
Abstract
One of the central issues in the understanding of early cellular evolution is the characterisation of the cenancestor. This includes the description of the chemical nature of its genome. The disagreements on this question comprise several proposals, including the possibility that AlkB-mediated methylation repair of alkylated RNA molecules may be interpreted as evidence of a cenancestral RNA genome. We present here an evolutionary analysis of the cupin-like protein superfamily based on tertiary structure-based phylogenies that includes the oxygen-dependent AlkB and its homologs. Our results suggest that the repair of methylated RNA molecules is the outcome of the enzyme substrate ambiguity, and doesn´t necessarily indicates that the last common ancestor was endowed with an RNA genome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Following the publication of Oparin's ideas on the heterotrophic origin of life (Oparin 1938), it was generally assumed that the evolution of cells could be described as a linear sequence of events in which microbes became increasingly complex. This somewhat simplistic narrative was challenged, first, with the proposal of the endosymbiotic origin of eukaryotes (Sagan 1967) and, secondly, when the evolutionary comparison of the sequences of the ribosomal small subunit RNA (16/18S rRNA) led to the construction of an unrooted, trifurcated phylogenetic tree, which all species can be grouped in one of three cell lineages or urkingdoms. These were originally termed eubacteria, archaebacteria and urkaryotes, which corresponded to the eukaryotic nucleocytoplasm (Woese and Fox 1977). Following a period of rather intense taxonomic debates (Mayr 1998; Woese 1998), these lineages were recognised as three major cell domains, now termed as Bacteria, Archaea and Eucarya (Woese et al. 1990).
The scientific impact of the use of rRNA as a phylogenetic marker can hardly be overestimated. It made it possible to recognise the three major cell lineages and, in particular, that prokaryotes are divided into two ancient, monophyletic groups. What is somewhat surprising is the persistence of the discussions on the nature of the last common ancestor of Bacteria, Archaea and Eucarya, for which Woese and Fox (1977) coined the term progenote. As demonstrated by the papers included in this volume, although nearly half a century has passed, the disagreements on the biological nature of the progenote and of the last common ancestor remain largely unresolved. These discussions, however, have led to fruitful debates on very early phases of cellular evolution which had been largely ignored.
What is the Progenote?
Although the basic patterns of DNA replication and gene expression are the same for Bacteria, Archaea and Eucarya, recognition of their differences led Woese and Fox (1977) to conclude that they were the evolutionary outcome of independent evolutionary refinements, i.e. that each of these three lineages had evolved independently from the progenote, a hypothetical entity in which phenotype and genotype still had an imprecise, rudimentary linkage relationship. While they recognised that eukaryotes had descended from prokaryotes, they wrote that this relationship of descent is defined “in the sense that the prokaryotic is an organisational, not a phylogenetic distinction. In analogous fashion prokaryotes arose from simpler entities. The latter are properly called progenotes, because they are still in the process of evolving the relationship between genotypes and phenotype” (Woese and Fox 1977; see also Kandler 1994; Gogarten 2019).
Since the 16/18 S rRNA-based phylogeny is an unrooted tree, in Woese and Fox (1977) proposal the progenote represents the last common ancestor, because it was the hypothetical ancestral entity located at the trifurcation that led to the Bacteria, Archaea and Eucarya. In Woese and Fox original scheme it also corresponds to the root of the tree, but they never claimed that it was the first living organism, and nor did they address the presence or absence of membranes, its metabolic abilities, or the environment in which it originated and thrived. According to the original definition however, it was an entity in which a primitive error-prone form of genetically encoded protein biosynthesis had already evolved.
The suggestion that the progenote was endowed with an RNA genome was implicit and well accepted, since the likelihood that in early stages of evolution RNA genetic polymers had preceded extant cellular DNA genomes had already gained wide acceptance (Lazcano 2012). Indeed, Woese (1967) had explicitly acknowledged the primeval role of RNA molecules in his influential book The genetic code: the molecular basis for gene expression, which can be read as an evolutionary account of translation based on the idea that the highly refined structures of extant ribosomes and specificities of aminoacyl tRNA synthases could have not existed during the early stages of evolution (Lazcano 2012).
The most significant breakthrough in the search for the cenancestor was the rooting of the tree of cellular life. The first to suggest the use of duplicated genes as outgroups to root a tree was Dayhoff (1972). This methodology was successfully employed by Gogarten et al. (1989) and Iwabe et al. (1989), and corroborated by numerous analyses including those of Brown and Doolittle (1995) and Gribaldo and Cammarano (1998), all of which placed the root of the rRNA tree in the Bacteria, i.e. the bacterial phenotype is the oldest one, which also implies that the eukaryotic nucleocytoplasm evolved from the archaea.
Placing the root in the Bacteria implies that the earliest recognisable evolutionary split occurred in prokaryotes, and by pushing back in time the progenote a clear distinction between it and the last common ancestor was established. Accordingly, the reconstruction of the gene complement of the LCA can be limited to the comparison of homologous trait shared by the Bacteria and the Archaea, and does not need to include the Eucarya. However, as summarised in Table 1, the characterisation of the last common ancestor, which is located in the earliest split in universal phylogenies, is far from solved. Indeed, the plurality of terms that have been coined to describe the last common ancestor (LCA), including cenancestor, urnacestor, commonote, last universal common ancestor, last universal cellular (LUCA,) and others, reflects the different and sometimes opposing views on its nature (Becerra et al. 2007). Here we adopt the term cenancestor proposed by Fitch and Upper (1987). The purpose of this paper is to address the issue of the chemical nature of the LUCA genome, i.e. whether the population ancestral to both Bacteria and Archaea was already endowed or not with a double-stranded DNA genome.
The Cenancestral Genome
To the best of our understanding, the first attempt to characterise LUCA was developed by Lazcano et al. (1992). The characteristics of an ancestral organism can be inferred from the distribution of homologous traits in its descendants, but 30 years ago only few genes present in the three cellular lineages had been sequenced and no completely sequenced microbial genomes were available. As a result of an extensive and painstakingly review work (Lazcano et al. 1992), it was possible to compile a catalog of traits common to the three cell lineages that indicated that the ancestral phenotype already possessed (a) a translational apparatus with an oligomeric RNA polymerase, complex ribosomes endowed with ribosomal proteins; (b) a membrane-associated H + ATPase engaged in active transport; and (c) a complex set of metabolic enzymes, including the histidine biosynthetic pathway. This inventory suggested that the last common ancestor was not a protocell or any other pre-life progenitor system, but a population of organisms very much like extant prokaryotes (Lazcano et al. 1992; Becerra et al. 2007). It also implies that the basic genetic processes and major subcellular molecular traits have remained largely unchanged since the early Precambrian, perhaps due to the intricate character of the networks of gene interactions and the complex relationships between deficient metabolic processes (Lazcano and Miller 1996).
The study of sets of paralogous sequences like the hydrophilic subunits of ATPases, elongation factors, and a rather large number of metabolic enzymes can also provide specific insights into the biological events that took place prior to the appearance of the cenancestor (Lazcano et al. 1992). As summarised elsewhere, the high numbers of these duplicates implies that the LCA descended from simpler cells in which only one copy of each of these genes existed (Lazcano 1995). The numerous paralogous gene families support the hypothesis that primitive enzymes were unspecific catalysts that could react with a wide range of chemically related substrates, which can be seen as a mechanism by which primitive cells with small genomes could overcome their limited coding abilities (cf. Becerra et al. 2007).
The similarities shared by the ancient proteins present in all three lines of descent also implies that considerable fidelity already existed in the last common ancestor genetic systems (Lazcano et al. 1992). No such preservation of sequence information is found in RNA genomes (Reanney 1982; Hernández-Morales et al. 2022), which suggests that double-stranded DNA genomes were a trait that had evolved prior to the divergence of the Bacteria and Archaea (Lazcano et al. 1992; Becerra et al. 2007). However, reconstruction of ancestral states is a treacherous terrain riddled by the vagaries of convergence, loss of sequences, lateral gene transfer, sample size and other methodological issues (Becerra et al. 1997, 2007; Estrada et al. 2022). It is therefore not surprising that not all accept the conclusion that the LCA population was endowed with DNA genome.
Confusion still abounds in the field, as shown by the relatively recent suggestion in which the LCA is equated with the first living entities (Weiss et al. 2016). Proposals that the LCA had a RNA or a hybrid RNA–DNA genome have been based on indirect evidence that include (a) the absence of ribonucleotide reductases in the theoretical reconstruction of minimal cells based on the comparison of two parasitic bacteria with highly streamlined genomes (Mushegian and Koonin 1996); (b) the apparent unrelatedness of the bacterial DNA polymerases and primases with their archaeal/eucaryal counterparts (Leipe et al 1999); (c) the absence of homologous DNA-replication machinery in LCA gene-sets (Koonin 2003); and (d) the eucaryal fragmented genome, as indicated by separate chromosomes, and intense intranuclear processing of RNA transcripts (Penny and Poole 1999), together with reports on enzyme-mediated repair of RNA adducts (Poole and Logan 2005). Prompted by reports on the in vivo repair of methylated adenine and cytosine in RNA molecules by AlkB oxidative demethylases, Poole and Logan (2005) have raised the possibility that LUCA was endowed with an RNA genome. The purpose of this paper is to critically analyse this hypothesis, and to argue that in some cases biogeochemical considerations may assist in the identification and proper interpretation of cenancestral traits. We also present three-dimensional structure-based phylogenies as a complementary and more robust approach to gain insights into the early evolution of O2-dependent DNA repair mechanisms, and their application for a better understanding of the chemical nature of the LCA/LUCA or cenancestral genome.
Oxygen and Nucleic Acid AlkB-Mediated Demethylation Repair
Following the emergence of life, the second most important biogeochemical event on Earth was the origin and accumulation of a permanent oxygen-rich atmosphere, a process that took place well after LUCA, i.e. long after the evolutionary split of Bacteria and Archaea ~ 3460 Gyr ago (Ueno et al. 2006; Becerra et al. 2007). Several major evolutionary consequences of the availability of free oxygen can be recognised, including (a) the development of defence and repair mechanisms such as MsrB (Delaye et al. 2007) and AlkB (Falnes et al. 2002) repair mechanisms; (b) the emergence of O2-dependent enzymes and routes that took advantage of the oxygen potential for new reactions; and (c) the evolutionary development of O2-sensing mechanisms like hypoxia induced factors (HIFs) and oxygen transport molecules like hemerythrins (Raymond and Segré 2006; Khademian and Imlay 2021; Alvarez-Carreno et al 2016; Khersonsky and Tawfik 2010).
One of the most successful folds that took advantage of biochemical possibilities opened by the availability of free oxygen is the cupin fold (also known as the double-stranded-beta-helix fold DSBH). The cupin fold caught the attention of researchers 40 years ago, when the key role of the AlkB protein in the repair of alkylated DNA (Falnes et al 2002) was identified in Escherichia coli (Kataoka et al. 1983). Based on iterative PSI-BLAST searches, Aravind and Koonin (2001) identified AlkB as a 2-oxoglutarate (2OG) and Fe (II)-dependent oxygenase.
The 2OG-Fe (II) oxygenases (hereinafter 2OGxs) are part of one of the most diverse pfam extant clans. As of today, the CL0029 “cupin” superfamily includes 72 protein families with thousands of different domain architectures. This remarkable diversity of sequences and architectures is reflected in a considerable number of functions (Ougland et al. 2015), and a low level of sequence identity/similarity among the 72 families (Agarwal et al. 2009).
This diversity represents a challenge for the study of the molecular evolution and phylogenetic distribution of the cupin fold. Attempts to overcome the limits of traditional alignments and phylogenies have led to other methodologies like sequence clustering (Mielecki et al. 2012), secondary structure prediction analysis and structure-based phylogenies. The outcome of 3D-structure analysis for AlkB homologues was reviewed by Dunwell et al. (2001), while Agarwal et al. 2009 performed a structure-bases phylogeny with proteins containing a cupin fold suggesting that, given the high divergence among the fold, a 3D-based analysis may help to assign new unknown functions for new homologs.
The Success and Complexity of the Cupin Fold
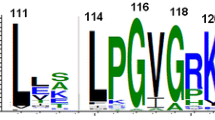
The cupin fold (also known as jumonji C (JmjC) or jelly-roll fold) is part of a protein superfamily that exhibits an extraordinary diversity of functions and a characteristic core with a double-stranded-beta-helix (DSBH) topology, that can exhibit some structural variations (Khuri et al. 2001; Islam et al. 2018) (Fig. 1). As of today 72 protein families have been described as members of the superfamily cupin, that includes a wide number of functions including isomerases or epimerases acting in cell wall carbohydrates, storage proteins in plants, transcription factors in animals (Dunwell et al. 2001), posttranslational modifications involved in collagen biosynthesis, lipid metabolism, nucleic acid repair, and chromatin and RNA splicing (Aik et al. 2012). It is well known that the variations in intermotifs in cupin folds (i.e. insertions in loops) make the identification of conserved residues a challenge (see, for instance, Fig. 1) (Khuri et al. 2001).

Structural comparison of five CL0029 pfam clan members representing the cupin fold as a typical DSBH topology. PBD codes: 1bk0_2OG-FeII_Oxy (pink), 5j3u_cNMP_binding (gold), 5nla_AraC_binding_2 (green), 6ip0_JmjC (cyan) and 6t8m_2OG-FeII_Oxy_3 (orange). The image was processed with UCSF-Chimera software using matchmaker algorithm with 5j3u_cNMP_binding structure as reference (Color figure online)
Since the fold comprises non-enzymatic as well as enzymatic proteins (Dunwell et al 2001), that catalyse between 50 and 100 different biochemical reactions, making this fold one of the largest and most catalytically diverse superfamily (Dunwell et al 2004), with approximately 5000 different architectures endowed with at least one cupin fold member, and over two million different proteins that have a cupin fold homolog (Paysan-Lafosse et al. 2023). In particular, the cupin proteins that act like DNA repair enzymes are all 2OGxs-like present in bacteria and some viruses (Hausinger 2015). In general, the reaction catalysed by 2OGxs involved in DNA repair depend on free molecular oxygen for oxidative catalytic reactions of SN2 alkylating agents in a direct reversal reaction (Begley and Samson 2003).
The cNMP_Binding Domain as the Ancestral Cupin-Like Fold
Ribonucleotide- and modified ribonucleotide-binding domains are an ancient feature whose origin may be traced to a stage in which RNA and ribonucleotides and modified ribonucleotide played a more conspicuous role in basic biological processes (Hernandez-Morales et al. 2019). Many folds that bind modified ribonucleotides (mono, di or cyclic-ribonucleotides), like the cNMP_binding domain, have a \(\beta\)-sheet fold that exhibits different interactions and binding patterns. Analysis of the evolution of lactate dehydrogenase, an enzyme with AMP/NMN binding units, led to the proposal that a \(\beta\)-sheet with three parallel strands corresponds in act to a primordial mononucleotide binding fold (Rossman et al. 1974; Rossmann and Argos 1978). Indeed, the cNMP_binding domain appears to be a reasonable candidate for an ancestral cupin ancient-like fold that reflects a conserved ancestral binding function that may have originated in a RNA/protein world.
The Biogeochemistry of O2-Dependent DNA-Repair Enzymes
It is well known than many enzymes require ions as co-factors to function. Accordingly, the relative abundances and the bioavailability of these ions can provide insights of the evolutionary history of specific enzymes, which is the case of AlkB and all the 2OGxs. The relative abundance of Fe2+ on Earth has varied across time and has a complex history coupled with that of free dioxygen. For instance, Fe2+ was abundant prior to the Great Oxidation Event ~ 2.4 Gyr ago, but the reaction of dissolved oxygen in oceans oxidised Fe2+ and the valence state changed to Fe3+, causing the precipitation of insoluble Fe3+ oxides. This is demonstrated by Moffett (2021) analysis, where the presence of Fe2+ is more abundant in oxygen deficient zones where O2 concentrations can actually reach zero.
However, the scarcity of Fe2+ did not cause an “iron crisis” (Khademian and Imlay 2021), perhaps because of the low concentration requirements of this element inside cells (Semsey et al. 2006). As discussed below, the complex geochemical history of transition metals specifically of iron, restricts the possibility of some enzymatic reactions which are relevant to theoretical reconstructions of the cenancestral gene complement.
The evolution of the cupin fold is a complex issue and remains an open question. Although there is no consensus of the evolutionary history of this fold, its well-known success is reflected on its wide distribution in the three domains of life and in some viral groups. As part of an effort to date the emergence of oxygen-dependent protein families, Jabłońska and Tawfik (2021) identified a list of O2-dependent enzymes that includes a DNA demethylase with the 2OG_FeII_Oxy_2 cupin domain that emerged at the hypothetical Last Universal Oxygen Ancestor (LUOA) node, i.e. long after cenancestral epochs 3.8–4.2 Gya ago (Moody et al. 2024).
Methodology
Tertiary Structure-Based Phylogeny
From the Protein Data Bank (December 2023/RCSB-PDB) a total of 76 structures containing CL0029 members were downloaded and trimmed to obtain only the homologous cupin domains regions with no mutations and a ligand present in the structure. In addition to the resolved crystal structures analysed, the sample was completed by adding top ranked AlphaFold database predictions (Jumper et al. 2021) for nine non-resolved or mutated structures of CL0029 pfam clan. The trimmed structures, including the predicted and complete resolved structures were aligned, and the TM-score was calculated by pairing comparison using mTM-align software (Dong et al. 2018) The output matrix was used by Fitch-Margoliash algorithm with global rearrangement optimization in the PHYLIP package (Felsenstein 2005). Two tertiary structure-based trees were constructed, one of them unrooted (Fig. 2) and a second one rooted using the cNMP_binding domain structure as an outgroup (Fig. 3). The tree was visualised and annotated in iTOL v6 (Letunic and Bork 2024). For each tree tip, a representative sequence at Interpro-pfam database were selected and their EC number was predicted with ECpred (Dalkiran et al. 2018).

Tertiary-structure based unrooted phylogeny for the 72 members of the pfam clan CL0029. Tips surrounded by the purple line lack oxidoreductase activity and do not require free oxygen and Fe2+ while the non-surrounded tips exhibit a strict requirement of O2 and Fe2+. The red shadowed area indicates the monophyletic character of 2OG dioxygenases (Color figure online)

Tertiary-based rooted phylogeny of all 72 domains/families endowed with the cupin fold. The red and blue arrows represent the incorporation of O2 and Fe2+ during the evolutionary history of the fold, and the origin of the fold’s oxidative activity. The orange arrow marks the later origin of the 2OG dioxygenases. Numbers at tips represent the EC class suggested by ECpred. Terminal tips annotated as “non enzyme” indicates that non-enzymatic activity was assigned by ECpred, suggesting a structural or binding function for the protein, while a “?” at tip indicates that ECpred was unable to assign an EC class number or a non-enzymatic function (Color figure online)
Structure Prediction and Validation of Two Procollagen Enzymes
Because no fully resolved procollagen-proline 4-dioxygenase from metazoa is available, Monosiga brevicollis (A9UV67_MONBE) and Mus musculus (P4HTM_MOUSE) procollagen-proline 4-dioxygenases tertiary structures were predicted using RoseTTAFold (Baek et al. 2021) and validated following the methodology developed by Martínez-Castilla and Rodríguez-Sotres (2010). The CL0029 members with non-resolved structure deposited in the PDB were retrieved from AlphaFold Protein Structure Database (Jumper et al. 2021).
Results
Tertiary Structure-Based Phylogeny and Structural Analysis
All the structural predictions of unsolved structures of cupin domains were included in the trees shown in Figs. 2 and 3. The significance of 2OGxs domain predictions is shown, for example, in the position 2OG-FeII_Oxy_3 domain in both trees. Because this domain is a quite important part of the architecture in the procollagen-proline 4-dioxygenase, the close relation of this domain prediction to other 2OGxs makes the prediction biologically valid. Figure 2 shows an unrooted tertiary structure-based phylogeny of all the 72 members of the cupin superfamily including the subunit \(\beta\) procollagen-proline 4-dioxygenase (2OG-FeII_Oxy_3 domain) for M. brevicollis and M. musculus.
Two main groups can be recognised in the unrooted phylogeny shown in Fig. 2. One of them is formed by domains that do not require neither oxygen nor iron, as the ECpred prediction suggest. Examples of these domains include the cNMP_binding domain, AraC_binding and Cupin_6 where transferase activity and a non-enzyme EC was assigned.
In the well-defined second group (Figs. 2 and 3), according to the EC number prediction, the majority of the cupin domains require molecular oxygen and iron for their enzymatic reactions. The 2OGxs forms a clearly defined monophyletic group (red shadowed area in Fig. 2), and appears to be closely related with other domains that partake in a huge diversity of reactions in which oxidoreductase is the prevalent enzymatic activity. In spite of the problems associated with the selection of an external group to root the phylogeny, Fig. 3 shows the same data but with a root in the cNMP_binding domain in an attempt to picture the evolutionary history of cupin fold evolution. Our hypothesis suggests that the first cupin domains had no oxidoreductase activity and, a cupin fold may have structural and transferases activity. The blue-red node shows where the oxygen and iron dependency first evolved and led to oxidoreductase activity, with the exception of EC 7 enzymes. The rooted phylogeny also shows consistently that all the EC numbers are part of the cupin superfamily.
Discussion and conclusions
The Evolutionary History of O2-Dependent Enzyme-Mediated Nucleic Acid Repair and the Chemical Nature of Cenancestor Genome
Several attempts based on different approaches to characterise the cenancestral genomic traits (Table 1) have concluded that there is no common DNA replication machinery. However, as shown here, important insights in the chemical nature of cenancestor genome can be based on additional information provided by repair enzymes to address this issue. An example of this approach relies on the use of the observed activity of the enzyme AlkB and its homologues.
There is a rather large amount of literature addressing the evolution of families with the cupin fold (Agarwal et al. 2009; Dunwell et al. 2001, 2004; Khuri et al. 2001). Given the complexity of the cupin superfamily evolution, it is difficult to establish the antiquity of the oxidative demethylation (Poole and Logan 2005). Fortunately, one of the most well-studied cupin superfamily members are precisely the dioxygenases, which are enzymes that have a strict catalytic requirement for both O2 and Fe2+ to carry out oxidoreductase activity (Aas et al. 2003; Falnes et al. 2004). As noted above, experimental data of AlkB activity on DNA and RNA demethylation reactions have been considered by Poole and Logan (2005) to raise the possibility that LUCA was in fact endowed with an RNA genome given the apparent RNA “repair” mediated by AlkB and other homologs.
Several arguments can be raised against this possibility. First, the experimental data have interpreted to argue that AlkB and its human homolog ALKBH3 are endowed with RNA “repair” activity. However, these experiments were performed with DNA and RNA homopolymers (Aas et al. 2003) that did not consider the complexity of the secondary structure of RNA present in mRNA and non-coding RNAs. Other assays have demonstrated lower efficiencies of AlkB and its homologs when the substrate is single-stranded or double-stranded RNA (van den Born et al. 2008). While the human homologue 3 (ALKBH3) and E coli’s AlkB have a strong preference for ssDNA, ALKBH3 (AlkB human homolog 3) also exhibits activity on viral single-stranded RNA genomes (Sedgwick et al. 2007; Falnes et al. 2004; Lee et al. 2005), but the recognition of the enzyme of the single-stranded nucleic acids is related to the regulation of mRNA and tRNA (Ougland et al. 2004, 2015). The differential preference and substate ambiguity may respond rather to the evolution of the homolog in a specific region of the cell like the nucleus (ALKBH2, ALKBH3, ALKBH4, ALKBH5), cytoplasm (ALKBH3) or even in mitochondria (ALKBH7) (Ougland et al. 2015).
In fact, the most likely explanation of AlkB and its homologs ambiguous recognition of substrates and the repair activity of RNA and DNA may lie in the chemical similarities of the single strand status of both RNA and DNA. RNA is typically a single-stranded molecule and DNA is momentarily single stranded during transcription and the replication process in phase S when it becomes highly susceptible to alkylation reactions (Falnes et al. 2002; Sedgwick 2004) and other chemical insults. Accordingly, the substrate ambiguity exhibited by AlkB and its homolog ALKBH3 may be a consequence of a similar substrate scaffold in single nucleic strands. The same phenomena probably explain the putative RNA replicase activity of a Betaflexiviridae virus (UNIPROT-A0A3G3LQ27_9VIRU) which includes a 2OG-FeII_Oxy_2 domain that recognises and binds the viral RNA during replications of these viruses.
The above arguments, combined with enzyme kinetics data of substrate preference in DNA instead RNA (Falnes et al. 2004), support our idea that the reported activity of AlkB (2OG-FeII_Oxy_2 domain) in methylated RNA is the outcome of an ambiguous substrate recognition. Our interpretation falls within the question raised by Sedgwick (2004) on the biological signification of RNA repair and support the possibility that RNA modifications may be related to complex RNA regulatory pathways of RNA molecules like mRNA and tRNA (Zheng et al. 2013; Roundtree et al. 2017; Boccaleto et al. 2022 and Wilkinson et al. 2022).
The evolutionary analyses of the structural superfamily cupin members (Figs. 2 and 3) demonstrates that the 2OGxs are a clearly defined monophyletic group (Fig. 3). The deeper node of these phylogenies suggest that the original cupin fold origin was not an oxidoreductase enzyme, but rather an anaerobic protein with transferase activity or with no enzymatic activity at all (see EC numbers in Fig. 3). A later node that indicates where the recruitment of O2 and Fe2+ may have evolved, represents an explosion of functions and divergence of the cupin fold. In particular, the oxidoreductase activity arises quickly in the neighbourhood of HgmA_N domain, but the diversification of enzymatic activity is not restricted only to oxidoreductases. Groups containing the JmjC domain and all the 2OGxs homologs reflect their late diversification of functions for eukaryotic histone (Klose et al. 2006) and demethylation of DNA.
The trees shown in Figs. 2 and 3 strongly suggests that the cupin fold domain may have emerged well before the free oxygen accumulation in the Earth atmosphere, and that the recruitment of molecular oxygen and iron necessary for an ancestral demethylation took place after the Great Oxidation Event ~ 2.4 Gyr once permanent concentrations of O2 was achieved (Lyons et al. 2014, 2024). In fact, the strict requirement of oxygen by dioxygenases allows insights on their emergence. Experimental data of the AlkB homolog 5 kinetics shows a 75% of reduced activity at 5.0% of atmosphere oxygen and a virtual absent activity at 1.0% of atmosphere oxygen (Thalhammer et al. 2011), so the requirement of free oxygen for its emergence and function may have had to be greater than 1% of atmospheric oxygen. Indeed, Jabłońska and Tawfik 2021 have suggested that enzymes that emerged after GOE they could have operated with in low oxygen levels.
The role performed by enzymes in pathways complexes provides an indirect way of relative dating of their emergence. For example, the role the 2OG-FeII_Oxy_3 and P4Ha_N domains in collagen posttranslational modifications in Metazoa is considered a milestone in the evolution and in fact, our results have shown that 2OG-FeII_Oxy_3 are in a very derivate node. Metazoan multicellularity requires the presence of collagen to maintain and create connective tissue. Collagen appears to be a strict requirement for the development of primordial interactions of cell aggregates in metazoa. Single-cell RNA sequencing has showed the expression of related enzymes of collagen posttranslational modifications in proline residues, like procollagen hydroxylase and collagen itself in larval and adult metacells of Amphimedon queenslandica (sponge), Mnemiopsis leidyi (ctenophore) and Trichoplax adhaerens (placozoa) (Sebé-Pedrós et al. 2018). By the same token, AlkB and its homologues functions should also be recent evolutionary development.
Proposals on the origin and early evolution of Metazoa rest on the idea that unicellular organisms with a complex life cycle evolved into multicellular organisms (Tikhonenkov et al. 2020; Sebé-Pedrós et al. 2018) but, until now, the origin of animals remains an open issue. A widely popular theory is the choanoblastaea theory, which assumes that the first animals were sponge-like entities, and that descended from a colony of choanoflagellate-like organisms, a possibility supported by the collar complex shared by choanoflagellates and sponges (Ruiz-Trillo et al. 2023). An alternative theory is the so-called synzoospore theory where the unicellular ancestor of animals was an organism with a complex life cycle and transitions between different life stages that fit with the current understanding of animal evolution (Ruiz-Trillo et al. 2023). Both of these explanations are based on the hypothesis of a common ancestor shared by choanoflagellates and the metazoa, but neither of them provides a detailed reconstruction of animal’s origins. As noted above, however, the metazoan ancestor was endowed with several key biological components required for multicellularity, including collagen biosynthetic encoding sequences.
As summarised by Sedgwick (2004), both AlkB and its homologue ABH3 can repair not only DNA but also RNA, albeit with a higher preference for the former. As she underlined, the same is true for other DNA repair mechanisms that have also been reported to act on RNA molecules, including O6-methylguanine-DNA-methyl transferase (Karran 1985) and the E. coli DNA-photolyase that repairs uracil cyclobutane dimers in poly(rU) molecules (Kim and Sancar 1991). These mechanisms simply reverse the damaged base and do not depend on the presence of a double-stranded nucleic acid to repair the excision of the modified nucleotide (Sedgwick 2004). Current estimates date the antiquity of LCA between 3.8 and 4.2 Gya ago (cf. Moody et al. 2024). As argued here, the available evidence strongly suggest that the AlkB-based repair mechanism could have not appeared during these early times, but are a much later evolutionary development once significant amount of free oxygen became available in the Precambrian environment. It is most likely that the use of RNA molecules as substrates for AlkB, the O6-methylguanine-DNA-methyl transferase (Karran 1985) and the E. coli DNA-photolyase (Kim and Sacar 1991) are the outcome of the lack of absolute specificity of these enzymatic mechanisms and cannot be used as indicating the presence of an RNA genome in the cenancestor.
References
Aas P, Otterlei M, Falnes, et al (2003) Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421(6925):859–863. https://doi.org/10.1038/nature01363
Agarwal G, Rajavel M, Gopal B et al (2009) Structure-Based phylogeny as a diagnostic for functional characterization of proteins with a cupin fold. PLoS ONE 4(5):e5736. https://doi.org/10.1371/journal.pone.0005736
Aik W, McDonough M, Thalhammer A, Chowdhury R, Schofield C (2012) Role of the jelly-roll fold in substrate binding by 2-oxoglutarate oxygenases. Curr Opin Struct Biol 22(6):691–700. https://doi.org/10.1016/j.sbi.2012.10.001
Alvarez-Carreno C, Becerra A, Lazcano A (2016) Molecular evolution of the oxygen-binding hemerythrin domain. PLoS ONE 11(6):e0157904. https://doi.org/10.1371/journal.pone.0157904
Aravind L, Koonin E (2001) The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate-and iron-dependent dioxygenases. Genome Biol. https://doi.org/10.1186/gb-2001-2-3-research0007
Baek M, DiMaio F, Anishchenko I et al (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373(6557):871–876. https://doi.org/10.1126/science.abj8754
Becerra A, Islas S, Leguina J et al (1997) Polyphyletic gene losses can bias backtrack characterizations of the cenancestor. J Mol Evol 45:115–117. https://doi.org/10.1007/PL00006209
Becerra A, Delaye L, Islas S et al (2007) The very early stages of biological evolution and the nature of the last common ancestor of the three major cell domains. Annu Rev Ecol Evol Syst 38:361–379. https://doi.org/10.1146/annurev.ecolsys.38.091206.095825
Begley T, Samson L (2003) AlkB mystery solved: oxidative demethylation of N1-methyladenine and N3-methylcytosine adducts by a direct reversal mechanism. Trends Biochem Sci 28(1):2–5. https://doi.org/10.1016/S0968-0004(02)00010-5
Boccaletto P, Stefaniak F, Ray A et al (2022) MODOMICS: a database of RNA modification pathways 2021 update. Nucleic Acids Res 50(D1):D231–D235. https://doi.org/10.1093/nar/gkab1083
Brown J, DoolittleW. (1995) Root of the universal tree of life based on ancient aminoacyl-tRNA synthetase gene duplications. Proc Natl Acad Sci 92(7):2441–2445. https://doi.org/10.1073/pnas.92.7.2441
Dalkiran A, Rifaioglu A, Martin M et al (2018) ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature. BMC Bioinform 19:1–13. https://doi.org/10.1186/s12859-018-2368-y
Dayhoff MO (1972) A model of evolutionary change in proteins. Atlas of Protein Seq Struct 5:89–99
Delaye L, Becerra A, Lazcano A (2005) The last common ancestor: what’s in a name? Orig Life Evol Biosph 35:537–554. https://doi.org/10.1007/s11084-005-5760-3
Delaye L, Becerra A, Orgel L et al (2007) Molecular evolution of peptide methionine sulfoxide reductases (MsrA and MsrB): on the early development of a mechanism that protects against oxidative damage. J Mol Evol 64:15–32. https://doi.org/10.1007/s00239-005-0281-2
Dong R, Pan S, Peng Z et al (2018) mTM-align: a server for fast protein structure database search and multiple protein structure alignment. Nucleic Acids Res 46(W1):W380–W386. https://doi.org/10.1093/nar/gky430
Dunwell J, Culham A, Carter C et al (2001) Evolution of functional diversity in the cupin superfamily. Trends Biochem Sci 26(12):740–746. https://doi.org/10.1016/S0968-0004(01)01981-8
Dunwell J, Purvis A, Khuri S (2004) Cupins: the most functionally diverse protein superfamily? Phytochemistry 65(1):7. https://doi.org/10.1016/j.phytochem.2003.08.016
Estrada A, Suárez-Díaz E, Becerra A (2022) Reconstructing the last common ancestor: epistemological and empirical challenges. Acta Biotheor 70(2):15. https://doi.org/10.1007/s10441-022-09439-1
Falnes P, Johansen R, Seeberg E (2002) AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 419(6903):178–182. https://doi.org/10.1038/nature01048
Falnes P, Bjørås M, Aas P et al (2004) Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res 32(11):3456–3461. https://doi.org/10.1093/nar/gkh655
Felsenstein J. (2005) PHYLIP (phylogeny inference package) version 3.6. distributed by author. Department of Genome Sciences, University of Washington, Seattle. http://evolution.genetics.washington.edu/phylip.html
Fitch W, Upper K (1987) The phylogeny of tRNA sequences provides evidence of ambiguity reduction in the origin of the genetic code. Cold Spring Harbor Symp Quant Biol 52:759–767. https://doi.org/10.1101/SQB.1987.052.01.085
Gogarten J (2019) The progenote, LUCA, and the root of the cellular tree of life. In: Kolb VM (ed) Handbook of Astrobiology. CRC Press, Boca Raton
Gogarten J, Kibak H, Dittrich P et al (1989) Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes. Proc Natl Acad Sci 86(17):6661–6665. https://doi.org/10.1073/pnas.86.17.6661
Gribaldo S, Cammarano P (1998) The root of the universal tree of life inferred from anciently duplicated genes encoding components of the protein-targeting machinery. J Mol Evol 47:508–516. https://doi.org/10.1007/PL00006407
Harris J, Kelley S, Spiegelman G et al (2003) The genetic core of the universal ancestor. Genome Res 13(3):407–412. https://doi.org/10.1101/gr.652803
Hausinger R (2015) Biochemical diversity of 2-oxoglutarate-dependent oxygenases. In: Hausinger RP, Schofield CJ (eds) 2-Oxoglutarate-Dependent Oxygenases. The Royal Society of Chemistry, London
Hernández-Morales R, Becerra A, Lazcano A (2019) Alarmones as vestiges of a bygone RNA world. J Mol Evol 15(87):37–51. https://doi.org/10.1007/s00239-018-9883-3
Hernández-Morales R, Becerra A, Campillo-Balderas J et al (2022) Structural biology of the SARS-CoV-2 replisome: evolutionary and therapeutic implications. Biomedical Innovations to Combat COVID-19. Academic Press, Cambridge
Islam M, Leissing T, Chowdhury R, Hopkinson R, Schofield C (2018) 2-Oxoglutarate-dependent oxygenases. Annu Rev Biochem 87(1):585–620. https://doi.org/10.1146/annurev-biochem-061516-044724
Iwabe N, Kuma K, Hasegawa M et al (1989) Evolutionary relationship of archaebacteria, eubacteria, and eukaryotes inferred from phylogenetic trees of duplicated genes. Proc Natl Acad Sci 86(23):9355–9359. https://doi.org/10.1073/pnas.86.23.9355
Jabłońska J, Tawfik D (2021) The evolution of oxygen-utilizing enzymes suggests early biosphere oxygenation. Nat Ecol Evol 5(4):442–448. https://doi.org/10.1038/s41559-020-01386-9
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–589. https://doi.org/10.1038/s41586-021-03819-2
Kandler O (1994) The early diversification of life. Early Life on Earth Nobel Symp 84:152–160
Karran P (1985) Possible depletion of a DNA repair enzyme in human lymphoma cells by subversive repair. Proc Natl Acad Sci 82(16):5285–5289. https://doi.org/10.1073/pnas.82.16.5285
Kataoka H, Yamamoto Y, Sekiguchi M (1983) A new gene (alkB) of Escherichia coli that controls sensitivity to methyl methane sulfonate. J Bacteriol 153(3):1301–1307. https://doi.org/10.1128/jb.153.3.1301-1307.1983
Khademian M, Imlay J (2021) How microbes evolved to tolerate oxygen. Trends Microbiol 29(5):428–440. https://doi.org/10.1016/j.tim.2020.10.001
Khersonsky O, Tawfik D (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu Rev Biochem 79:471–505. https://doi.org/10.1146/annurev-biochem-030409-143718
Khuri S, Bakker F, Dunwell J (2001) Phylogeny, function, and evolution of the cupins, a structurally conserved, functionally diverse superfamily of proteins. Mol Biol Evol 18(4):593–605. https://doi.org/10.1093/oxfordjournals.molbev.a003840
Kim S, Sancar A (1991) Effect of base, pentose, and phosphodiester backbone structures on binding and repair of pyrimidine dimers by Escherichia coli DNA photolyase. Biochemistry 30(35):8623–8630. https://doi.org/10.1021/bi00099a019
Klose R, Kallin E, Zhang Y (2006) JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 7(9):715–727. https://doi.org/10.1038/nrg1945
Koonin E (2003) Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol 1(2):127–136. https://doi.org/10.1038/nrmicro751
Koonin E, Martin W (2005) On the origin of genomes and cells within inorganic compartments. Trends Genet 21(12):647–654. https://doi.org/10.1016/j.tig.2005.09.006
Lazcano A (1995) Cellular evolution during the early Archean: what happened between the progenote and the cenancestor? Microbiología SEM 11:185–198
Lazcano A (2012) The origin and early evolution of life: where when and how? Evolution 5:334–336
Lazcano A, Miller S (1996) The origin and early evolution of life: prebiotic chemistry the pre-RNA world and time. Cell 85(6):793–798. https://doi.org/10.1016/S0092-8674(00)81263-5
Lazcano A, Fox G, Oró J (1992) Life before DNA, the origin and evolution of early Archaean cells. In: Mortlock RP (ed) The Evolution of Metabolic Function. CRC Press, Boca Raton, pp 237–295
Lee D, Jin S, Cai S, Chen Y, Pfeifer G, O’Connor T (2005) Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem 280(47):39448–39459. https://doi.org/10.1074/jbc.M509881200
Leipe D, Aravind L, Koonin E (1999) Did DNA replication evolve twice independently? Nucleic Acids Res 27(17):3389–3401. https://doi.org/10.1093/nar/27.17.3389
Letunic I, Bork P (2024) Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. https://doi.org/10.1093/nar/gkae268
Lyons T, Reinhard C, Planavsky N (2014) The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506(7488):307–315. https://doi.org/10.1038/nature13068
Lyons T, Tino C, Fournier G et al (2024) Co-evolution of early earth environments and microbial life. Nat Rev Microbiol. https://doi.org/10.1038/s41579-024-01044-y
Martínez-Castilla L, Rodríguez-Sotres R (2010) A score of the ability of a three-dimensional protein model to retrieve its own sequence as a quantitative measure of its quality and appropriateness. PLoS ONE 5(9):e12483. https://doi.org/10.1371/journal.pone.0012483
Mayr E (1998) Two empires or three? Proc Natl Acad Sci 95(17):9720–9723. https://doi.org/10.1073/pnas.95.17.9720
Mielecki D, Zugaj DŁ, Muszewska A (2012) Novel AlkB dioxygenases: alternative models for in silico and in vivo studies. PLoS ONE 7(1):e30588. https://doi.org/10.1371/journal.pone.0030588
Moffett J (2021) Iron (II) in the world’s oxygen deficient zones. Chem Geol 580:120314. https://doi.org/10.1016/j.chemgeo.2021.120314
Moody E, Álvarez-Carretero S, Mahendrarajah T, Clark J, Betts H, Dombrowski N, Donoghue P (2024) The nature of the last universal common ancestor and its impact on the early earth system. Nat Ecol Evol. https://doi.org/10.1038/s41559-024-02461-1
Mushegian A, Koonin E (1996) A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc Natl Acad Sci 93(19):10268–10273. https://doi.org/10.1073/pnas.93.19.10268
Oparin AI (1938) The origin of life. MacMillan, New York
Ougland R, Zhang C, Liiv A et al (2004) AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell 16(1):107–116. https://doi.org/10.1016/j.molcel.2004.09.002
Ougland R, Rognes T, Klungland A et al (2015) Non-homologous functions of the AlkB homologs. J Mol Cell Biol 7(6):494–504. https://doi.org/10.1093/jmcb/mjv029
Paysan-Lafosse T, Blum M, Chuguransky S et al (2023) InterPro in 2022. Nucleic Acids Res 51(D1):D418–D427. https://doi.org/10.1093/nar/gkac993
Penny D, Poole A (1999) The nature of the last universal common ancestor. Curr Opin Genet Dev 9(6):672–677. https://doi.org/10.1016/S0959-437X(99)00020-9
Poole A, Logan D (2005) Modern mRNA proofreading and repair: clues that the last universal common ancestor possessed an RNA genome? Mol Biol Evol 22(6):1444–1455. https://doi.org/10.1093/molbev/msi132
Raymond J, Segrè D (2006) The effect of oxygen on biochemical networks and the evolution of complex life. Science 311(5768):1764–1767. https://doi.org/10.1126/science.1118439
Reanney D (1982) The evolution of RNA viruses. Ann Rev Microbiol 36(1):47–73. https://doi.org/10.1146/annurev.mi.36.100182.000403
Rossmann M, Argos P (1978) The taxonomy of binding sites in proteins. Mol Cell Biochem 21:161–182. https://doi.org/10.1007/BF00240135
Rossmann M, Moras D, Olsen K (1974) Chemical and biological evolution of a nucleotide-binding protein. Nature 250(5463):194–199. https://doi.org/10.1038/250194a0
Roundtree I, Evans M, Pan T et al (2017) Dynamic RNA modifications in gene expression regulation. Cell 169(7):1187–1200. https://doi.org/10.1016/j.cell.2017.05.045
Ruiz-Trillo I, Kin K, Casacuberta E (2023) The origin of metazoan multicellularity: a potential microbial black swan event. Annu Rev Microbiol 77(1):499–516. https://doi.org/10.1146/annurev-micro-032421-120023
Sagan L (1967) On the origin of mitosing cells. J Theor Biol. https://doi.org/10.1016/0022-5193(67)90079-3
Sebé-Pedrós A, Chomsky E, Pang K et al (2018) Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat Ecol Evol 2(7):1176–1188. https://doi.org/10.1038/s41559-018-0575-6
Sedgwick B (2004) Repairing DNA-methylation damage. Nat Rev Mol Cell Biol 5(2):148–157. https://doi.org/10.1038/nrm1312
Sedgwick B, Bates P, Paik J, Jacobs S, Lindahl T (2007) Repair of alkylated DNA: recent advances. DNA Repair 6(4):429–442. https://doi.org/10.1016/j.dnarep.2006.10.005
Semsey S, Andersson A, Krishna S (2006) Genetic regulation of fluxes: iron homeostasis of Escherichia coli. Nucleic Acids Res 34(17):4960–4967. https://doi.org/10.1093/nar/gkl627
Thalhammer A, Bencokova Z, Poole R (2011) Human AlkB homologue 5 is a nuclear 2-oxoglutarate dependent oxygenase and a direct target of hypoxia-inducible factor 1α (HIF-1α). PLoS ONE 6(1):e16210. https://doi.org/10.1371/journal.pone.0016210
Tikhonenkov D, Hehenberger E, Esaulov A et al (2020) Insights into the origin of metazoan multicellularity from predatory unicellular relatives of animals. BMC Biol 18:1–24. https://doi.org/10.1186/s12915-020-0762-1
Ueno Y, Yamada K, Yoshida N et al (2006) Evidence from fluid inclusions for microbial methanogenesis in the early Archaean era. Nature 440(7083):516–519. https://doi.org/10.1038/nature04584
van den Born E, Omelchenko M, Bekkelund A et al (2008) Viral AlkB proteins repair RNA damage by oxidative demethylation. Nucleic Acids Res 36(17):5451–5461. https://doi.org/10.1093/nar/gkn519
Weiss M, Sousa F, Mrnjavac N et al (2016) The physiology and habitat of the last universal common ancestor. Nat Microbiol 1(9):1–8. https://doi.org/10.1038/nmicrobiol.2016.116
Weiss M, Preiner M, Xavier J et al (2018) The last universal common ancestor between ancient earth chemistry and the onset of genetics. PLoS Genet 14(8):e1007518. https://doi.org/10.1371/journal.pgen.1007518
Wilkinson E, Cui Y, He Y (2022) Roles of RNA modifications in diverse cellular functions. Front Cell Dev Biol 10:828683. https://doi.org/10.3389/fcell.2022.828683
Woese CR (1987) Bacterial evolution. Microbiol Rev 51(2):221–271. https://doi.org/10.1128/mr.51.2.221-271.1987
Woese CR (1998) Default taxonomy: Ernst Mayr’s view of the microbial world. Proc Natl Acad Sci 95(19):11043–11046. https://doi.org/10.1073/pnas.95.19.11043
Woese C, Fox G (1977) Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc Natl Acad Sci 74(11):5088–5090. https://doi.org/10.1073/pnas.74.11.5088
Woese C, Kandler O, Wheelis M (1990) Towards a natural system of organisms: proposal for the domains archaea, bacteria, and eucarya. Proc Natl Acad Sci 87(12):4576–4579. https://doi.org/10.1073/pnas.87.12.4576
Woose CR (1967) The genetic code, the molecular basis for gene expression. Modern Perspectives in Biology, New York, Harper and Row
Zheng G, Dahl J, Niu Y et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49(1):18–29. https://doi.org/10.1016/j.molcel.2012.10.015
Acknowledgements
This paper is part of the requirements for obtaining a Ph.D. degree from Posgrado en Ciencias Biológicas UNAM of WCS. This work was supported during its early stages by a fellowship from Consejo Nacional de Ciencia y Tecnología (CONACYT) México (2020-2021; CVU 815057). We are indebted to Ricardo Hernández-Morales and José Alberto Campillo-Balderas for discussion enrichment and technical assistance in the preparation of the manuscript. Support from INFOCAB-DGAPA PB202223 and INFOCAB-DGAPA PB201624 is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Aaron Goldman.
Supplementary Information
Below is the link to the electronic supplementary material.
239_2024_10194_MOESM1_ESM.png
Supplementary file1 (PNG 2481 KB) Fig. S1. Tertiary-based rooted phylogeny of all 72 domains/families endowed the cupin fold showing their biological distribution among the three cellular domains of life and some viruses. Tips marked with “E” indicates that this domain is present in the Eukarya, “B” in Bacteria, “A” in Archaea, and finally “V” indicates that the domain is present in some viruses
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Cottom-Salas, W., Becerra, A. & Lazcano, A. RNA or DNA? Revisiting the Chemical Nature of the Cenancestral Genome. J Mol Evol (2024). https://doi.org/10.1007/s00239-024-10194-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00239-024-10194-9