. Am J Physiol 225(1):119–124. https://doi.org/10.1152/ajplegacy.1973.225.1.119
Wang WH (1994) Two types of K+ channel in thick ascending limb of rat kidney. Am J Physiol 267(4 Pt 2):F599–F605. https://doi.org/10.1152/ajprenal.1994.267.4.F599
Lu M, Wang T, Yan Q, Wang W, Giebisch G, Hebert SC (2004) ROMK is required for expression of the 70-pS K channel in the thick ascending limb. Am J Physiol Ren Physiol 286(3):F490–F495. https://doi.org/10.1152/ajprenal.00305.2003
Greger R, Schlatter E (1981) Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle’s loop of rabbit kidney. Pflugers Arch 392(1):92–94
Palmer LG, Frindt G (2006) Cl− channels of the distal nephron. Am J Physiol Ren Physiol 291(6):F1157–F1168. https://doi.org/10.1152/ajprenal.00496.2005
Mercado A, Song L, Vazquez N, Mount DB, Gamba G (2000) Functional comparison of the K+–Cl− cotransporters KCC1 and KCC4. J Biol Chem 275(39):30326–30334. https://doi.org/10.1074/jbc.M003112200
Hebert SC, Andreoli TE (1986) Ionic conductance pathways in the mouse medullary thick ascending limb of Henle. The paracellular pathway and electrogenic Cl− absorption. J Gen Physiol 87(4):567–590
de Baaij JH, Hoenderop JG, Bindels RJ (2015) Magnesium in man: implications for health and disease. Physiol Rev 95(1):1–46. https://doi.org/10.1152/physrev.00012.2014
Moor MB, Bonny O (2016) Ways of calcium reabsorption in the kidney. Am J Physiol Ren Physiol 310(11):F1337–F1350. https://doi.org/10.1152/ajprenal.00273.2015
Gong Y, Hou J (2017) Claudins in barrier and transport function-the kidney. Pflugers Arch 469(1):105–113. https://doi.org/10.1007/s00424-016-1906-6
Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP (1999) Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285(5424):103–106
Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S (2006) Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79(5):949–957. https://doi.org/10.1086/508617
Hou J, Shan Q, Wang T, Gomes AS, Yan Q, Paul DL, Bleich M, Goodenough DA (2007) Transgenic RNAi depletion of claudin-16 and the renal handling of magnesium. J Biol Chem 282(23):17114–17122. https://doi.org/10.1074/jbc.M700632200
Hou J, Renigunta A, Gomes AS, Hou M, Paul DL, Waldegger S, Goodenough DA (2009) Claudin-16 and claudin-19 interaction is required for their assembly into tight junctions and for renal reabsorption of magnesium. Proc Natl Acad Sci USA 106(36):15350–15355. https://doi.org/10.1073/pnas.0907724106
Plain A, Alexander RT (2018) Claudins and nephrolithiasis. Curr Opin Nephrol Hypertens 27(4):268–276. https://doi.org/10.1097/MNH.0000000000000426
Gong Y, Renigunta V, Himmerkus N, Zhang J, Renigunta A, Bleich M, Hou J (2012) Claudin-14 regulates renal Ca(+)(+) transport in response to CaSR signalling via a novel microRNA pathway. EMBO J 31(8):1999–2012. https://doi.org/10.1038/emboj.2012.49
Breiderhoff T, Himmerkus N, Stuiver M, Mutig K, Will C, Meij IC, Bachmann S, Bleich M, Willnow TE, Muller D (2012) Deletion of claudin-10 (Cldn10) in the thick ascending limb impairs paracellular sodium permeability and leads to hypermagnesemia and nephrocalcinosis. Proc Natl Acad Sci USA 109(35):14241–14246. https://doi.org/10.1073/pnas.1203834109
Breiderhoff T, Himmerkus N, Drewell H, Plain A, Gunzel D, Mutig K, Willnow TE, Muller D, Bleich M (2018) Deletion of claudin-10 rescues claudin-16-deficient mice from hypomagnesemia and hypercalciuria. Kidney Int 93(3):580–588. https://doi.org/10.1016/j.kint.2017.08.029
Toka HR, Pollak MR, Houillier P (2015) Calcium sensing in the renal tubule. Physiology (Bethesda) 30(4):317–326. https://doi.org/10.1152/physiol.00042.2014
Wang WH, Lu M, Hebert SC (1996) Cytochrome P-450 metabolites mediate extracellular Ca(2+)-induced inhibition of apical K+ channels in the TAL. Am J Physiol 271(1 Pt 1):C103–C111. https://doi.org/10.1152/ajpcell.1996.271.1.C103
Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, Bruneval P, Mandet C, Christensen EI, Faure H, Cheval L, Laghmani K, Collet C, Eladari D, Dodd RH, Ruat M, Houillier P (2012) PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Investig 122(9):3355–3367. https://doi.org/10.1172/JCI57407
Dimke H, Desai P, Borovac J, Lau A, Pan W, Alexander RT (2013) Activation of the Ca(2+)-sensing receptor increases renal claudin-14 expression and urinary Ca(2+) excretion. Am J Physiol Ren Physiol 304(6):F761–F769. https://doi.org/10.1152/ajprenal.00263.2012
Gong Y, Hou J (2014) Claudin-14 underlies Ca(+)(+)-sensing receptor-mediated Ca(+)(+) metabolism via NFAT-microRNA-based mechanisms. J Am Soc Nephrol 25(4):745–760. https://doi.org/10.1681/ASN.2013050553
Toka HR, Al-Romaih K, Koshy JM, DiBartolo S III, Kos CH, Quinn SJ, Curhan GC, Mount DB, Brown EM, Pollak MR (2012) Deficiency of the calcium-sensing receptor in the kidney causes parathyroid hormone-independent hypocalciuria. J Am Soc Nephrol 23(11):1879–1890. https://doi.org/10.1681/ASN.2012030323
Devuyst O, Olinger E, Rampoldi L (2017) Uromodulin: from physiology to rare and complex kidney disorders. Nat Rev Nephrol 13(9):525–544
Trudu M, Janas S, Lanzani C, Debaix H, Schaeffer C, Ikehata M, Citterio L, Demaretz S, Trevisani F, Ristagno G, Glaudemans B, Laghmani K, Dell’Antonio G, team S, Loffing J, Rastaldi MP, Manunta P, Devuyst O, Rampoldi L (2013) Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19(12):1655–1660. https://doi.org/10.1038/nm.3384
Mutig K, Kahl T, Saritas T, Godes M, Persson P, Bates J, Raffi H, Rampoldi L, Uchida S, Hille C, Dosche C, Kumar S, Castaneda-Bueno M, Gamba G, Bachmann S (2011) Activation of the bumetanide-sensitive Na+, K+, 2Cl− cotransporter (NKCC2) is facilitated by Tamm–Horsfall protein in a chloride-sensitive manner. J Biol Chem 286(34):30200–30210. https://doi.org/10.1074/jbc.M111.222968
Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S (2011) Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J Biol Chem 286(3):2224–2235. https://doi.org/10.1074/jbc.M110.149880
Mo L, Huang HY, Zhu XH, Shapiro E, Hasty DL, Wu XR (2004) Tamm–Horsfall protein is a critical renal defense factor protecting against calcium oxalate crystal formation. Kidney Int 66(3):1159–1166. https://doi.org/10.1111/j.1523-1755.2004.00867.x
Liu Y, Mo L, Goldfarb DS, Evan AP, Liang F, Khan SR, Lieske JC, Wu XR (2010) Progressive renal papillary calcification and ureteral stone formation in mice deficient for Tamm–Horsfall protein. Am J Physiol Ren Physiol 299(3):F469–F478. https://doi.org/10.1152/ajprenal.00243.2010
Garimella PS, Bartz TM, Ix JH, Chonchol M, Shlipak MG, Devarajan P, Bennett MR, Sarnak MJ (2017) Urinary uromodulin and risk of urinary tract infections: the cardiovascular health study. Am J Kidney Dis 69(6):744–751
Roy A, Al-bataineh MM, Pastor-Soler NM (2015) Collecting duct intercalated cell function and regulation. Clin J Am Soc Nephrol 10(2):305–324. https://doi.org/10.2215/CJN.08880914
Sampogna RV, Al-Awqati Q (2013) Salt and pepper distribution of cell types in the collecting duct. J Am Soc Nephrol 24(2):163–165. https://doi.org/10.1681/ASN.2012121183
Madsen KM, Verlander JW, Tisher CC (1988) Relationship between structure and function in distal tubule and collecting duct. J Electron Microsc Tech 9(2):187–208. https://doi.org/10.1002/jemt.1060090206
Wall SM, Weinstein AM (2013) Cortical distal nephron Cl(−) transport in volume homeostasis and blood pressure regulation. Am J Physiol Ren Physiol 305(4):F427–F438. https://doi.org/10.1152/ajprenal.00022.2013
Smith AN, Lovering RC, Futai M, Takeda J, Brown D, Karet FE (2003) Revised nomenclature for mammalian vacuolar-type H+-ATPase subunit genes. Mol Cell 12(4):801–803
Miller RL, Zhang P, Smith M, Beaulieu V, Paunescu TG, Brown D, Breton S, Nelson RD (2005) V-ATPase B1-subunit promoter drives expression of EGFP in intercalated cells of kidney, clear cells of epididymis and airway cells of lung in transgenic mice. Am J Physiol Cell Physiol 288(5):C1134–C1144
Breton S, Brown D (2013) Regulation of luminal acidification by the V-ATPase. Physiology (Bethesda) 28(5):318–329. https://doi.org/10.1152/physiol.00007.2013
Brochard K, Boyer O, Blanchard A, Loirat C, Niaudet P, Macher MA, Deschenes G, Bensman A, Decramer S, Cochat P, Morin D, Broux F, Caillez M, Guyot C, Novo R, Jeunemaitre X, Vargas-Poussou R (2009) Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome. Nephrol Dial Transplant 24(5):1455–1464. https://doi.org/10.1093/ndt/gfn689
Stechman MJ, Loh NY, Thakker RV (2009) Genetic causes of hypercalciuric nephrolithiasis. Pediatr Nephrol 24(12):2321–2332. https://doi.org/10.1007/s00467-008-0807-0
Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, Sanjad SA, Lifton RP (1996) Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet 14(2):152–156
Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP (1996) Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na–K–2Cl cotransporter NKCC2. Nat Genet 13(2):183–188
Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, Schurman S, Nayir A, Alpay H, Bakkaloglu A, Rodriguez-Soriano J, Morales JM, Sanjad SA, Taylor CM, Pilz D, Brem A, Trachtman H, Griswold W, Richard GA, John E, Lifton RP (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet 17(2):171–178
Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T (2002) Association between activating mutations of calcium-sensing receptor and Bartter’s syndrome. Lancet 360(9334):692–694
Birkenhager R, Otto E, Schurmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, Milford DV, Jeck N, Konrad M, Landau D, Knoers NV, Antignac C, Sudbrak R, Kispert A, Hildebrandt F (2001) Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet 29(3):310–314
Besbas N, Ozaltin F, Jeck N, Seyberth H, Ludwig M (2005) CLCN5 mutation (R347X) associated with hypokalaemic metabolic alkalosis in a Turkish child: an unusual presentation of Dent’s disease. Nephrol Dial Transplant 20(7):1476–1479. https://doi.org/10.1093/ndt/gfh799
Okamoto T, Tajima T, Hirayama T, Sasaki S (2012) A patient with Dent disease and features of Bartter syndrome caused by a novel mutation of CLCN5. Eur J Pediatr 171(2):401–404. https://doi.org/10.1007/s00431-011-1578-3
Bogdanovic R, Draaken M, Toromanovic A, Dordevic M, Stajic N, Ludwig M (2010) A novel CLCN5 mutation in a boy with Bartter-like syndrome and partial growth hormone deficiency. Pediatr Nephrol 25(11):2363–2368. https://doi.org/10.1007/s00467-010-1615-x
Laghmani K, Beck BB, Yang SS, Seaayfan E, Wenzel A, Reusch B, Vitzthum H, Priem D, Demaretz S, Bergmann K, Duin LK, Gobel H, Mache C, Thiele H, Bartram MP, Dombret C, Altmuller J, Nurnberg P, Benzing T, Levtchenko E, Seyberth HW, Klaus G, Yigit G, Lin SH, Timmer A, de Koning TJ, Scherjon SA, Schlingmann KP, Bertrand MJ, Rinschen MM, de Backer O, Konrad M, Komhoff M (2016) Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 Mutations. N Engl J Med 374(19):1853–1863
Colussi G, De Ferrari ME, Tedeschi S, Prandoni S, Syren ML, Civati G (2002) Bartter syndrome type 3: an unusual cause of nephrolithiasis. Nephrol Dial Transplant 17(3):521–523
Puricelli E, Bettinelli A, Borsa N, Sironi F, Mattiello C, Tammaro F, Tedeschi S, Bianchetti MG, Italian Collaborative Group for Bartter S (2010) Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant 25(9):2976–2981. https://doi.org/10.1093/ndt/gfq119
Jeck N, Konrad M, Hess M, Seyberth HW (2000) The diuretic- and Bartter-like salt-losing tubulopathies. Nephrol Dial Transplant 15 Suppl 6:19–20
Peters M, Jeck N, Reinalter S, Leonhardt A, Tonshoff B, Klaus GG, Konrad M, Seyberth HW (2002) Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med 112(3):183–190
Lee BH, Cho HY, Lee H, Han KH, Kang HG, Ha IS, Lee JH, Park YS, Shin JI, Lee DY, Kim SY, Choi Y, Cheong HI (2012) Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant 27(4):1516–1521. https://doi.org/10.1093/ndt/gfr475
Seys E, Andrini O, Keck M, Mansour-Hendili L, Courand PY, Simian C, Deschenes G, Kwon T, Bertholet-Thomas A, Bobrie G, Borde JS, Bourdat-Michel G, Decramer S, Cailliez M, Krug P, Cozette P, Delbet JD, Dubourg L, Chaveau D, Fila M, Jourde-Chiche N, Knebelmann B, Lavocat MP, Lemoine S, Djeddi D, Llanas B, Louillet F, Merieau E, Mileva M, Mota-Vieira L, Mousson C, Nobili F, Novo R, Roussey-Kesler G, Vrillon I, Walsh SB, Teulon J, Blanchard A, Vargas-Poussou R (2017) Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol 28(8):2540–2552. https://doi.org/10.1681/ASN.2016101057
Li D, Tian L, Hou C, Kim CE, Hakonarson H, Levine MA (2016) Association of mutations in SLC12A1 encoding the NKCC2 cotransporter with neonatal primary hyperparathyroidism. J Clin Endocrinol Metab 101(5):2196–2200
Wongsaengsak S, Vidmar AP, Addala A, Kamil ES, Sequeira P, Fass B, Pitukcheewanont P (2017) A novel SLC12A1 gene mutation associated with hyperparathyroidism, hypercalcemia, nephrogenic diabetes insipidus, and nephrocalcinosis in four patients. Bone 97:121–125
Weber S, Schneider L, Peters M, Misselwitz J, Ronnefarth G, Boswald M, Bonzel KE, Seeman T, Sulakova T, Kuwertz-Broking E, Gregoric A, Palcoux JB, Tasic V, Manz F, Scharer K, Seyberth HW, Konrad M (2001) Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 12(9):1872–1881
Praga M, Vara J, Gonzalez-Parra E, Andres A, Alamo C, Araque A, Ortiz A, Rodicio JL (1995) Familial hypomagnesemia with hypercalciuria and nephrocalcinosis. Kidney Int 47(5):1419–1425
Wolf MT, Dotsch J, Konrad M, Boswald M, Rascher W (2002) Follow-up of five patients with FHHNC due to mutations in the Paracellin-1 gene. Pediatr Nephrol 17(8):602–608
Zimmermann B, Plank C, Konrad M, Stohr W, Gravou-Apostolatou C, Rascher W, Dotsch J (2006) Hydrochlorothiazide in CLDN16 mutation. Nephrol Dial Transplant 21(8):2127–2132. https://doi.org/10.1093/ndt/gfl144
Tasic V, Dervisov D, Koceva S, Weber S, Konrad M (2005) Hypomagnesemia with hypercalciuria and nephrocalcinosis: case report and a family study. Pediatr Nephrol 20(7):1003–1006
Toka HR, Genovese G, Mount DB, Pollak MR, Curhan GC (2013) Frequency of rare allelic variation in candidate genes among individuals with low and high urinary calcium excretion. PLoS One 8(8):e71885. https://doi.org/10.1371/journal.pone.0071885
Guha M, Bankura B, Ghosh S, Pattanayak AK, Ghosh S, Pal DK, Puri A, Kundu AK, Das M (2015) Polymorphisms in CaSR and CLDN14 genes associated with increased risk of kidney stone disease in patients from the eastern part of India. PLoS One 10(6):e0130790. https://doi.org/10.1371/journal.pone.0130790
Ure ME, Heydari E, Pan W, Ramesh A, Rehman S, Morgan C, Pinsk M, Erickson R, Herrmann JM, Dimke H, Cordat E, Lemaire M, Walter M, Alexander RT (2017) A variant in a cis-regulatory element enhances claudin-14 expression and is associated with pediatric-onset hypercalciuria and kidney stones. Hum Mutat 38(6):649–657. https://doi.org/10.1002/humu.23202
Vezzoli G, Tanini A, Ferrucci L, Soldati L, Bianchin C, Franceschelli F, Malentacchi C, Porfirio B, Adamo D, Terranegra A, Falchetti A, Cusi D, Bianchi G, Brandi ML (2002) Influence of calcium-sensing receptor gene on urinary calcium excretion in stone-forming patients. J Am Soc Nephrol 13(10):2517–2523
Ding Q, Fan B, Shi Y, Fan Z, Ding L, Li F, Tu W, Jin X, Qin C, Cao Q, Yuan Q, Wang J, Ouyang J (2017) Calcium-sensing receptor genetic polymorphisms and risk of developing nephrolithiasis in a Chinese population. Urol Int 99(3):331–337
Vezzoli G, Terranegra A, Arcidiacono T, Gambaro G, Milanesi L, Mosca E, Soldati L (2010) Calcium kidney stones are associated with a haplotype of the calcium-sensing receptor gene regulatory region. Nephrol Dial Transplant 25(7):2245–2252
Vezzoli G, Terranegra A, Aloia A, Arcidiacono T, Milanesi L, Mosca E, Mingione A, Spotti D, Cusi D, Hou J, Hendy GN, Soldati L, Paloschi V, Dogliotti E, Brasacchio C, Dell’Antonio G, Montorsi F, Bertini R, Bellinzoni P, Guazzoni G, Borghi L, Guerra A, Allegri F, Ticinesi A, Meschi T, Nouvenne A, Lupo A, Fabris A, Gambaro G, Strazzullo P, Rendina D, De Filippo G, Brandi ML, Croppi E, Cianferotti L, Trinchieri A, Caudarella R, Cupisti A, Anglani F, Del Prete D (2013) Decreased transcriptional activity of calcium-sensing receptor gene promoter 1 is associated with calcium nephrolithiasis. J Clin Endocrinol Metab 98(9):3839–3847
Shakhssalim N, Kazemi B, Basiri A, Houshmand M, Pakmanesh H, Golestan B, Eilanjegh AF, Kashi AH, Kilani M, Azadvari M (2010) Association between calcium-sensing receptor gene polymorphisms and recurrent calcium kidney stone disease: a comprehensive gene analysis. Scand J Urol Nephrol 44(6):406–412
Hamilton DC, Grover VK, Smith CA, Cole DE (2009) Heterogeneous disease modeling for Hardy–Weinberg disequilibrium in case–control studies: application to renal stones and calcium-sensing receptor polymorphisms. Ann Hum Genet 73(2):176–183
Chou YH, Woon PY, Chen WC, Hsu YW, Chang JM, Hwang DY, Chiu YC, Kuo HC, Chang WP, Hou MF, Liu ME, Chang JG, Chang WC (2011) A genetic polymorphism (rs17251221) in the calcium-sensing receptor gene (CASR) is associated with stone multiplicity in calcium nephrolithiasis. PLoS One 6(9):e25227
Gudbjartsson DF, Holm H, Indridason OS, Thorleifsson G, Edvardsson V, Sulem P, de Vegt F, d’Ancona FC, den Heijer M, Wetzels JF, Franzson L, Rafnar T, Kristjansson K, Bjornsdottir US, Eyjolfsson GI, Kiemeney LA, Kong A, Palsson R, Thorsteinsdottir U, Stefansson K (2010) Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet 6(7):e1001039
Lightwood R (1935) Communication no. 1. Arch Dis Child 10:205
Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP (1999) Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21(1):84–90
Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE (2000) Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26(1):71–75
Enerback S, Nilsson D, Edwards N, Heglind M, Alkanderi S, Ashton E, Deeb A, Kokash FEB, Bakhsh ARA, Van’t Hoff W, Walsh SB, D’Arco F, Daryadel A, Bourgeois S, Wagner CA, Kleta R, Bockenhauer D, Sayer JA (2018) Acidosis and deafness in patients with recessive mutations in FOXI1. J Am Soc Nephrol 29(3):1041–1048
Karet FE, Gainza FJ, Gyory AZ, Unwin RJ, Wrong O, Tanner MJ, Nayir A, Alpay H, Santos F, Hulton SA, Bakkaloglu A, Ozen S, Cunningham MJ, di Pietro A, Walker WG, Lifton RP (1998) Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis. Proc Natl Acad Sci USA 95(11):6337–6342
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE (1983) Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA 80(9):2752–2756
Sly WS, Whyte MP, Sundaram V, Tashian RE, Hewett-Emmett D, Guibaud P, Vainsel M, Baluarte HJ, Gruskin A, Al-Mosawi M et al (1985) Carbonic anhydrase II deficiency in 12 families with the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. N Engl J Med 313(3):139–145
Mohebbi N, Wagner CA (2018) Pathophysiology, diagnosis and treatment of inherited distal renal tubular acidosis. J Nephrol 31(4):511–522
Palazzo V, Provenzano A, Becherucci F, Sansavini G, Mazzinghi B, Orlandini V, Giunti L, Roperto RM, Pantaleo M, Artuso R, Andreucci E, Bargiacchi S, Traficante G, Stagi S, Murer L, Benetti E, Emma F, Giordano M, Rivieri F, Colussi G, Penco S, Manfredini E, Caruso MR, Garavelli L, Andrulli S, Vergine G, Miglietti N, Mancini E, Malaventura C, Percesepe A, Grosso E, Materassi M, Romagnani P, Giglio S (2017) The genetic and clinical spectrum of a large cohort of patients with distal renal tubular acidosis. Kidney Int 91(5):1243–1255
Vargas-Poussou R, Houillier P, Le Pottier N, Strompf L, Loirat C, Baudouin V, Macher MA, Dechaux M, Ulinski T, Nobili F, Eckart P, Novo R, Cailliez M, Salomon R, Nivet H, Cochat P, Tack I, Fargeot A, Bouissou F, Kesler GR, Lorotte S, Godefroid N, Layet V, Morin G, Jeunemaitre X, Blanchard A (2006) Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J Am Soc Nephrol 17(5):1437–1443
Rysava R, Tesar V, Jirsa M Jr, Brabec V, Jarolim P (1997) Incomplete distal renal tubular acidosis coinherited with a mutation in the band 3 (AE1) gene. Nephrol Dial Transplant 12(9):1869–1873
Bruce LJ, Cope DL, Jones GK, Schofield AE, Burley M, Povey S, Unwin RJ, Wrong O, Tanner MJ (1997) Familial distal renal tubular acidosis is associated with mutations in the red cell anion exchanger (Band 3, AE1) gene. J Clin Investig 100(7):1693–1707
Wrong O, Davies HE (1959) The excretion of acid in renal disease. Q J Med 28(110):259–313
Escobar LI, Simian C, Treard C, Hayek D, Salvador C, Guerra N, Matos M, Medeiros M, Enciso S, Camargo MD, Vargas-Poussou R (2016) Mutations in ATP6V1B1 and ATP6V0A4 genes cause recessive distal renal tubular acidosis in Mexican families. Mol Genet Genom Med 4(3):303–311
Berrettini S, Neri E, Forli F, Panconi M, Massimetti M, Ravecca F, Sellari-Franceschini S, Bartolozzi C (2001) Large vestibular aqueduct in distal renal tubular acidosis. High-resolution MR in three cases. Acta Radiol 42(3):320–322
Berrettini S, Forli F, Franceschini SS, Ravecca F, Massimetti M, Neri E (2002) Distal renal tubular acidosis associated with isolated large vestibular aqueduct and sensorineural hearing loss. Ann Otol Rhinol Laryngol 111(5 Pt 1):385–391
Berrettini S, Forli F, Bogazzi F, Neri E, Salvatori L, Casani AP, Franceschini SS (2005) Large vestibular aqueduct syndrome: audiological, radiological, clinical, and genetic features. Am J Otolaryngol 26(6):363–371
Batlle D, Haque SK (2012) Genetic causes and mechanisms of distal renal tubular acidosis. Nephrol Dial Transplant 27(10):3691–3704
Alper SL (2010) Familial renal tubular acidosis. J Nephrol 23(Suppl 16):S57–S76
Bruce LJ, Wrong O, Toye AM, Young MT, Ogle G, Ismail Z, Sinha AK, McMaster P, Hwaihwanje I, Nash GB, Hart S, Lavu E, Palmer R, Othman A, Unwin RJ, Tanner MJ (2000) Band 3 mutations, renal tubular acidosis and South-East Asian ovalocytosis in Malaysia and Papua New Guinea: loss of up to 95% band 3 transport in red cells. Biochem J 350(Pt 1):41–51
Awad M, Al-Ashwal AA, Sakati N, Al-Abbad AA, Bin-Abbas BS (2002) Long-term follow up of carbonic anhydrase II deficiency syndrome. Saudi Med J 23(1):25–29
Goodman AD, Lemann J Jr, Lennon EJ, Relman AS (1965) Production, excretion, and net balance of fixed acid in patients with renal acidosis. J Clin Investig 44:495–506
Preminger GM, Sakhaee K, Pak CY (1987) Hypercalciuria and altered intestinal calcium absorption occurring independently of vitamin D in incomplete distal renal tubular acidosis. Metabolism 36(2):176–179
Bushinsky DA, Frick KK (2000) The effects of acid on bone. Curr Opin Nephrol Hypertens 9(4):369–379
Alexander RT, Cordat E, Chambrey R, Dimke H, Eladari D (2016) Acidosis and urinary calcium excretion: insights from genetic disorders. J Am Soc Nephrol 27(12):3511–3520
Daudon M, Bouzidi H, Bazin D (2010) Composition and morphology of phosphate stones and their relation with etiology. Urol Res 38(6):459–467
Dessombz A, Letavernier E, Haymann JP, Bazin D, Daudon M (2015) Calcium phosphate stone morphology can reliably predict distal renal tubular acidosis. J Urol 193(5):1564–1569
Domrongkitchaiporn S, Pongsakul C, Stitchantrakul W, Sirikulchayanonta V, Ongphiphadhanakul B, Radinahamed P, Karnsombut P, Kunkitti N, Ruang-raksa C, Rajatanavin R (2001) Bone mineral density and histology in distal renal tubular acidosis. Kidney Int 59(3):1086–1093
Disthabanchong S, Domrongkitchaiporn S, Sirikulchayanonta V, Stitchantrakul W, Karnsombut P, Rajatanavin R (2004) Alteration of noncollagenous bone matrix proteins in distal renal tubular acidosis. Bone 35(3):604–613
Domrongkitchaiporn S, Pongskul C, Sirikulchayanonta V, Stitchantrakul W, Leeprasert V, Ongphiphadhanakul B, Radinahamed P, Rajatanavin R (2002) Bone histology and bone mineral density after correction of acidosis in distal renal tubular acidosis. Kidney Int 62(6):2160–2166
Nash MA, Torrado AD, Greifer I, Spitzer A, Edelmann CM Jr (1972) Renal tubular acidosis in infants and children. Clinical course, response to treatment, and prognosis. J Pediatr 80(5):738–748
McSherry E, Morris RC Jr (1978) Attainment and maintenance of normal stature with alkali therapy in infants and children with classic renal tubular acidosis. J Clin Investig 61(2):509–527
Preminger GM, Sakhaee K, Pak CY (1988) Alkali action on the urinary crystallization of calcium salts: contrasting responses to sodium citrate and potassium citrate. J Urol 139(2):240–242
Greenberg AJ, McNamara H, McCrory WW (1966) Metabolic balance studies in primary renal tubular acidosis: effects of acidosis on external calcium and phosphorus balances. J Pediatr 69(4):610–618
Wikstrom B, Backman U, Danielson BG, Fellstrom B, Johansson G, Ljunghall S (1983) Ambulatory diagnostic evaluation of 389 recurrent renal stone formers. A proposal for clinical classification and investigation. Klin Wochenschr 61(2):85–90
Ito H, Kotake T, Suzuki F (1993) Incidence and clinical features of renal tubular acidosis-1 in urolithiasis. Urol Int 50(2):82–85
Gambaro G, Croppi E, Coe F, Lingeman J, Moe O, Worcester E, Buchholz N, Bushinsky D, Curhan GC, Ferraro PM, Fuster D, Goldfarb DS, Heilberg IP, Hess B, Lieske J, Marangella M, Milliner D, Preminger GM, Reis Santos JM, Sakhaee K, Sarica K, Siener R, Strazzullo P, Williams JC (2016) Metabolic diagnosis and medical prevention of calcium nephrolithiasis and its systemic manifestations: a consensus statement. J Nephrol 29(6):715–734
Dhayat NA, Gradwell MW, Pathare G, Anderegg M, Schneider L, Luethi D, Mattmann C, Moe OW, Vogt B, Fuster DG (2017) Furosemide/fludrocortisone test and clinical parameters to diagnose incomplete distal renal tubular acidosis in kidney stone formers. Clin J Am Soc Nephrol 12(9):1507–1517
Pak CY, Poindexter JR, Adams-Huet B, Pearle MS (2003) Predictive value of kidney stone composition in the detection of metabolic abnormalities. Am J Med 115(1):26–32
Pak CY, Poindexter JR, Peterson RD, Heller HJ (2004) Biochemical and physicochemical presentations of patients with brushite stones. J Urol 171(3):1046–1049
Zhang J, Fuster DG, Cameron MA, Quinones H, Griffith C, Xie XS, Moe OW (2014) Incomplete distal renal tubular acidosis from a heterozygous mutation of the V-ATPase B1 subunit. Am J Physiol Ren Physiol 307(9):F1063–F1071
Dhayat NA, Schaller A, Albano G, Poindexter J, Griffith C, Pasch A, Gallati S, Vogt B, Moe OW, Fuster DG (2016) The vacuolar H+-ATPase B1 subunit polymorphism p.E161K associates with impaired urinary acidification in recurrent stone formers. J Am Soc Nephrol 27(5):1544–1554
Mohebbi N, Vargas-Poussou R, Hegemann SC, Schuknecht B, Kistler AD, Wuthrich RP, Wagner CA (2013) Homozygous and compound heterozygous mutations in the ATP6V1B1 gene in patients with renal tubular acidosis and sensorineural hearing loss. Clin Genet 83(3):274–278
Fuster DG, Zhang J, Xie XS, Moe OW (2008) The vacuolar-ATPase B1 subunit in distal tubular acidosis: novel mutations and mechanisms for dysfunction. Kidney Int 73(10):1151–1158
Acknowledgements
DF was supported by the Swiss National Centre of Competence in Research NCCR TransCure and the Swiss National Science Foundation (Grants # 31003A_135503, 31003A_152829 and 33IC30_166785/1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest. DF has served as a consultant for Otsuka Pharmaceuticals. DF has received unrestricted research funding from Novartis, Abbvie and Otsuka Pharmaceuticals.
Rights and permissions
About this article

Cite this article
Faller, N., Dhayat, N.A. & Fuster, D.G. Nephrolithiasis secondary to inherited defects in the thick ascending loop of henle and connecting tubules. Urolithiasis 47, 43–56 (2019). https://doi.org/10.1007/s00240-018-1097-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00240-018-1097-z