
Abstract
DNA repair refers to a collection of processes by which a cell identifies and corrects damage to genomic DNA molecules. DNA repair processes significantly overcome DNA damage and restore the normal nucleotide sequence and DNA structure. This study focuses on the evolution of the endonuclease III gene/protein family, which plays a key role in the base excision repair pathway. We analyzed 463 homologs of the endonuclease III protein and compared them with the corresponding gene and 16S/18S rRNA sequences to understand the evolutionary processes of this protein family. The sequence analysis and comparison reveal consensus sequence motifs within the ENDO3c and iron–sulfur cluster loop domains that are functionally and structurally important. On the basis of phylogenetic analysis, we propose an evolutionary model of the endonuclease III protein family. Horizontal gene transfer was identified as the key event among bacteria, archaea, and eukaryotic organisms that occurred during the evolution of the endonuclease III gene family among bacteria, archaea, and eukaryotic organisms. This analysis may be exploited to achieve a better prediction of the endonuclease III family gene/protein in unannotated organisms or families of organisms that are completely sequenced as well as in those for which sequencing is ongoing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA repair is a vital event in a cell that interplays between the integrity of the genetic information of an organism and the evolution of organisms. The molecular mechanisms of DNA repair can be classified into four main categories direct repair: (DR), nucleotide excision repair (NER), mismatch repair (MMR), and base excision repair (BER) (Dmitry and Kosuke 1997). In the DR mechanism, the unusual chemical bonds of bases, nucleotides, or substituents are directly modified with the help of repair enzymes. Ultraviolet (UV)-induced DNA damage is repaired by this mechanism. The DNA photolyase, photoproduct photolyase, spore photoproduct lyase, and the O6-methylguanine DNA methyl transferase families are the major enzymes that catalyze direct DNA repair (Aziz 1995; Lindahl 1982; Richard 2001). In the case of NER, the phosphodiester bonds of both sides of the damaged base are cleaved, and the damaged base containing the nucleotide is removed. Exonucleases and excision nucleases are two major groups of enzymes that perform NER (Aziz 1996). In the MMR mechanism, the repair of occasional mismatches during DNA replication and recombination is initiated with the help of the MutS, MutH, and MutL protein families (Thomas and Dorothy 2005). Among the different repair mechanisms, BER is the prominent pathway for the repair of small DNA lesions resulting from exposure to either environmental agents or cellular metabolic processes that produce alkylating agents, reactive oxygen species, or reactive metabolites. In the first step of the BER mechanism, relatively small, monomeric, and mono-functional glycosylases recognize the damaged base with high specificity and catalyze the breakage of the glycosyl bond between the damaged base and the DNA sugar-phosphate backbone. At the end of this action, a spontaneous apurinic/apyrimidinic (AP) site is generated in place of the damaged base (Hegde et al. 2008). This AP site is subsequently hydrolyzed by AP endonuclease, which catalyzes the incision of the damaged strand, leaving a 3′hydroxyl (OH) and a 5′deoxyribose-phosphate moiety (5′dRP) at the margins. However, in the case of oxidized base damage, bi-functional glycosylases cleave the damaged base from the sugar moiety and process the AP site via a β elimination reaction. Finally, in most cases, DNA polymerase ß (pol ß) hydrolyzes the 5′dRP moiety and fills the single nucleotide gap. The repaired strand is then ligated either by a DNA Ligase I or by a DNA Ligase complex (Hegde et al. 2008).
AP endonucleases are a group of enzymes that act on abasic site and break the phosphodiester bond. Based on the position of the phosphodiester cleavages, all AP endonucleases are classified into two major categories. Class I AP endonucleases cleave DNA at the 3′ side of the AP site, whereas class II AP endonuclease enzymes cleave at the 5′ side. In both cases, a 5′phosphate and a 3′OH group are generated at the cleavage site (Shida et al. 1999). Escherichia coli endonuclease III as well as E. coli exonuclease III and endonuclease IV are the most well-studied enzymes of classes I and II, respectively.
Endonuclease III or nth family proteins belongs to the helix-hairpin-helix (HhH) superfamily along with the OggI, MutY/Mig, AlkA, MpgII, and OggII gene families. These specifically recognize and excise varying spectra of the damaged bases and base pairing mismatches (Yang et al. 2000; Denver et al. 2003). The proteins from this superfamily have an HhH (Thayer et al. 1995) structural element followed by a Gly/Pro-rich loop and a conserved aspartic acid residue (Nash et al. 1996; Labahn et al. 1996). Among these gene families, the endonuclease III protein is a bi-functional enzyme with AP endonuclease as well as glycosylase activity. Endonuclease III has a broad specificity for DNA BER, removing numerous forms of modified thymine and cytosine bases from DNA (Thayer et al. 1995). The endonuclease III family of proteins repair damaged bases and mismatched bases, whereas DNA glycosylase hydrolyses the N-glycosylic bond between the target base and the sugar moiety, thus releasing the free damaged base. This process produces an AP site. These enzymes are also classified as mono-functional and bi-functional DNA glycosylases. Mono-functional glycosylases possess only glycosylase activity, whereas bi-functional DNA glycosylases exhibit both AP lyase and glycosylase activity. The enzymatic mechanism of endonuclease III is reviewed in detail by Dodson et al. (Dobson et al. 1994).
This study aims to provide an insight into the evolution of the endonuclease III gene/protein family among all lineages of life. In this study, we focus on the insertion/deletion of domains/motifs and the conservation of important amino acids during the course of evolution. Furthermore, our phylogenetic analyses based on gene and protein sequences of endonuclease III examine the evolution of the endonuclease III genes/proteins homologs in all five kingdoms of life, and these data are then compared with 16S/18S rRNA sequence-based species evolution. Evolutionary studies have identified a few horizontal gene transfer events. Based on these events, we propose a model of the evolutionary history of the entire endonuclease III protein family.
Materials and Methods
Retrieval of Endonuclease III Protein Homologs
The endonuclease III protein sequence (NCBI Acc. No. NP_416150.1) of E. coli was used as a query to retrieve homologs from the National Center for Biotechnology Information (NCBI) protein sequence database. BLASTP (Altschul et al. 1990) with default parameters was used to retrieve the homologous sequences of this protein from the NCBI non-redundant (NR) protein database (with an E value cut off of ≤1 × 10−5). In addition to BLASTP, two rounds of PSI-BLAST search with a default parameter (Altschul et al. 1997) were also conducted using the same query sequence (E. coli endonuclease III protein sequence, Acc No. NP_416150.1) to identify additional homologs.
cDNA sequences of endonuclease III protein homologs were retrieved from the NCBI sequence database and were used to construct a gene-based phylogenetic tree. The mean G+C content and the G+C content at the 3rd codon position of the cDNA sequences of endonuclease III were calculated by CAIcal server (Puigbo et al. 2008). The G+C content of the genomes of the species was retrieved from the NCBI genome database. We retrieved 16S ribosomal RNA gene sequences of bacterial and archaeal species from the ribosomal sequence database (Cole et al. 2009), and 18S ribosomal RNA gene sequences of eukaryotic species were obtained from the NCBI nucleotide database.
Sequence Alignment and Domain Search
Pairwise sequence alignments between E. coli endonuclease III and the other selected endonuclease III homologs were performed using the EMBOSS Needle program (Rice et al. 2000). Multiple sequence alignments of endonuclease III protein homologs were conducted using the M-Coffee (Moretti et al. 2007) and ClustalW servers (Thompson et al. 1994) with default parameter/s. The domains and motif of the endonuclease III homologs were analyzed using the conserved domain (CD) search tools available at the NCBI (Marchler and Bryant 2004).
The conservation patterns of the amino acid positions, which were based on the protein sequence and structure in the endonuclease III protein family, were analyzed by sequence logos. The sequence logos of multiple sequence alignments of the endonuclease III protein family were generated by Weblogos 3.2 (Crooks et al. 2004) without any compositional adjustment. Sequence logos are a graphical representation of the multiple sequence alignment of a protein and/or DNA sequences. Each logo consists of stacks of symbols, one stack for each position in the sequence. The overall height of the stack indicates the sequence conservation at that position, whereas the height of the symbols within the stack indicates the relative frequency of each amino or nucleic acid at that position (Crooks et al. 2004).
Phylogenetic Analysis
All the maximum likelihood (ML) and neighbor-joining (NJ) phylogenetic trees were generated by Mega 5.1 (Tamura et al. 2011). The evolutionary distances for the gene-based trees were constructed using the maximum composite likelihood method (Tamura et al. 2004), whereas the protein-based tree was constructed using the Jones Taylor Thornton method (Jones et al. 1992). The reliability of the interior branches was assessed with 1000 bootstrap re-samplings of the amino acid/cDNA/16S–18S sequences in the phylogenetic trees (Felsenstein 1985).
Results and Discussion
Data Curation
After removing the redundant hits, global pairwise sequence alignment was performed with the remaining hits from the BLAST and PSI-BLAST searches (Rice et al. 2000). The protein sequences with less than 15 % identity against the corresponding E. coli endonuclease III protein were removed. Finally, hits with at least one common domain of the endonuclease III protein family were selected. A set of 463 homologs of the endonuclease III family proteins was considered for evolutionary study (Tables 1, 2). Of the 463 endonuclease III homologs, 44 belonged to eukaryotes, 42 to archaea, and 377 to bacteria.
All homologs of endonuclease III were divided into five kingdoms, namely, monera (comprising both bacteria and archaea), protista, fungi, plantae, and animalia (Whittaker 1969). Due to a large number of bacterial homologs, sequences from bacterial species were divided into various divisions (Garrity and Holt 2001). Phylogenetic trees were generated for endonuclease III homologs of various bacterial divisions and four additional kingdoms using the NJ algorithm. The phylogenetic tree of each group was inspected visually, and representative homologs from different clades of each tree were chosen. The phylogenetic trees of all bacterial divisions and other phyla are available in Supplementary Fig. S1a–n. Finally, a set of 54 endonuclease III homologs was selected to study the evolution of the endonuclease III protein family. This set of protein homologs belongs to 22 bacterial, two archeal, and four eukaryotic divisions. Supplementary Table S1 provides the NCBI accession numbers of the 54 endonuclease III protein homologs and the length of the protein sequences.
Domains and Motifs of Endonuclease III Family
Domains and motifs are the two most important entities that are conserved during evolution, and these factors are important parameters for gaging the evolution process (Kanchan et al. 2014). By applying a CD search, we identified ENDO3c (smart00478) and Fe-S (iron–sulfur domain; smart00525) as two major domains. ENDO3c is the main domain that spans residues 30 to 183 of the E. coli endonuclease III protein. The ENDO3c domain spans almost the entire protein in all endonuclease III homologs. A 19-residue HhH segment within the ENDO3c domain is responsible for non-specific DNA binding through hydrogen bonds between the N-atoms of the protein backbone and the phosphate groups of DNA. Multiple sequence analysis (MSA) of the 54 homologs suggests the presence of a consensus L111X2LP115GVG118XK120TA122 sequence (Fig. 1) within the HhH motif. Among these highly conserved residues, K120 is the main catalytic residue that is responsible for the AP lyase activity (Thayer et al. 1995). The mutation of K120 in seven AP endonuclease III homologs (AP endonucleases of Gemmatimonas aurantiaca, Methylacidiphilum infernorum, Oceanobacillus iheyensis, and Caldivirga maquilingensis; MutY of E. coli, Homo sapiens and Thermomicrobium roseum) suggests a loss of AP lyase activity. All these homologs are within the mono-functional AP endonuclease protein family that exclusively exhibits glycosylase activity. Conserved L111, L114, and A122 residues are a part of the hydrophobic core structure, which stabilizes the fold of the HhH motif. The beta turn is formed by residues from P115 to G118 and is a key structure in the HhH motif. The V117 side chain within the beta turn region is oriented toward the hydrophobic core. The interaction among the hydrophobic residues is critical for the overall architecture of the HhH motif, and subsequently aids in positioning the catalytically important K120 residue toward the active site cavity (Fig. 2). The D138 residue within the ENDO3c domain is also conserved among the 53 homologs. This residue initiates nucleophilic attack during the glycosylase activity (Manuel et al. 2004). The absence of both lysine and aspartic acid residues at positions 120 and 138 in Caldivirga indicates that this protein may not have glycosylase or lyase activity; however, it contains the ENDO3c domain. Approximately 100 amino acid N-terminus insertions are observed in the endonuclease III protein of plant species. The detailed sequence comparison predicts (Emanuelsson et al. 2007) the existence of a possible signal peptide within the inserted region, which is targeted to the chloroplast.

Sequence conservation within the HhH motif from residues 111–122 is shown by the sequence logo. A bit score of 3.2 and above corresponds to more than 80 % sequence conservation

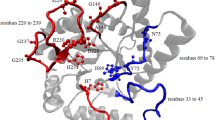
The HhH motif from E. coli AP endonuclease III crystal structure (PDB ID: 2ABK) is presented by the ribbon diagram. Hydrophobic core-forming residues are shown by spacefill representation, whereas the orientation of the catalytic L120 side chain is demonstrated by the stick representation
The Fe-S domain (smart00525) at the C-terminal end of the AP endonuclease III protein is present in most of the homologs that contain a 21 residues iron–sulfur cluster loop (FCL) motif. The iron–sulfur cluster plays an important role in orienting the positively charged residues of the FCL motif for DNA binding (Lukianova and David 2005). The MSA of 54 homologs suggests the presence of a consensus G183X3C187X6C194X2C197X5C203 sequence in the Fe-S domain. In the case of E. coli MutY, the mutation of highly conserved cysteine residues reduces the stability of the iron–sulfur cluster and the extent of destabilization is position dependent (Golinelli et al. 1999). Out of the total 54 selected homologs, all four cysteine residues are conserved in the 47 homologs. These four cysteine residues are absent in Gemella haemolysans and Gramella forsetii, whereas the other five homologs contain at least two conserved cysteine residues. These data suggest that the FCL motif may be absent in G. haemolysans and G. forsetii. In addition, two cysteine residues are sufficient for the formation as well as the stabilization of the Fe-S cluster as indicated in previous mutational studies (Golinelli et al. 1999).
The structure of the E. coli endonuclease III protein (Thayer et al. 1995) contains six helix barrel and FCL cluster domains. The crystal structure (PDB ID: 2ABK) also reveals that helices 2–7 are located within the helix barrel domain, whereas helix 1 and helices 8–10 belong to the FCL cluster. MSA reveals that substantial sequence conservation exists within helix 2 and 3, which contains the helix-turn-helix region (shown in Fig. 3a). Among the conserved residues, Q41 plays a key role in substrate recognition as it penetrates through the major groove of DNA and interacts with the AP site of DNA (Fromme and Verdine 2003), whereas S39 and D44 form polar interactions with other segments of the protein, thus providing segmental flexibility. L33 and L38 are part of the hydrophobic core that stabilizes this particular fold. In the case of the Fe-S domain, most of the residues of helix 8 (Fig. 3b), four cysteine residues (C187, C194, C197, and C203), L33, and L38 are highly conserved. The conserved T139, H140, R143, and R147 residues form a hydrophilic/charge surface within helix 8 that interacts with the DNA backbone and provides stability during DNA binding.

a The consensus sequence between residues 33 and 44 is shown by sequence logo representation. L33, L38, S39, Q41, and D44 are conserved for greater than 80 % of sequences. b The conservation of residues from 137 to 147 is shown by the sequence logo. V137, D138, T139, H140, R143, and R147 are conserved in greater than 80 % of sequences
In the endonuclease III protein family, the efficiency and specificity of the enzymes are highly dependent on the architecture of the DNA binding the HhH and FCL motifs. These motifs bind to the DNA substrate with the help of positively charged amino acid residues. Sequence analysis of all 54 homologs demonstrates that the important basic residues are conserved throughout the evolution. Among all the homologs, 3.74 positively charged residues are present on average within the FCL motif and are strategically positioned to interact with the negatively charged DNA backbone. In the case of G. haemolysans and G. forsetii, the DNA binding activity of the enzyme is entirely dependent on the HhH motif because this protein does not contain the FCL motif. Hence, we hypothesize that enzyme activity and specificity is reduced in these species.
Endonuclease III Gene-Based Phylogenetic Tree
The phylogenetic tree of the gene sequences of endonuclease III family was constructed from alignment using both the ML and NJ methods. The topologies of both trees generated using the ML and NJ methods are very similar. Bootstrap analysis reveals that most of the clades in both trees are robust, and the majority of clades are supported by ≥50 % bootstrap value. Therefore, only one tree (ML tree) is discussed. The endonuclease III gene tree was compared with the 16S/18S rRNA gene sequence-based species tree. A total of 14 distinct clades were formed by the 16S/18S rRNA gene sequences of these species (Fig. 4). Out of these 14 clades, eight clades contain species from the same phylum/division. Given that only a few bacterial divisions contain single species, the remainder of the phylogenetic clades contains species from different bacterial divisions. Details of the 16S/18S rRNA sequence-based species tree reveal that almost all species in the different bacterial divisions as well as species of archaea and eukaryotes form a distinct clade, with the sole exception of Thermosinus carboxydivorans. As a Firmicutes bacterium, T. carboxydivorans shares a clade with the cyanobacterial species.

16S/18S rRNA-based maximum likelihood tree. Bootstrap support values are presented next to the tree branches for each clade with ≥50. The tree is generated from a clustalw-based multiple sequence alignment. Organisms from the same division are represented with the same symbol and color code. Details are provided in Supplementary Table S2
A total of 11 distinct clades are formed by the homologs of the endonuclease III genes (Fig. 5). All eukaryote homologs (clade 8) and the homologs of the three bacterial divisions (Cyanobacteria clade 4, Thermotogae clade 11 and Chlorobi clade 1) are close to each other within the phylogenetic clade. Interestingly, four out of five mono-functional endonuclease III genes of different species are clustered together (clade 9), indicating a co-evolution of mono-functional endonuclease III genes. Each of the remaining seven distinct clades contains homologs from different bacterial divisions. The endonuclease III gene-based phylogeny tree suggests that the gene and species evolution of Cyanobacterial, Thermotogal, Chlorobial, and eukaryotic organisms may exhibit a similar pattern, as these classes of organisms remain within a distinct clade in both the species and gene-based phylogenetic trees. Interestingly, the endonuclease III genes of the four archaeal species are located within different lineages (shown as a red branch in Fig. 5), although all archaeal species are within the same clade in the species tree (Fig. 4). This observation indicates that the endonuclease III gene of these archaeal species evolved differently, suggesting that environmental pressure might have played an important role in shaping the endonuclease III gene. For example, the endonuclease III gene of Methanobrevibacter smithii (archaea) shares the same clade (clade 11) with two bacterial species, Petrotoga mobilis and Thermotoga lettingae, which are anaerobic and associated with fermentation.

AP endonuclease III gene-based maximum likelihood tree. Bootstrap support values are presented next to the tree branches for each clade with ≥50. The tree is generated from a clustalw-based multiple sequence alignment
The endonuclease III gene-based phylogeny analysis indicates that a large number of species share a clade with species from different divisions. To explain this anomaly of sharing different clades by the species belonging to the same division, we calculated the mean G+C content of the gene, the G+C content at the 3rd codon position, and the G+C content of the species, which could be an appropriate tool to explain the clustering pattern in the phylogenetic tree (Brochier et al. 2000). Table 3 presents the G+C content of the endonuclease III gene, the 3rd codon position of the endonuclease III gene, and the genome of the corresponding organism. With the exception of H. sapiens, the G+C content of endonuclease III gene and the G+C content of all other organisms are similar. In H. sapiens, the G+C content of the species is approximately 24 % less than the G+C content of the endonuclease III gene. The average G+C content and the G+C content at the 3rd codon position of Endonuclease III gene of Dictyoglomus thermophilum, Acholeplasma laidlawii, G. haemolysans, Borrelia recurrentis, and Fusobacterium nucleatum (clade 7) are 30.8 and 18.1 %, respectively (standard deviation of 3.3 and 6.3), which could possibly explain why these species share the same clade. Similarly, the archaeal species M. smithii, two species from the Thermotogae division, and one species from each of the Aquificae, Firmicutes, Chlamydiae, and mono-functional Verrucomicrobia phyla share the same clade (clade 11) in the tree due to the similar G+C content at the gene level and the G+C content at the 3rd codon position (36.7 and 45.1 %, respectively, with a standard deviation of 4.8 and 5.1, respectively). This observation also suggests that the mono-functional endonuclease III gene from a species of the Verrucomicrobia division shares different clades unlike other mono-functional genes (Clade 9) in which the G+C content and the G+C content at the 3rd base of endonuclease III gene are 62.4 and 66.8 %, respectively. G. forsetii, Parabacteroides distasonis, and Salinibacter ruber from the Bacteroidetes division share three different clades as their G+C contents and G+C content at the 3rd codon position of endonuclease III gene differ significantly (36.5 and 30.6 %; 46.1 and 50.5 %; 65.6 and 68.3 %, respectively). These species reside within the clade that contains species with similar G+C contents. A similar trend is observed in the case of species within the Spirochaetes and Chloroflexi divisions. As a member of the bacterial division, Roseiflexus castenholzii shares a clade with the human AP endonuclease III gene because of its G+C content (62 %), which is very similar to that of humans (64 %). The endonuclease III gene-based phylogeny tree of 54 taxa suggests that the G+C content of the gene contributes significantly to the position of the taxa within the tree and to species evolution. In addition, endonuclease III gene evolution shapes up differently in most of the cases. Given that the AP endonuclease III gene in all living organisms diverges over a long evolutionary period, synonymous nucleotide substitution of the G+C content at the 3rd codon position as well as the overall G+C content of the gene makes gene sequence-based phylogenetic tree construction noisy.
Protein-Based Phylogenetic Tree
Overall, seven distinct branches were identified from the AP endonuclease III protein-based phylogenetic tree. Among them, three branches are divided further to form ten clades (Fig. 6). The bi-functional AP endonuclease III homologs of all the eukaryotic organisms (except Penicillum marneffei), its mono-functional homologs, and the homologs from the Thermotogal and Actinobacterial divisions form four homogeneous distinct clades. We observe that organisms from the Cyanobacterial, Chlorobial, and Proteobacterial (except Saccharophagus degradans) divisions also cluster together in the phylogenetic tree. Other clades contain proteins from species belonging to different divisions. The proteins from Desulfurococcus kamchatkensis and P. marneffei as well as O. iheyensis and C. maquilingensis form two distinct clades that are out-grouped from the remainder of the species. A closer inspection of the multiple sequence alignment of the AP endonuclease III protein demonstrated three insertions within the helix barrel domain of P. marneffei along with the ~150-amino acid extra N-terminal region. This insertion (of ~57 amino acids) distinguishes P. marneffei from other eukaryotic organisms. The comparison of protein sequences from C. maquilingensis and O. iheyensis reveals that the catalytic K120 residue is absent in both the species. In addition, D138 is absent in C. maquilingensis, which supports the idea that the proteins from these two organisms belong to the HhH superfamily (as both of these proteins contain HhH motif) but not the endonuclease III sub-family. All the archaeal species occupy different clades in the protein-based tree, as observed in the case of the gene-based tree. Four archaeal species (Halogeometricum borinquense, C. maquilingensis, D. kamchatkensis, and M. smithii) share clades with D. thermophilum (Dictyoglomi), O. iheyensis (Firmicutes), P. marneffei (Fungi), and P. mobilis (Thermotogae), respectively. This observation reaffirms that the evolution of the endonuclease III protein within archaeal species is highly influenced by the environmental factors. Note that the endonuclease III proteins of bacterial species R. castenholzii and Synechococcus sp. are close to those of the eukaryotic species, indicating a probable HGT event. From this analysis, it is evident that the G+C content and the G+C content at the 3rd base have influenced the location of species within a tree. Thus, compared with a gene-based tree, protein-based analysis provides a better view of the evolutionary history of a protein.

AP endonuclease III protein sequence-based tree. The evolutionary distances are computed using JTT as the substitution model. The bootstrap values are provided in Fig. 4
A Model for the Evolutionary History of Endonuclease III Protein Family
We seek to reconstruct the possible evolutionary history of the endonuclease III family on the basis of sequence and phylogenetic tree analysis. Proteins of the endonuclease III family belong to the HhH-GPD superfamily and are close to the proteins of the MutY family. As shown in Tables 1 and 2, proteins from the HhH-GPD superfamily and the MutY family are retrieved along with the endonuclease III family proteins. These proteins are grouped together and are placed as out-group relatives to the other clades in the phylogenetic tree. The proposed model (Fig. 7) suggests that the eukaryotic lineage of the endonuclease III gene family diverged from eubacteria and archeabacteria lineages during early evolution. One of the important features in this evolutionary model is that a few HGT events from bacteria to eukaryotes and/or archaea have occurred in the endonuclease III gene family. The HGT events can be inferred from the observation that the clades of Roseiflexus (Chlorofexi), Synechococcus (Cyanobacteria) and eukaryotes are closely associated. Similarly, the clade for Thermotogae bacteria is shared with archaeal species. The phylogenetic tree of all bacterial and archaeal species reveals a mixed association of endonuclease III proteins. Another possible event of importance in the endonuclease III protein family is the endosymbiotic transfer of endonuclease III genes from bacteria (Cyanobacteria) to plants. Plant chloroplasts are derived from an ancestral endosymbiosis related to cyanobacteria. The Synechococcus clade (Cyanobacteria) is placed near the eukaryotic clade along with Oryza sativa and Arabidopsis thaliana, suggesting a possible endosymbiotic transfer event from bacteria (Cyanobacteria) to plants. Moreover, the insertion of a signal peptide at the N-terminal region of the endonuclease III protein appears to have occurred during the evolution of the plant homologs.

A model for the evolutionary history of the endonuclease III gene family is schematically shown. The archeal endonuclease III genes likely originated from Thermotogae by HGT. Eukaryotic endonuclease III genes likely originated from either Chlorofexi or Cyanobacteriae by HGT. Eukaryotic endonuclease III in plants contains an insertion at the N-terminal, which is targeted to the chloroplast
Conclusions
This study provides an overall picture of the evolutionary history of the endonuclease III gene family that plays a crucial role in BER of DNA. Based on the conservation of the amino acid positions, two consensus sequences have been identified for the helix-hairpin motif and the Fe-S domain. Sequence analysis has also identified that quite a few residues within helix 2 and helix 8 are crucial for the structure and function. The endonuclease III gene-based phylogenetic tree reveals few HGT events. Endosymbiosis events are also predicted from this evolutionary model. This evolutionary analysis may be exploited to understand the functions of uncharacterized genes in species.
Abbreviations
- DR:
-
Direct repair
- NER:
-
Nucleotide excision repair
- MMR:
-
Mismatch repair
- BER:
-
Base excision repair
- AP:
-
Apurinic/apyrimidinic
- 5′dRP:
-
5′Deoxy ribose phosphate
- HhH:
-
Helix-hairpin-helix
- NCBI:
-
National Center for Biotechnology Information
- EMBOSS:
-
European Molecular Biology Open Software Suite
- ML:
-
Maximum likelihood
- NJ:
-
Neighbor joining
- PDB:
-
Protein Data Bank
- FCL:
-
Iron–sulfur cluster loop
- HGT:
-
Horizontal gene transfer
References
Altschul SF, Gish W, Miller W et al (1990) Basal local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Aziz S (1995) DNA repair in humans. Annu Rev Genet 29:69–105
Aziz S (1996) DNA excision repair. Annu Rev Biochem 65:43–81
Brochier C, Philippe H, Moreira D (2000) The evolutionary history of ribosomal protein RpS14: horizontal gene transfer at the heart of the ribosome. Trends Genet 16:529–533
Cole JR, Wang Q, Cardenas E et al (2009) The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37(1):D141–D145
Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14:1188–1190
Denver DR, Swenson SL, Lynch M (2003) An evolutionary analysis of the helix-hairpin-helix superfamily of DNA repair glycosylases. Mol Biol Evol 20:1603–1611
Dmitry GV, Kosuke M (1997) DNA repair enzymes. Curr Opin Struct Biol 7:103–109
Dodson ML, Michaels Mark L, Lloyd RS (1994) Unified catalytic mechanism for DNA glycosylase. J Biol Chem 269:32709–32712
Emanuelsson O, Brunak S, Von HG, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Fromme JC, Verdine GL (2003) Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J 22:3461–3471
Garrity GM, Holt JG (2001) The road map to the manual. In: Boone, Castenholz RW, Garrity GM (eds) Bergey’s manual of systematic bacteriology. The archaea and the deeply branching and phototrophic bacteria, vol 1, 2nd edn. Springer, New York, pp 119–166
Golinelli MP, Chmiel NH, David SS (1999) Site-directed mutagenesis of the cysteine ligands to the [4Fe 4S] cluster of Escherichia coli MutY. Biochemistry 38:6997–7007
Hegde ML, Hazra TK, Mitra S (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 18:27–47
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci 8:275–282
Kanchan S, Mehrotra R, Chowdhury S (2014) Evolutionary pattern of four representative DNA repair proteins across six model organisms: an in silico analysis. Netw Model Anal Health Inform Bioinform 3:70
Labahn J, Schärer OD, Long A et al (1996) Structural basis for the excision repair of alkylation-damaged DNA. Cell 86:321–329
Lindahl T (1982) DNA repair enzymes. Annu Rev Biochem 51:61–87
Lukianova OA, David SS (2005) A role for iron–sulfur clusters in DNA repair. Curr Opin Chem Biol 9:145–151
Manuel RC, Hitomi K, Arvai AS et al (2004) Reaction intermediates in the catalytic mechanism of Escherichia coli MutY DNA glycosylase. J Biol Chem 279:46930–46939
Marchler BA, Bryant SH (2004) CD Search: protein domain annotations on the fly. Nucleic Acids Res 32(Web Server issue):W327–W331
Moretti S, Armougom F, Wallace IM et al (2007) The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res 35(Web Server issue):W645–W648
Nash HM, Bruner SD, Schärer OD et al (1996) Cloning of a yeast 8-oxoguanine DNA glycosylase reveals the existence of a baseexcision DNA-repair protein superfamily. Curr Biol 6:968–980
Puigbo P, Bravo IG, Garcia-Vallve S (2008) CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3:38
Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277
Richard DW (2001) Human DNA repair genes. Science 291:1284–1289
Shida T, Kaneda K, Ogawa T, Sekiguchi J (1999) Abasic site recognition mechanism by the Escherichia coli exonuclease III. Nucleic Acids Symp Ser 42:195–196
Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA 101:11030–11035
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Thayer MM, Ahern H, Xing D et al (1995) Novel DNA binding motifs in the DNA repair enzyme endonuclease Ill crystal structure. EMBO J 14:4108–4120
Thomas AK, Dorothy AE (2005) DNA mismatch repair. Annu Rev Biochem 74:681–710
Thompson JD, Higgins DG, Gibson TJ (1994) Clustalw: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Whittaker RH (1969) New concepts of kingdoms or organisms. Evolutionary relations are better represented by new classifications than by the traditional two kingdoms. Science 163:150–160
Yang H, Fitz-Gibbon S, Marcotte EM et al (2000) Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum. J Bacteriol 182:1272–1279
Acknowledgments
SK is thankful to the University Grant Commission (UGC), Govt. of India, New Delhi for providing the financial support in the form of the Basic Science Research Fellowship. The authors are thankful to Manoj Kannan for critical reading of the manuscript. The authors would also like to acknowledge the infrastructural facilities provided by Department of Biological Sciences, Birla Institute of Technology and Science, Pilani, Rajasthan, India.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kanchan, S., Mehrotra, R. & Chowdhury, S. In Silico Analysis of the Endonuclease III Protein Family Identifies Key Residues and Processes During Evolution. J Mol Evol 81, 54–67 (2015). https://doi.org/10.1007/s00239-015-9689-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-015-9689-5