
Abstract
Kinesins are eukaryotic microtubule motor proteins subdivided into conserved families with distinct functional roles. While many kinesin families are widespread in eukaryotes, each organismal lineage maintains a unique kinesin repertoire composed of many families with distinct numbers of genes. Previous genomic surveys indicated that land plant kinesin repertoires differ markedly from other eukaryotes. To determine when repertoires diverged during plant evolution, we performed robust phylogenomic analyses of kinesins in 24 representative plants, two algae, two animals, and one yeast. These analyses show that kinesin repertoires expand and contract coincident with major shifts in the biology of algae and land plants. One kinesin family and five subfamilies, each defined by unique domain architectures, emerged in the green algae. Four of those kinesin groups expanded in ancestors of modern land plants, while six other kinesin groups were lost in the ancestors of pollen-bearing plants. Expansions of different kinesin families and subfamilies occurred in moss and angiosperm lineages. Other kinesin families remained stable and did not expand throughout plant evolution. Collectively these data support a radiation of kinesin domain architectures in algae followed by differential positive and negative selection on kinesins families and subfamilies in different lineages of land plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Kinesins are a conserved superfamily of eukaryotic microtubule motor proteins instrumental in multiple essential processes including spindle formation, chromosome segregation, locomotion, morphogenesis, and vesicle trafficking (Vale 2003; Hirokawa et al. 2009; Ali and Yang 2020). The structural diversity of kinesins facilitates their function in myriad cellular activities (Hirokawa and Takemura 2004; Miki et al. 2005; Ali and Yang 2020). All kinesins bind microtubules and hydrolyze ATP through a kinesin motor head comprised of an ATPase and microtubule binding domain (Vale 2003; Miki et al. 2005; Kato et al. 2018). Through ATP hydrolysis, many kinesins walk along microtubules carrying cargo tethered to the motor domain by elongated stalks of alpha-helices (Vale 2003; Wang et al. 2015). Additional sequences and domains outside the conserved kinesin motor help define distinct kinesin families and enable them to participate in specific cellular roles (Lee and Liu 2004; Hirokawa et al. 2009; Welburn 2013).
The kinesin repertoire of the last eukaryotic common ancestor (LECA) is thought to contain 11 kinesin families (Wickstead et al. 2010). Since divergence from LECA, different organismal lineages evolved specific kinesin repertoires by retention, elaboration, and loss of unique combinations of kinesin families (Dagenbach and Endow 2004; Wickstead et al. 2010; Vicente and Wordeman 2015). Initial phylogenetic surveys of kinesin sequences identified 14 families in eukaryotes (Reddy and Day 2001; Dagenbach and Endow 2004; Miki et al. 2005; Richardson et al. 2006; Wickstead and Gull 2006). Successive examinations included more diverse eukaryotes, recognized 17 monophyletic kinesin families, and observed lineage-specific kinesin repertoires and domain architectures (Wickstead et al. 2010).
Notable differences in kinesin repertoires exist between photosynthetic organisms and other eukaryotes, and among plant and algal groups (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006; Wickstead et al. 2010). The initial kinesin surveys were understandably confined by early genome sequencing technology, and so our understanding of evolutionary trends in plant kinesin repertoires is growing along with sequencing technologies (Reddy and Day 2001; Dagenbach and Endow 2004; Richardson et al. 2006; Wickstead et al. 2010; Ye et al. 2022). Initial reports of plant and algal kinesin repertoires included five angiosperms and one bryophyte, and these reports identified kinesins with novel domain architectures in plants and green algae genomes (Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010). Kinesin families-7 and -14 clearly expanded in surveyed plant and algal kinesin repertoires in comparison to other eukaryotes, while other kinesin families appeared absent in some land plants (Reddy and Day 2001; Richardson et al. 2006; Shen et al. 2012). Initial observations led to much speculation about the function of expanded plant kinesin families (Li et al. 2012; Gicking et al. 2018; Ali and Yang 2020). Hypotheses have been challenging to test as few plant kinesins have been functionally characterized genetically or biochemically (Lee et al. 2015; Nebenführ and Dixit 2018; Ali and Yang 2020). This comprehensive phylogenetic analysis of kinesin sequences from diverse and updated land plant genomes will comprise a framework for focused experimentation of plant kinesin evolution and function.
Multiple hypotheses seek to explain the inflation of the kinesin-14 family in plants (Lee et al. 2015; Schneider and Persson 2015; Yamada and Goshima 2017; Gicking et al. 2018). The expanded kinesin-14 family has been hypothesized to compensate for dynein loss in land plants, as both dyneins and some animal kinesin-14 proteins can walk toward microtubule minus ends (Reddy and Day 2001; Schneider and Persson 2015; Yamada and Goshima 2017; Tseng et al. 2018). Animal kinesin-14 proteins contain C-terminal motors and exhibit minus-end movement, while most other kinesins are plus-end directed with N-terminal motors (She and Yang 2017). Indeed, some plant kinesin-14 are minus-end directed (Yamada et al. 2017; Tseng et al. 2018; Li et al. 2021), but most plant kinesin-14 proteins do not contain C-terminal motors (Reddy and Day 2001; Lee and Liu 2004; Zhu and Dixit 2012). Furthermore, cytoplasmic dynein was lost in the ancestor to green algae, while axonemal dyneins exist in many land plants (Wickstead and Gull 2007; Hodges et al. 2012; Kollmar 2016; Lucas and Geisler 2022). Previous plant kinesin surveys did not address whether the loss of cytoplasmic or axonemal dyneins corresponded with the expansion of kinesin-14 (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006; Wickstead et al. 2010; Shen et al. 2012).
Novel protein domain architectures exist in plant and algal kinesins, and the origin and expansion of these novel kinesins is unclear (Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010). Unique domains in plant kinesins include armadillo repeat (ARM), actin-binding calponin-homology (CH), myosin-like MyTH4, plasma membrane localizing FERM, and malectin-binding sequences (Reddy and Reddy 1999; Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010; Nebenfuhr and Dixit 2018). These domains increase the richness of plant kinesin repertoires and likely add functionality to kinesins (Lee and Liu 2004; Richardson et al. 2006; Wickstead et al. 2010), but their evolutionary origin and overall presence across plant diversity have not yet been explored.
To address multiple hypotheses and knowledge gaps, we performed comprehensive phylogenetic analyses of kinesins in 24 diverse land plants, two representative sister algal groups, two animals, and one yeast. These data demonstrated that novel protein domain architectures appeared in green algae, and these new subfamilies of kinesin-7, 14, and 19 expanded throughout land plant evolution. However, expansions were not uniform across taxa, nor did expansions strongly correlate with genome duplications or dynein loss. Our analysis placed the previously ungrouped armadillo repeat-domain kinesins within a kinesin-19 subclade and sister to kinesin-1. C-terminal kinesin-14C and 14D were phylogenetically distinct from the other kinesin-14 sequences in plants. Multiple kinesin groups were lost in ancestors to pollen-bearing plant groups along with flagella. This phylogenetic analysis of kinesins in diverse land plants furthers our understanding of plant cellular evolution.
Results
Kinesin Families in Land Plants
To test hypotheses regarding kinesin evolution in plants, sequences were collected from 24 land plants that span the evolutionary history of embryophytes, two green algae, and three non-photosynthetic outgroups. Land plants from significant and distinct evolutionary branches were surveyed, including five bryophytes and 19 tracheophytes (two seed-free vascular plants, five gymnosperms, and 12 angiosperms). To anchor the phylogenetic analyses, kinesins sequences were obtained from two green algae: (1) Klebsormidium flaccidum, a filamentous member of the Charophycean algal group considered a closely related sister group to land plants, and (2) Chlamydomonas reinhardtii, a more distantly related unicellular Chlorophycaean green algae (Donoghue and Paps 2020). Kinesin sequences from Homo sapiens, Drosophila melanogaster, and Saccharomyces cerevisiae were also included as non-photosynthetic, more ancestrally divergent outgroups. 1499 kinesin sequences were collected from public datasets of the 29 complete genome assemblies based on the presence of a kinesin motor domain (PFAM PF00225) (Supplementary Table 1, Supplementary File 1).
Next, maximum likelihood analyses were generated to evaluate molecular evolutionary relationships using the 1499 sequences from all surveyed organisms. Maximum likelihood analyses were produced from full-length protein sequences (Fig. 1 and Supplementary Fig. 1) and two sets of trimmed protein sequences that minimize alignment gaps at 10 and 80% minimum presence (Supplementary Fig. 4A, B and Supplementary Files 2 and 3). Most kinesin sequences readily assembled into clades with strong bootstrap support in all analyses (Fig. 1 and Supplementary Figs. 1, 4). We observed no major differences in clade grouping after analyses of these data sets.

Families of kinesin domain-containing proteins in plants. Maximum likelihood analysis of 1519 protein sequences including 1499 that contained kinesin domain (PFAM 00225) from 29 species. Clades with high bootstrap support (< 66 B.S.) were identified by the position of known A. thaliana, P. patens, and animal reference sequences and labeled according to nomenclature of Reddy and Day (2001), Lawrence et al. (2004), and Wickstead et al. (2010). Kinesin 14C and 14D subfamilies were not strongly supported to be monophyletic with kinesin 14A and B in our multiple phylogenic analyses. A Kinesin 14A/B sister clade contained C. reinhardtii and gymnosperm sequences on long branches (‘Kinesin 14A/B sister’, orange, see Supplementary Fig. 1 for sequence identifiers). Several plant and algal kinesins with long branches could not be assigned to an established numbered clade (‘Unassigned’, black). 20 gymnosperm sequences with high BLAST similarity to Arabidopsis thaliana kinesins and do not contain kinesin domains are labeled “non-kinesins.” Supplementary Fig. 1A presents a linear version of this tree without the 20 non-kinesins with bootstrap support values, branch lengths, and protein sequence identifiers
We present kinesin clades and relationships supported with high bootstrap values (above 66) in multiple analyses. Kinesin families are defined as clades with independent origins from the last common ancestor with algae and one or more of the distant outgroups H. sapiens, D. melanogaster, and S. cerevisiae. We followed nomenclature of Wickstead et al. (2010) to classify kinesin clades, as that naming convention updated a previous codified system (Lawrence et al. 2004) and was based upon rigorous phylogenetic analysis using numerous diverse organisms. Tree topology and clades were remarkably consistent among the full-length and trimmed sequences (Fig. 1 and Supplementary Figs. 1, 4, 5).
To evaluate the robustness of the predicted phylogenetic relationships and family assignments, maximum likelihood and parsimony analyses were performed on subsets of kinesin sequences focused on angiosperms (Supplementary Fig. 2) or more even sampling of taxa (Supplementary Fig. 3). Family assignments were unchanged among the different analyses and sampling strategies. A few sequences were not strongly associated with any numbered kinesin family or appeared sister to more than one family, and these sequences are listed as ‘unassigned’ in Fig. 2.

Kinesin families and subfamilies in 29 taxa. The number of kinesins per family and subfamily for each surveyed organism based upon phylogenetic analyses. The presence or absence of flagella during the life cycle and taxonomic relationships are designated on left side of table. Plants and yeast that do not form flagella lack kinesin families 19A, 4/10A, 9, and 16 (Blue cells mark kinesin groups in taxa with no members). Many kinesin families and subfamilies expanded in land plants. Kinesins outside of established families or sister groups quantified as “unassigned” (Color figure online)
The 15 families identified in this study (kinesins-1, 2, 3, 4/10, 5, 6, 7, 8, 9, 13, 14, 15, 16, 18, and 19) largely agreed with the published Bayesian analysis by Wickstead et al. (2010). This and previous analyses concurred that kinesin families-4 and 10 represent a single monophyletic clade, and therefore labeled as kinesin 4/10 (Figs. 1, 2) (Wickstead et al. 2010). These analyses also agreed that kinesin-12 was a polyphyletic group containing both kinesin families-15 and 16, and so we resolved these into two monophyletic families kinesin-12/15 and 16 (Figs. 1, 2) (Wickstead et al. 2010). Land plants in this study lacked kinesin families-17 and 20, and pollen-producing gymnosperms and angiosperms did not contain families-2, 3, 9, or 16. Our analyses confirmed tracheophytes do, but bryophytes do not, encode kinesin-6 (Wickstead et al. 2010). This study revealed that most surveyed tracheophyte genomes contained a single kinesin-6 gene. These kinesin-6 data also support the hypothesis that bryophytes are monophyletic (Su et al. 2021; Wang et al. 2022a).
These analyses demonstrated that some kinesin families contain well-supported “subfamilies” clades with high bootstrap support. We named subfamilies that contain animal sequences “subfamily A”. Kinesin families- 4/10 and 19 each divided into A and B subfamilies, and pollen-bearing plants contained neither kinesin-4/10A nor 19A sequences. Kinesin-7 divided into three distinct clades (subfamilies A, B, and C). Kinesin-14 split into subfamilies A and B, and B contained four independent subgroups.
These analyses clarified the origin and individuality of some plant kinesin groups. Our multiple analyses invariantly placed kinesin-14C and 14D sequences each as separate monophyletic clades separate from other kinesin-14 sequences in green organisms (bootstrap 100 for each clade, Fig. 1, Supplementary Figs 1–4). Thus, kinesin-14C and 14D appear of distinct evolutionary origin from a common algal or earlier ancestor. Our analyses also identified kinesin family-19 as a sister group to kinesin-1 in photosynthetic organisms (Figs. 1, 5, Supplementary Figs. 1–4).
Hypotheses Regarding Genome Size and Kinesin Expansion in Plants
Land plant genomes contain an unusually large number of kinesins (Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010). Larger, more complex organisms have been hypothesized to require more genes, including kinesins (Brawley et al. 2017), and so we assessed the total number of kinesins per genome to body size and tissue complexity. In land plants, the total number of kinesins ranged from 29 to 86 (Fig. 2). No clear relationship emerged between morphological complexity and total kinesin number (Table 1). For example, thalloid hornworts and liverworts with distinct tissues contained fewer kinesins (30 and 29, respectively) than the unicellular algae (C. reinhardtii, 39) and filamentous algae (K. flaccidum, 42). Furthermore, the early angiosperm sister groups, Amborella trichocarpa and Nymphaea colorata, contain similar numbers of kinesins (41 and 42, respectively) to algae despite the larger and more complex bodies of those flowering plants. Additionally, some seed-free tracheophytes and small statured moss encoded 70 or more kinesins, which is much more than seed-bearing A. trichocarpa and N. colorata (Table 1 and Fig. 2). Collectively, these data do not support the hypothesis that kinesins increase in number with body size.
We next tested the hypothesis that total kinesin number increased coincident with chromosome numbers as kinesins can facilitate mitotic spindle formation and chromosome movement. Several kinesin families play conserved roles in mitosis (Lee and Liu 2004; Gicking et al. 2018; Ali and Yang 2020; Liu and Lee 2022). Chromosome numbers varied from five in Anthoceros angustus and Arabidopsis thaliana to 39 in Ceratoperis richardii (Table 1). No correlation emerged between chromosome numbers and total kinesin number (Table 1). Increased spindle kinetochore fibers are needed to organize increased numbers of chromosomes, and so we next tested the hypothesis that chromosome number related to the size of kinesin families that are hypothesized to be ancestral spindle components in LECA (kinesins-5, 8, 13, and 14) (Wickstead et al. 2010). While the kinesin-14 family diversified and expanded in land plants (Fig. 2) (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2007), kinesin families-5, 8, and 13 minimally expanded from algae to angiosperms (Fig. 2). No significant correlation was found between kinesin quantities in individual families, chromosome number, or genome size (Table 1).
To test the hypothesis that plant kinesin gene families increased in size due to polyploidization or whole genome duplication, the total number of kinesins in each surveyed species was compared to two genome size measurements, C-value and total protein-coding genes (Table 1). In this survey, total kinesin gene number varied considerably from the unicellular yeast S. cerevisiae (6) to Populus trichocarpa (86), with a range of 29–86 (median 56) kinesin genes per land plant genome (Table 1 and Fig. 2). Strong correlations emerged between the number of kinesin genes and total protein-coding genes but not C-value, a measure total DNA (Pellicer and Leitch 2020) (Table 1). The number of chromosomes did not correlate with number of genes or C-values (Table 1). Total protein-coding genes and C-value did correlate across genomes. Phylogenetically independent contrast (PIC) analyses supported the positive correlation between the total number of kinesins and genes and the association between C-value and total number of genes. PIC analysis also revealed a negative correlation between C-value and number of chromosomes. These data indicated that total kinesin numbers correlated with total protein-coding genes but neither chromosome number nor C-value.
Overall, these data support the hypothesis that kinesin expansion was driven by protein-coding gene duplications, hybridization, and polyploidization. Indeed, plants with the largest kinesin numbers are documented polyploids and/or recent hybrids. Populus trichocarpa is a tetraploid and contains 86 kinesins (Liu et al. 2017; Wang et al. 2022b). Recent hybrids Helianthus annuus (Baack et al. 2005), Selaginella moellendorffii (Wang et al. 2020), Physcomitrium patens (Lang et al. 2018), and Zea mays (Hufford et al. 2021) all encode more than 73 kinesins (Table 1, Fig. 2).
Total Kinesin Quantity Independent of Repertoire Richness
The above data prompted further examination of kinesin repertoires, defined as the number of families and subfamilies within a species along with the number of sequences in each kinesin category. Kinesin repertoires differ among eukaryotic lineages (Reddy and Day 2001; Lawrence et al. 2004; Wickstead et al. 2010). Land plant repertoire richness varied from 11 to 19 kinesin families and subfamilies, ranged in size from 29 to 86 total kinesins, and the size of individual kinesin families did not correlate with total protein-coding genes (Fig. 2).
In plants, repertoire richness did not correspond to the total kinesin number nor body complexity (Fig. 2). For example, the richest repertoires composed of 19 kinesin families and subfamilies were encoded by the alga K. flaccidum with 42 kinesins and the more anatomically elaborate fern Ceratopteris richardii with 68 kinesins (Fig. 3). Overall, bryophyte repertoires contained 15, 16, or 18 families and subfamilies, but displayed a large variation of total kinesin number from 29 in Marchantia polymorpha to 73 in Physcomitrium patens (Figs. 2 and 3). Angiosperms held shallower repertoires despite routinely higher total kinesin numbers (Fig. 2). Angiosperms Populus trichocarpa, Helianthus annuus, and Zea mays contained the most kinesins, but their repertoires included only 14, 13, or 12 families and subfamilies, respectively (Figs. 2 and 3). The variation in repertoire richness and total kinesin number among plant lineages prompted investigation of kinesin expansions and contractions.

Richness and size of kinesin repertoires vary among plant and algae taxa. The total number of kinesins in the genomes of green alga Klebsormidium flaccidum and three land plants represented by diameter of pie chart. The three land plants shown encode similar numbers of kinesins, yet the kinesin repertoires different depending upon the presence of kinesins associated with flagella ( ~) and monophyletic expansions of specific kinesin groups (*). The size of kinesin families and subfamilies is indicated by the width of pie wedges. The thinnest wedges represent single kinesins, expanded kinesin groups are marked with *, and groups specific to organisms with flagella are marked with ~
Examinations of kinesin repertoires by plant evolutionary lineage indicated that some kinesin expansions and losses were restricted to specific plant groups (Figs. 2 and 3). Physcomitrium patens, Ceratopteris richardii, and Helianthus annuus (a bryophyte, fern, and angiosperm) all have comparably large total kinesin numbers which arose from independent expansions and losses of kinesin families and subfamilies. In comparison with the algae that share a common ancestor with land plants, kinesin-15 expanded both in tracheophytes and in moss. Within bryophytes, all three mosses contained more than ten kinesin family-15 sequences, while thalloid bryophytes and algae contained three or less kinesin-15 sequences (Fig. 2). All tracheophytes except W. mirabilis and S. tuberosum contained more kinesin-15 sequences than algae and fewer than moss (Fig. 2). In relation to algal, bryophyte and tracheophyte common ancestors, kinesin subfamily-19B expanded independently in the moss P.patens, fern C. richardii, and rosid P. trichocarpa (Fig. 2). Within the angiosperm lineage, kinesin subfamilies-7C and 14B with CH domain appeared expanded in most rosids and monocots (Fig. 2).
Despite increases in genome sizes and total kinesin numbers, some kinesin families remained small in all plant genomes. Kinesins-1, 2, 3, and 16 contained a single member in most plant genomes (Fig. 2). Additionally, kinesin-6 appeared in both algae and tracheophytes as a single copy per genome but was absent in bryophytes. In combination, these analyses suggested that genome duplications during plant evolution likely increased total kinesin numbers, and subsequent positive and negative selection for duplicated kinesin genes, respectively, expanded and contracted specific kinesins families. The presence of singleton kinesins suggests that additional copies of some kinesins are detrimental, yet those kinesins are important for survival.
Loss of Four Kinesin Families and Two Subfamilies Coincident with Flagella Loss
Several kinesin families (2, 3, 9, and 16) and subfamilies (4/10A, 19A) were identified in only algae, plants, and animals that produce flagella during their life cycle. These kinesin groups were entirely absent in plants that make pollen (Fig. 2). For example, Kinesin-2 was not found in yeast or plants that produce pollen, including four gymnosperms and all 12 angiosperms surveyed (Fig. 2). Orthologues of kinesin-2 and 9 mediate intraflagellar transport and flagellar beating (Scholey 2003; Yokoyama et al. 2004; Wickstead et al. 2010). Flagellated sperm were independently lost twice in seed plants, once within gymnosperms and once in ancestors to angiosperms (Hodges et al. 2012). Kinesin families 6, 9 and subfamily 4/10A appear to have been lost in ancestors to seed plants, while kinesin families 2, 3 and subfamily 19A were lost in the ancestors to angiosperms (Fig. 2).
Most kinesin groups associated with flagella contained few genes in each species. A single kinesin-2 gene was identified in all surveyed algae and bryophytes, the fern C. richardii, and gymnosperm Ginkgo biloba (Fig. 2). Selaginella moellendorffii, H. sapiens, and D. melanogaster contained three or four kinesin-2 copies (Fig. 2). One or two members of kinesin families-3, 16 and subfamily-19A were identified in algae and most plants with flagella but absent in plants without flagella (Fig. 2). Two or three copies of kinesin family-9 and subfamily-4/10A were common in flagellated plants, and both families were missing in Ginkgo biloba and all pollen-bearing plants (Fig. 2).
Some kinesins families and subfamilies associated with flagella were occasionally lost in specific plant species. We observed no consistent or shared pattern of kinesin loss in flagellated plants that lacked kinesins-3, 4/10A, 16 or 19A (Fig. 2). Kinesin-4/10A was absent in the surveyed liverwort and hornwort, though both species contained sequences belonging in subfamily-19A, while two moss (C. purpureus and P. patens) lost kinesin 19A sequences (Fig. 2). No expansions of kinesins correlated with the loss of flagella and production of pollen (Fig. 2).
Kinesin-19 and ARM Domain Kinesins are Specific to Plants
Kinesins with armadillo repeats (ARM) domains have been found only in plants and have been considered “ungrouped” (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006; Shen et al. 2012). Wider phylogenetic surveys designated these ARM-containing sequences as kinesin family-19 (Wickstead et al. 2010). Our phylogenetic analyses with full-length and trimmed sequences revealed kinesin-19 shared a common ancestor with kinesin-1 (Figs. 1 and 4, and Supplementary Figs. 1, 4). This common ancestry was observed in evaluations of all 1499 trimmed and full-length kinesin sequences (Supplementary Figs. 1, 4) and sequence subsets to balance taxonomic sampling (Fig. 4, Supplementary Figs. 2, 3). Both algae C. reinhardtii and K. flaccidum contain a single kinesin-1 and two kinesin-19 sequences (Fig. 2). Both algae also encoded one sequence sister to both kinesin-1 and 19 families, and no other surveyed species encoded kinesins in these phylogenetic positions (Figs. 2 and 4, Supplementary Fig. 1). These data suggested that kinesin-19 derived from a duplication of an earlier kinesin-1 among or before the divergence green algae, followed by the loss of the duplicated sister gene in the ancestor to land plants.

Kinesin-19 is sister to kinesin-1. A Subsection of a maximum likelihood tree demonstrates the sister relationship between kinesin-1 and 19 (BS = 96 to separate the kinesin families). Kinesin-19 further separates into clades with and without ARM domains. Entire maximum likelihood tree shown in Supplementary Fig. 2. B Structural predictions of kinesin-1 members. To the left, K. flaccidum and A. thaliana kinesin-19 sequences are 426 and 465 amino acids, respectively, and retain the kinesin motor domain (black bracket). To the right, predicted models of two H sapiens kinesin-1 proteins are 1032 and 860 amino acids in length, respectively, and contain long coiled regions adjacent to motor domain (black bracket). C Predicted structures of kinesin 19B subfamily. K. flaccidum and A. thaliana kinesin-19B models show the ARM domain (red bracket) adjacent to the motor (black bracket) and opposite the elongated coils. Models of predicted structures for kinesin-19A proteins not available from AlphaFold at this point (Color figure online)
These analyses further split kinesin-19 into two distinct subclades, supported with 99 bootstrap values, that correlated with the appearance of an armadillo repeat (ARM) domain (Fig. 4). In this survey, kinesin-19A subfamily sequences lacked ARM domains and were identified in both algae and most land plants with flagellated sperm (Figs. 2 and 4). However, kinesin-19A was missing in all pollen-bearing plants and two of three surveyed mosses (Fig. 2). All kinesin subfamily-19B sequences contained ARM domains and were found in all land plants except S. tuberosum, K. flaccidum, but not C. reinhardtii. Therefore, this kinesin-19B architecture with an ARM domain is unique to land plants and charophycean algae.
AlphaFold structural predictions of A. thaliana kinesin-19B proteins showed the C-terminal ARM domain opposite to the kinesin tail and adjacent to the N-terminal motor domain (Fig. 4C) (Jumper et al. 2021). K. flaccidum and A. thaliana kinesin-1 are shorter proteins than H. sapiens, and K. flaccidum and A. thaliana kinesin-1 models differed from both H. sapiens kinesin-1 (Fig. 4B) and kinesin-19B models (Fig. 4C). All available kinesin-19B and the H. sapiens kinesin-1 (KIF5A and KIF5C) structures contained longer alpha helical regions with helix hairpin-like structures (Figs. 4B, C). No structural models for kinesin-19A proteins were available for comparison.
Expansion and Diversification of Kinesin-14 Subtypes in Algae and Plants
The kinesin-14 family diversified into subfamilies with unique domains and architectures during land plant evolutionary history (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006; and Wickstead et al. 2010). This analysis supported the separation of kinesin-14 into A and B subfamilies, and further division of kinesin-14B into four subgroups (Fig. 5 and Supplementary Fig. 1). Kinesins in the 14A subfamily contained C-terminal motor domains, while the kinesin-14B subfamilies contained N-terminal or internal motor domains. Kinesin-14B resolved into four well-supported subgroups (bootstrap values > 94) that also correlated with unique domain architectures (Fig. 5 and Supplementary Figs. 1, 2). This phylogenetic grouping was retained when sequence alignments were trimmed to 80% presence which removed all but 526 informative amino acids found in almost all plant kinesins (Supplementary Fig. 4, Supplementary File 3, and see “Methods” section). Members of each kinesin-14B subgroup contained either an actin-binding calponin-homology (CH) domain, malectin-binding domain, KCA-like domains, or a unique disordered region (Fig. 5).

Diversification and expansion of plant and algal kinesin-14 family. Maximum likelihood tree of all kinesin-14 sequences of 29 species in this analysis. Majority of plant and algal kinesin-14 sequences belong within four subgroups of kinesin-14B. Representative Arabidopsis thaliana AlphaFold structural models for each subgroup are shown with the conserved kinesin motor domain (bracket) positioned at the bottom with coils of tether domain upright, and subgroup-specific domains highlighted. Blue colors within AlphaFold models indicate high-confidence structures. At1g55550 represents the disordered subgroup as much of the protein structure is unstructured (low-confidence AlphaFold models are shown in yellow). The malectin-binding domain of At1g72250 predicted to contain beta-sheets separate from motor and adjacent to coils of tether domain. KCA proteins (represented by At5g10470) predicted to contain multiple unnamed domains with alpha-helices that may facilitate function in organelle movement. The CH-domain subgroup of kinesin-14B expanded more than other subgroups in plants. Nearly all Arabidopsis thaliana CH-domain proteins contain a CH-domain with alpha-helices and a coiled region jutting away from the motor domain, as seen in At2g47500. At1g73850 is a member of the CH-domain subgroup but does not encode a CH-domain. Plant and animal C-terminal kinesin-14A proteins showed similar AlphaFold structures, as shown in side-by-side comparisons of At4g21270 (middle 14A structure) and H. sapiens KIF1C (right). Biochemical studies indicated that Kinesin-14A At5g54670 contain a second microtubule binding site outside of the motor down (arrow) (Color figure online)
AlphaFold models of A. thaliana kinesin-14 tertiary structures showed consistent differences between the kinesin-14A and all four 14B subgroups (Fig. 5). A. thaliana kinesin-14A structures appeared similar to the H. sapiens kinesin-14 KIFC1 orthologue (Fig. 5). Structural predictions indicated that the CH, malectin-binding, and KCA domains were positioned near the elongated stalk and separated from the motor domain on A. thaliana proteins (Fig. 5). One A. thaliana kinesin-14B in the CH subgroup lacked the CH domain and retained a coil that jutted away from the nearby motor domain (Fig. 5). AlphaFold structural models suggested three uncharacterized globular domains adjacent to the stalk in KCA-like sequences (Fig. 5). Collectively, these structures highlight the differences among the kinesin-14B subgroups.
The expansion of the kinesin-14 family in plants is enigmatic (Gicking et al. 2018; Nebenfuhr and Dixit 2018). To further understand the expansion of the kinesin-14 family, we assessed family growth by identifying phylogenetic nodes where duplication events likely occurred. Overall, in land plants, the kinesin-14B subfamily expanded more than kinesin-14A (Figs. 5, 6A). Three kinesin-14B subgroups are represented by one or two copies each in algae or thalloid bryophyte giving the hypothetical land plant ancestor approximately three to six kinesin-14B genes (Fig. 2). Both algae possessed sequences sister to the kinesin-14B subfamily and three kinesin-14B subgroups, but neither animals nor yeast encoded sequences in these positions. Algal and animal outgroups encoded additional sequences sister to the full plant kinesin-14 family (Figs. 2, 5). Chlamydomonas reinhardtii encoded 10 sequences sister to the kinesin-14A/B clade (Fig. 2, Supplementary Figs. 1, 2). In general, K. flaccidum and thalloid land plants contained the same kinesin-14B subfamilies (Figs. 6A). Together, these data suggest that kinesin-14B subgroups arose in the green algae and became more numerous in land plants.

Expansion of kinesin-7 and -14 subfamilies in land plants. A The majority of kinesin-14’s in plants belong within the kinesin-14B subfamily. Species names shown as abbreviated versions of full name and displayed in the same order as Table 1. The kinesin-14 family expanded in many land plants, except the thalloid hornworts and liverworts, and the expansion did not correlate with the complete loss of dyneins. In comparison of kinesin-14B, few copies of kinesin-14A and -14C are found in land plants. B Within the kinesin-14B subfamily, the groups containing CH or malectin-binding domains expanded in many plants. The largest CH and malectin subgroups are found in monocots and dicots. KCA and disordered kinesin-14B subgroups remained in low copy number in land plants. C Kinesin-7B and -7C expanded in monocots and dicots in comparison to the kinesin 7A subfamily. Smaller kinesin-7A expansions appear in the bryophytes and kinesin-7B enlarged in fern and Selaginella
Specific subgroups of kinesin-14B sequences duplicated in moss and tracheophytes, with additional duplication events evident in monocots and dicots (Fig. 6A). The CH domain kinesin-14B subgroup increased more than other subgroups during plant evolution and they account for nearly half of the kinesin-14B sequences. Three kinesin-14B CH duplication events appear to have occurred first in the ancestor to all tracheophytes, secondly in angiosperms, and last in rosids. The next-largest kinesin-14B subgroup contained a malectin-binding domain. A malectin-binding domain kinesin-14B duplication appeared in ancestors to tracheophytes, and a second single copy duplication just one phylogenetic branch (Fig. 6B). The KCA-like and disordered kinesin-14B subgroups remained small in all surveyed land plants and did not show expansion in bryophyte or tracheophyte plant genomes (Fig. 6B).
Kinesin-14A is represented by a single copy in K. flaccidum and thalloid land plants, and therefore, the ancestor to land plants likely had only one kinesin-14A gene (Fig. 2). Duplication of kinesin-14A sequences appears to have occurred independently in mosses and tracheophytes. Based on the position of nodes yielding four kinesin-14A genes in most species, a duplication may have occurred prior to the common ancestor of gymnosperms and angiosperms. However, angiosperms may have experienced a loss of kinesin-14A genes as both sister taxa A. trichocarpa and N. colorata have either one or two genes. While most angiosperms and gymnosperms contain four or more kinesin-14A copies, additional copies appear within the angiosperm clade. Kinesin-14A expanded both in the common ancestor of dicots and in the common ancestor of Brassicaceae.
The kinesin-14 expansion in plants has been hypothesized to compensate for loss of dynein proteins, a eukaryotic class of minus-directed microtubule motors (Reddy and Day 2001; Schneider and Persson 2015; Yamada and Goshima 2017; Tseng et al. 2018). Only kinesin-14A orthologues contain C-terminal motor domains, and therefore predicted to be minus-directed motors capable to substitute for dyneins (Lee and Liu 2004; Zhu and Dixit 2012). In land plants, dynein heavy chain proteins were found only in plants that reproduce with flagellated sperm (Lucas and Geisler 2022), and so we predicted an increase in kinesin-14A sequences coincident with the loss of flagella. However, we found no significant differences between the number of kinesin-14A sequences in flagellated (average 2.2 per genome, N = 10) and non-flagellated (3.3, N = 16) photosynthetic organisms (p = 0.68). Additionally, no kinesin-14B subgroup expanded in correlation with the complete loss of dyneins (Fig. 6A). In combination, the expansion of the kinesin-14 gene family did not appear related to the loss of axonemal dyneins.
Kinesin-14C and 14D Distinct Phylogenetic Clades
Kinesins-14C sequences have only been identified in photosynthetic organisms (Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010). These C-terminal motor kinesins have been considered within the kinesin-14 family and contain several domains uncommon in kinesins including actin-binding MyTH4, calmodulin-binding, homodimerization, and lipid-binding FERM domains (Reddy and Day 2001; Richardson et al. 2006; Wickstead et al. 2010). Due to the calmodulin-binding domain, these kinesins are also called Kinesin-like Calmodulin-Binding Proteins (KCBPs) (Song et al. 1997; Vinogradova et al. 2009). Kinesin-14D is also called PAKRP2 and KINID1 (Lee et al. 2007; Hiwatashi et al. 2008), and these sequences have been classified as “orphan” (Shen et al. 2012), kinesin-10 (Richardson et al. 2006; Zhu and Dixit 2012), and kinesin-14 (Wickstead et al. 2010).
Here, multiple maximum parsimony and nearest neighbor analyses indicated kinesin-14C and 14D sequences each formed clades distinct from other kinesin families (Fig. 1, Supplementary Figs. 1–4). Maximum parsimony and nearest neighbor analyses were performed on all 1519 retrieved full-length and trimmed kinesin sequences, and two different subsets of sequences to control for taxon sampling. These analyses consistently showed that kinesin-14C and -14D sequences formed distinct clades outside of the kinesin-14 family, each with well-supported and resolved internal branches (Supplementary Figs. 1–4).
In phylogenetic trees, kinesin-14C consistently separated from other kinesin families on a long branch (indicating high sequence divergence from other kinesins) with strong support, bootstrap 99% (Fig. 1 and Supplementary Figs. 1, 2). The domain architecture of kinesin-14C is unique to algae and plants (Wickstead et al. 2010). AlphaFold structural models align with crystal structure studies (Fig. 8A) (Vinogradova et al. 2009). The N-terminal MyTH4 and FERM domains fold near the C-terminal motor (Fig. 7A). The calmodulin-binding and homodimerization regions formed short alpha-helixes adjacent the motor domain and opposite of the actin and lipid-binding domains (Fig. 7A). The predicted structures of C. reinhardtii and A. thaliana appeared similar, and different than other kinesin-14 models (Fig. 7A inset). Cumulatively, these structural data bolster the phylogenetic distinctness of kinesin-14C sequences.

Kinesin-14C and -14D structural predictions. A Kinesin-14C contains FERM and MyTH4 binding domains (circled) near the N-terminus. The calmodulin-binding region appears as an alpha-helix adjacent to the homodimerization domain and kinesin motor (black bracket). These domains and structures appear conserved in both C. reinhardtii and A. thaliana kinesin-14C proteins (inset). However, kinesin-14A and -14B do not contain similar domains, as shown within inset and in Fig. 5. B Predicted models of A. thaliana (At4g14330) and P. patens (PpKINID1A) kinesin-14D protein structures show motor domain (black brackets), two elongated coils, and a large region of uncertain structure. Yellow, orange, and red regions of AlphaFold models indicate low-confidence folds. These kinesin-14D structures are unlike other kinesin structures, as shown in inset with comparisons to S. cerevisiae SMY1 and KIP1, and A. thaliana kinesin-5 (At3g45650) (Color figure online)
Depending upon analysis, the kinesin-14D clade was adjacent to either kinesin-13 or kinesin-5 with modest support (bootstrap 66 and 63, respectively, Fig. 1 and Supplementary Figs. 1, 2). Saccharomyces cerevisiae SMY1 was poorly associated with the photosynthetic kinesin-14D clade in all analyses (Shen et al. 2012) (Fig. 1 and Supplementary Fig. 1). The kinesin-14D clade contained land plant and algal sequences (Fig. 2), and no other domain for kinesin-14D has been annotated yet besides the N-terminal motor domain (Gicking et al. 2018). AlphaFold models of A. thaliana and P. patens kinesin-14D structures appeared similar and included similar structural features (Fig. 7B). Models displayed two antiparallel helices oriented at an acute angle near the motor domain and a globular structure of short helices which might represent an undocumented domain (Fig. 7B). However, the plant kinesin-14D structures did not resemble structural models of SMY1, kinesin-5, or kinesin-13 (Fig. 7B, inset).
Both kinesin-14C and -14D sequences appeared first in green algae and persisted in land plants (Fig. 2), Unlike the kinesin-14A and B subgroups that expanded in land plants, most plant genomes contained one or two kinesin-14C and 14D copies each (Figs. 2, 6A). We observed no lineage-specific pattern of expansion for either kinesin-14C or 14D, and any absence or duplication appeared species specific (Fig. 2 and Supplementary Fig. 1).
Expansion of Kinesin 7 Subfamilies in Plants
Within the kinesin-7 family, three well-supported clades emerged: 7A, 7B, and 7C (Figs. 1, 8 and Supplementary Figs. 1, 2). Phylogenetic analyses placed an algal, animal, or fungal sequence ancestral within each kinesin-7 subfamily (Fig. 8 and Supplementary Fig. 1). The kinesin-7A subfamily contained H. sapiens, D. melanogaster, algal, and plant orthologues (Fig. 2 and Supplementary Figs. 1, 2). Plant, algal, and S. cerevisiae sequences comprised the kinesin-7B subfamily (Fig. 2 and Supplementary Fig. 8). Kinesin-7C contained only plant and algal sequences (Fig. 2 and Supplementary Figs. 1, 2), and thus, kinesin-7C appears unique to photosynthetic organisms in this survey.

Kinesin-7 subfamily phylogeny and structures. Subsection of maximum likelihood tree focused upon the subfamilies within kinesin-7 (full tree shown in Supplementary Fig. 1), and AlphaFold structural predictions shown for representative subfamily members. Motor domains (black brackets) of A. thaliana kinesin-7A and D. melanogaster CENP were similarly located within comparable protein structures. Predicted kinesin-7B and 7C protein structures are distinct from kinesin-7A. All A. thaliana kinesin-7B sequences contain N-terminal signal sequences (arrow) predicted to target proteins to mitochondria or plastids, and two A. thaliana kinesin-7B sequences also contain Zinc-finger domains (arrow). The S. cerevisiae kinesin-7B KIP2 also contains a polar N-terminal peptide which may function as a signal sequence. Kinesin-7C subfamily sequences are folded in motor domains and multiple helixes. Helices clustered into units (red bracket) in AlphaFold models of kinesin-7C proteins, and the function of these potential domains is not yet known. Three plant sequences fell outside of the three subfamilies on long branches (yellow). Black dots within the tree indicate location of animal sequences (Color figure online)
The kinesin-7 family expanded in land plants (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006; and Wickstead et al. 2010), and kinesin-7 subfamilies expanded unequally multiple times during plant evolution (Fig. 6C). While the surveyed algae contained four and five kinesin-7 sequences, independent expansions occurred in ancestors of moss, seed-free tracheophytes, and angiosperms (Figs. 2, 6C). Within the kinesin-7 family, subfamily-7A minimally expanded, while subfamily-7C expanded the most (Figs. 2, 6C, Supplementary Figure 1, 2). Kinesin-7A may have doubled from one to two copies in the ancestor to land plants (Figs. 2, 6C). Kinesin-7C appeared to double between ancestors of thalloid bryophytes and mosses, and independently expanded in ancestors of C. richardii, monocots, and rosids (Fig. 6C). The kinesin-7B subfamily enlarged in ancestors to the seed-free vascular plants (S. moellendorffii and C. richardii), angiosperms H. annuus, Z. mays, and rosids. Kinesin-7B was lost from S. tuberosum ancestors (Fig. 6C, Supplementary Figs. 1, 2).
H. sapiens and S. cerevisiae Kinesin-7A and 7B proteins are involved in mitosis (Wickstead et al. 2010), and they share structural similarities with A. thaliana predicted protein structures (Fig. 8). Plant and animal kinesin-7A protein models exhibited similar overall appearance, with the N-terminal motor domain centrally positioned relative to several elongated alpha-helix hairpins (Fig. 8). All A. thaliana kinesin-7B sequences contain predicted N-terminal signal sequences for targeting proteins to plastids by SUBA3 consensus (Tanz et al. 2013). Yeast and A. thaliana kinesin-7B predicted protein structures appeared similar, with multiple alpha-helices and disordered loops opposite to the motor domain and signal peptide (Fig. 8). Two out of the five A. thaliana kinesin-7B sequences also contain a zinc finger domain (Fig. 8).
Kinesins within subfamily-7C did not contain UniProt recognized domains outside of the N-terminal motor. However, AlphaFold structural predictions of A. thaliana kinesin-7C sequences contained clustered alpha-helices that may represent a functional unit distinct from the motor domain (red bracket, Fig. 8). The genetically redundant A. thaliana HINKEL and TETRASPORE structural models are involved in pollen cytokinesis and shared structural similarities (Oh et al. 2008).
Discussion
This comprehensive phylogenetic analysis clarified evolutionary changes in plant kinesin repertoires by using updated genomes from an increased number of diverse plants compared to prior studies. Kinesin repertoires reflect evolutionary histories of lineage and species. Despite lineage-specific differences, many overarching trends regarding kinesin evolution were clear (Fig. 9). Nine novel kinesin domain architectures emerged in green algae, and then six kinesin groups were lost coincident with the loss of flagellated sperm in ancestors to pollen-bearing plants. Kinesins-14C and -14D each form distinct clades, separate from the canonical kinesin-14 family, and thus indicated that C-terminal kinesins may have evolved more than once. Kinesin with ARM domains rose from the kinesin-1/19 family complex. Kinesin-14B and kinesin-7 subgroups enlarged in land plants, while other kinesin groups remained in low copy numbers regardless of genome-wide duplication. The phylogeny presented here represents only the extant genes after multiple rounds of gene duplication and subsequent pruning of redundant paralogs. The gene copies that persisted likely were under independent selection and provide some unique benefit. Overall, this phylogenomic evaluation of kinesin repertoire evolution in land plants suggested that novel kinesin architectures enabled new plant cell functions and terrestrial life.

Summary of key cytoskeletal and kinesin repertoire events. 1. Before the divergence of chlorophytes from other algal lineages, cytoplasmic dynein and outer arm dynein alpha were lost, while kinesin families- 19, 14C, and subfamilies 7C and 14B emerged. 2. During green algal evolution when the phragmoplast and preprophase band appears, four types of kinesin emerged: kinesin-14D, 19B, and two subgroups of 14B (KCA and malectin-binding). Axonemal dyneins were also lost (beta and gamma outer arms and DHC 3 and 4) during green algal evolution. 3. All land plants build phragmoplasts and preprophase bands (PPBs) without centrosomes and contain kinesin-14B disordered subgroup. Kinesins-4A, 4B, and 12/15 expanded in the moss lineage (a), kinesin-7B expanded in lycophytes (b). Pink spots represent sequential loss of some dynein light chains, IFT and DHC12 heavy chains (Lucas and Geisler 2022). 4. Flagella were lost in both gymnosperms and angiosperms, along with multiple kinesin families. Inner arm dyneins were lost sequentially in plants that reproduce with flagellated plants, and all dynein heavy chains were completely lost with flagella. Within angiosperms, kinesins-7C and -14B CH domains expanded in rosids (c)
Diversification of Kinesin Repertoires in Algae
Previous genomic surveys identified unique domain architectures in plant kinesins (Reddy and Day 2001; Richardson et al. 2006; Wickstead and Gull 2006). Our study indicated domain addition in algae leads to diversification of kinesin families into subfamilies that then expanded in land plants. Kinesin-7C and three distinct kinesin-14B subfamilies contain additional domains in algae, appeared in algal ancestors, and expanded in ancestors to modern land plants. Also in algal ancestors, the kinesin-19 family separated from kinesin-1 and then ARM domains were added in a subset of algal kinesin-19 sequences. Furthermore, kinesin-14C and-14D both with distinctive domains appeared in algal ancestors, and the kinesin-14B subfamily with disordered domains emerged in land plants. Our results were also consistent with previous phylogenetic studies that kinesin-10 and 12 are not monophyletic and therefore reassigned sequences into evolutionarily related groups (Wickstead and Gull 2006; Wickstead et al. 2010). Kinesin-12 was separated into kinesin families-12/15 and 16, which revealed that kinesin-16 sequences were specific to organisms with flagella (this paper; Wickstead and Gull 2006; Wickstead et al. 2010).
Green algal kinesin diversification may be related to algal cellular and genomic innovations. In green algae, multiple microtubule-based mechanisms exist to complete cytokinesis (Buschmann and Zachgo 2016). The appearance of novel algal kinesins may be related to different modes of cytokinesis. Genomic sequencing of more diverse algae in both Viridiplantae and Rhodophyta will help to elucidate algal and kinesin evolution (Bowles et al. 2023). A more complete understanding of algal genome evolution will help clarify when distinctive kinesins appeared during evolution, and thereby aid our functional understanding of distinctive kinesins.
Notably, all green algae surveyed lack a cytoplasmic dynein heavy chain and most lack centrosomes (Hodges et al. 2012). Without cytoplasmic dynein or centrosomes, other microtubule-associated proteins likely organize microtubules. The expanded plant kinesin-14 family has been suggested to compensate for dynein loss, because both cytoplasmic dynein and some C-terminal kinesin-14 proteins are minus-directed microtubule motors (Reddy and Day 2001; Schenider and Persson 2015; Yamada and Goshima 2017; Gicking et al. 2018; Tseng et al. 2018). Indeed, plant kinesin-14A and 14C proteins have demonstrated minus-directed activity and therefore could functionally substitute for cytoplasmic dynein (Jonsson et al. 2015). However, kinesin-14B proteins account for the vast kinesin 14 expansion in plants, and minus-directed motility has not yet been demonstrated for any full-length plant kinesin-14B protein (Lee and Liu 2004; Jonsson et al. 2015; Schneider and Persson 2015; Tseng et al. 2018). Therefore, it is unclear whether kinesin-14B proteins could compensate for cytoplasmic dynein. Kinesin-14C likely does not solely compensate for cytoplasmic dynein because mutations are viable, despite showing defects in trichome morphogenesis (Oppenheimer et al. 1997). Further functional and genomic surveys focusing upon algal kinesins and dyneins are needed to elucidate when kinesin-14B, C, and D subgroups appear in relation to cytoplasmic dynein loss.
Loss of Flagella and Kinesin Families
Kinesin repertoires sharply contracted when flagella were lost in gymnosperm and angiosperm lineages (Fig. 9). Kinesin families-2, 3, 4/10, 9, 16 were present in the surveyed fungal, animal, and most plant genomes that produce flagella during their life cycle, and the absence of these kinesins from pollen-bearing land plants implies these kinesins retained their ancestral roles in flagella and they were not repurposed during evolution (Wickstead et al. 2010; Hodges et al. 2012). Only kinesin-2 was found in all flagellated plants, and this kinesin-2 functions in anterograde intraflagellar transport in many other organisms with flagella (Ali and Yang 2020). Curiously, other microtubule motor proteins that facilitate intraflagellar transport, dynein-16 heavy chain and two associated light chains, are found in algae, hornworts, and liverworts, but lost from other flagellated land plants (Lucas and Geisler 2022). The other kinesins (families 3, 4/10, 9, 16) were occasionally missing from flagellated plants, which suggested these proteins were not necessary for plant flagella or that plant flagella are the proteins lost some functionality.
The sharp loss of flagella-associated kinesins contrasts the sequential loss of axonemal dyneins in land plants (Lucas and Geisler 2022). Collectively, the different patterns of kinesin and axonemal dynein loss suggest that dyneins were interchangeable or dispensable, while kinesins were necessary with distinct functions during flagellar development and/or function. An alternative explanation for differences in kinesin and dynein loss could be that kinesin-2, 3, 9, 4/10A, and 19 without ARM domains could be functionally divergent in plants and operate outside of flagella, yet still be associated with sperm-based reproduction or hinder pollen production.
Expansion of Novel Domain Kinesins Architectures in Land Plants
The appearance and expansion of unique kinesin domain architectures in algae and land plants may have enabled plant-specific processes (Hamada 2007; Li et al. 2012; Zhu and Dixit 2012; Lee et al. 2015; Gicking et al. 2018). In plants, the microtubule cytoskeleton collaborates with other organelle systems to orchestrate elaborate cellular activities, and the numerous actin-binding kinesin-14B CH and kinesin-14C proteins could coordinate crosstalk among the cytoskeletal and membrane. For example, plant cytokinesis requires phragmoplasts and preprophase bands which are large assemblages of actin, microtubules, and endoplasmic reticulum (Schneider and Persson 2015; Nebenführ and Dixit 2018). Multiple unique kinesins localize to the phragmoplast and/or preprophase band. A. thaliana kinesin-15 members associate with the preprophase band and phragmoplast (Lee et al. 2007; Hiwatashi et al. 2008; Oh et al. 2012; Li et al. 2015; Herrmann et al. 2021), a kinesin-19B ARM localize to preprophase bands (Malcos and Cyr 2011), and kinesin-14D associates with phragmoplasts (Lee et al. 2007). Additionally, the cytokinesis in pollen grains in Arabidopsis thaliana require a plant-specific kinesin-7C action (Oh et al. 2008).
Interphase growth, morphogenesis, and cell wall deposition also require coordination between actin and microtubule cytoskeletal networks (Schneider and Persson 2015; Nebenführ and Dixit 2018). Again, the numerous actin-binding kinesin-14’s could facilitate crosstalk within cells. Members of the kinesin-19B ARM subfamily function in germination, trafficking, and interphase tip growth in root hairs by binding endoplasmic reticulum and microtubules (Eng et al. 2017; Sun et al. 2020; Lan et al. 2023). Malectin-binding kinesin-14B proteins have been implicated in pollen and embryo development (Galindo-Trigo et al. 2020). Members of kinesin-14B KCA subgroup also associate with cytokinetic division planes and mediate actin-based chloroplast positioning in interphase cells (Vanstraelen et al. 2006; Suetsugu et al. 2010; Shen et al. 2015). The kinesin-4/10 FRA1 mediates cell wall formation through vesicle trafficking to the plasma membrane (Ganguly et al. 2020).
Processes Leading to Kinesin Families
All deep phylogenic studies of gene families using genomic data from extant species examine the outcome of multiple rounds of small-scale gene and large-scale whole genome duplication events. Extant genes survived due to positive selection pressure, or they were recently obtained and have not yet been eliminated. Overall, kinesin family expansions reflected genome-wide duplications, hybridizations, and unique evolutionary histories. For example, a monophyletic expansion of kinesin-12/15 occurred in moss (this paper and Shen et al. 2012). Analysis of branching patterns in our phylogenetic trees often showed distinct splits between bryophytes and tracheophytes, in compliance that these two groups are each monophyletic (Su et al. 2021; Wang et al. 2022a).
Despite genome duplications, kinesin-14C and 14D did not expand indicating negative selection on additional copies of these kinesin proteins. Other kinesin subfamilies and groups minimally or rarely expanded (for example, kinesin-1, 6, and 14B KCA). These cases may represent a large ancestral gene family slowly being eliminated with little selection on the remaining single genes. Indeed, the predicted A. thaliana kinesin-1 sequence is only 469 amino acids, while animal kinesin-1 are 900–1000 amino acids in length. However, in other small kinesin families, one or two full-length genes exist in remain in most taxa, which suggests negative selection pressure on multiple gene copies. Copy number variation (CNV) leads to negative fitness due to altered expression levels in animals and fungi (Ionita-Laza et al. 2009; Robinson et al. 2023). In plants, copy number variation has been linked to advantageous traits (e.g., Bai et al. 2016), but it is also likely that a select number of genes are deleterious when duplicated.
Our separation of kinesin-4/10 and 19 into subgroups showed that specific types were lost during land plant evolution, while others were retained. In combination with our result that kinesin-19 is a sister group to kinesin-1, our data suggest an evolutionary process which involved gene duplication followed by sequence changes and neofunctionalization that culminated in distinct kinesin family subtypes that were differentially selected during evolution.
Methods
Data Sources and Kinesin Identification
Kinesins were identified in complete and nearly complete genomes with BUSCO scores over 90%. BUSCO score is a measure of genome completeness based on presence and quality of orthologs common to all eukaryotes (Supplementary Table 1). Most BUSCO scores for genomes in this study were above 90%. However, Ginkgo biloba, Gnetum montanum, and Welwitschia mirabilis BUSCO scores were less than 90% and so additional gymnosperm genomes with higher BUSCO scores were included. Some taxonomically relevant species were excluded based on low BUSCO scores; the absence of a kinesin gene sequence could be due to genome incompleteness.
Proteins sequences with predicted kinesin domains (PFAM 00225) from the 29 genomes were collected for phylogenetic assessment. Sequences that encoded peptides of less than 100 amino acids in length were also removed as they likely represented gene fragments. Two other sequences were less than 200 amino acids in length and may be remnants on its way to deletion (GAQ87587 a kinesin-18 from K. flaccidum and Fb00052318 a kinesin-5 from D. melanogaster). Most all other sequences ranged in length from approximately 300 to 5000 amino acids, and all sequences contained a single motor domain. A total of 1499 kinesin sequences remained and were subsequently used in phylogenetic analyses.
Phylogenetic Analysis
A total sequence alignment of 1499 kinesin domain-containing proteins gathered from 29 species was generated using MUSCLE (muscle3.8.425_i86linux64) with default settings in Aliview (version 2.8). This alignment was tested for phylogenetic relationships using a maximum likelihood method with model selection and ultrafast bootstrap validation in IQtree (version 1.6). The input data contained 1519 protein sequences with 1499 protein sequences that contained kinesin domains, 20 others were recovered from blast searches in 4 gymnosperms but not removed. There were 9391 amino acid sites for full-length alignment, with 1406 constant sites (15%), 5530 parsimony informative sites, and 8032 distinctive patterns. The VT + R10 model gave the most likely tree with a log-likelihood of − 3085081 and total tree length of 3191, with 21% being internal branches. 50 branches were considered too long (> 9.8 default setting) and proteins at these branches were moved to the unassigned by subfamily category (Fig. 2). The presented trees were mid-point rooted in Figtree (version 1.4.4). Initial phylogenetic analyses were also performed using neighbor joining and maximum parsimony methods using default settings in MEGA (version 10).
Additional analyses were performed with a subset of species to evaluate whether taxonomic distribution impacted clade relationships apparent in the master tree containing 29 species. An additional tree (Supplementary Fig. 2) was constructed for a more even sampling per major lineage by including fewer angiosperm and gymnosperm taxa and all sequences from all other taxa. Maximum likelihood analysis of 12 angiosperm taxa and three gymnosperm taxa as roots (Supplementary Fig. 3) was compared to the master tree of all 29 species to ensure key findings within angiosperms were not affected by long branches that appear in some of the algal and bryophyte kinesins. This was compared to the master tree to determine if the slight oversampling of angiosperms affected the position of the deeper nodes and thus the position of early events in kinesin evolution and diversification. No major differences among the three trees were observed. Trees and subtrees presented in figures were taken from the master tree (all 29 species included) and drawn using Figtree (version 1.4).
Furthermore, all 1499 kinesin domain-containing sequences were trimmed using Trimal (version 1.2) with seven different settings of increasing stringency for the presence of gaps (gap threshold parameter) from minimum 10% presence to minimum 80% presence. Trimming progressively narrowed sequences to the motor domains especially at 80% minimum presence. Trees were constructed for all using IQ-TREE (with settings as above). Alignments and resulting trees representing 10% presence and 80% presence minima are presented as Supplementary Fig. 4. These two trees had 1765 and 526 parsimony informative sites, respectively, but produced trees with similar placement of proteins into clades representing the kinesin families in the untrimmed full-length alignment. Kinesin-14C and kinesin 14-D both remained monophyletic and distinct from other kinesin families, and the relative positions of kinesin-14C and kinesin-14D to other families differed in trimmed and untrimmed analyses.
Grouping and Numbering of Kinesin Families and Subfamilies
Clades with high bootstrap support were labeled by kinesin family using a modification the numbering system proposed by Wickstead et al. (2010). Clades were identified by determining the position of known A. thaliana, P. patens proteins, and animal reference sequences. A table of mapped family numbers to A. thaliana is presented in Supplementary Table 2. In some cases, algal and human kinesins appeared as sister to more than one numbered kinesin family and these were thus indicated. Several kinesins with long branches appeared as sister to the well-defined clades with poor bootstrap support, and thus could not be assigned to a numbered clade as the placement was likely the result of long branch attraction. These kinesins were declared as unassigned by family in Fig. 2 and scored in terms of total kinesins per species. Another small group of kinesins had strong support as sister to the kinesin-14A and -14B clades. Kinesin-14C and -14D subfamilies were not strongly supported to be monophyletic with kinesin-14A and -14B using this method, but rather appeared as separate distinct families. The position and possible origin of these kinesins are discussed in this work. A linear version of this tree showing bootstrap support values and branch lengths along with protein sequence accession numbers is presented in Supplementary Fig. 1. A FASTA format protein sequence file with all protein accession numbers is presented as Supplementary File 1.
Correlation and Phylogenetic Independent Contrast Comparative Analysis
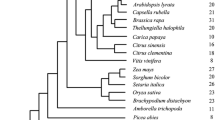
Across 29 species, the total number of kinesins was compared to the C-value (Kew database), number of haploid chromosomes, and protein-coding genes determined from published literature. This was initially done using Pearson correlation, comparing test statistics, and p values between Kinesins (K), C-value (C), and chromosome number (Chr), and number of genes (genes) were performed in Excel. However these data could represent correlation based on both functional adaptation and phylogenetic relatedness of species. To remove the covariance caused by species relatedness, a phylogenetically independent contrast was performed using the method of Felsenstein (1985) implemented through the R package “Analyses of Phylogenetics and Evolution” (ape; Paradis et al. 2004). This method provided correction for the non-independence of data points (species) which is assumed in linear correlations. A species tree with relationships and evolutionary distances of all taxa (or substitutes in the same genus for Sphagnum, Marchantia, Zea, and Oryza as indicated with asterisks) was obtained from the TimeTree 5 database (Supplementary Fig. 5) (Kumar et al. 2022). This was exported in newick format and loaded into R using the ape::read.tree function. A phylogenetic independence vector was made using the ape::pic function for each measured value set (number of kinesins K; nuclear DNA quantity C; number of genes, number of chromosomes) and the species tree. These vectors were then compared pairwise using Pearson’s (product moment) correlation to determine if a correlation between variables occurred independently of their taxonomic relationship.
References
Ali I, Yang WC (2020) The functions of kinesin and kinesin-related proteins in eukaryotes. Cell Adh Migr 14(1):139–152. https://doi.org/10.1080/19336918.2020.1810939
Baack EJ, Whitney KD, Rieseberg LH (2005) Hybridization and genome size evolution: timing and magnitude of nuclear DNA content increases in Helianthus homoploid hybrid species. New Phytol 167(2):623–630. https://doi.org/10.1111/j.1469-8137.2005.01433.x
Bai Z, Chen J, Liao Y et al (2016) The impact and origin of copy number variations in the Oryza species. BMC Genomics 17:261. https://doi.org/10.1186/s12864-016-2589-2
Bowles AMC, Williamson CJ, Williams TA, Lenton TM, Donoghue PCJ (2023) The origin and early evolution of plants. Trends Plant Sci 28(3):312–329. https://doi.org/10.1016/j.tplants.2022.09.009
Brawley SH, Blouin NA, Ficko-Blean E, Wheeler GL, Lohr M, Goodson HV, Jenkins JW, Blaby-Haas CE, Helliwell KE, Chan CX, Marriage TN, Bhattacharya D, Klein AS, Badis Y, Brodie J, Cao Y, Collén J, Dittami SM, Gachon CMM, Green BR, Karpowicz SJ, Kim JW, Kudahl UJ, Lin S, Michel G, Mittag M, Olson BJSC, Pangilinan JL, Peng Y, Qiu H, Shu S, Singer JT, Smith AG, Sprecher BN, Wagner V, Wang W, Wang ZY, Yan J, Yarish C, Zäuner-Riek S, Zhuang Y, Zou Y, Lindquist EA, Grimwood J, Barry KW, Rokhsar DS, Schmutz J, Stiller JW, Grossman AR, Prochnik SE (2017) Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta). Proc Natl Acad Sci USA 114(31):E6361–E6370. https://doi.org/10.1073/pnas.1703088114
Buschmann H, Zachgo S (2016) The evolution of cell division: from streptophyte algae to land plants. Trends Plant Sci 21(10):872–883. https://doi.org/10.1016/j.tplants.2016.07.004
Dagenbach EM, Endow SA (2004) A new kinesin tree. J Cell Sci 117(Pt 1):3–7. https://doi.org/10.1242/jcs.00875
Donoghue P, Paps J (2020) Plant evolution: assembling land plants. Curr Biol 30(2):R81–R83. https://doi.org/10.1016/j.cub.2019.11.084
Eng RC, Halat LS, Livingston SJ, Sakai T, Motose H, Wasteneys GO (2017) The ARM domain of ARMADILLO-REPEAT KINESIN 1 is not required for microtubule catastrophe but can negatively regulate NIMA-RELATED KINASE 6 in Arabidopsis thaliana. Plant Cell Physiol 58(8):1350–1363. https://doi.org/10.1093/pcp/pcx070
Felsenstein J (1985) Phylogenies and the comparative method. Am Nat 125(1):1–15. https://doi.org/10.1086/284325
Galindo-Trigo S, Grand TM, Voigt CA, Smith LM (2020) A malectin domain kinesin functions in pollen and seed development in Arabidopsis. J Exp Bot 71(6):1828–1841. https://doi.org/10.1093/jxb/eraa023
Ganguly A, Zhu C, Chen W, Dixit R (2020) FRA1 kinesin modulates the lateral stability of cortical microtubules through cellulose synthase-microtubule uncoupling proteins. Plant Cell 32(8):2508–2524. https://doi.org/10.1105/tpc.19.00700
Gicking AM, Swentowsky KW, Dawe RK, Qiu W (2018) Functional diversification of the kinesin-14 family in land plants. FEBS Lett 592(12):1918–1928. https://doi.org/10.1002/1873-3468.13094
Hamada T (2007) Microtubule-associated proteins in higher plants. J Plant Res 120(1):79–98. https://doi.org/10.1007/s10265-006-0057-9
Herrmann A, Livanos P, Zimmermann S, Berendzen K, Rohr L, Lipka E, Müller S (2021) KINESIN-12E regulates metaphase spindle flux and helps control spindle size in Arabidopsis. Plant Cell 33(1):27–43. https://doi.org/10.1093/plcell/koaa003
Hirokawa N, Takemura R (2004) Kinesin superfamily proteins and their various functions and dynamics. Exp Cell Res 301(1):50–59. https://doi.org/10.1016/j.yexcr.2004.08.010
Hirokawa N, Noda Y, Tanaka Y, Niwa S (2009) Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol 10(10):682–696. https://doi.org/10.1038/nrm2774
Hiwatashi Y, Obara M, Sato Y, Fujita T, Murata T, Hasebe M (2008) Kinesins are indispensable for interdigitation of phragmoplast microtubules in the moss Physcomitrella patens. Plant Cell 20(11):3094–3106. https://doi.org/10.1105/tpc.108.061705
Hodges ME, Wickstead B, Gull K, Langdale JA (2012) The evolution of land plant cilia. New Phytol 195(3):526–540. https://doi.org/10.1111/j.1469-8137.2012.04197.x
Hufford MB, Seetharam AS, Woodhouse MR, Chougule KM, Ou S, Liu J, Ricci WA, Guo T, Olson A, Qiu Y, Della Coletta R, Tittes S, Hudson AI, Marand AP, Wei S, Lu Z, Wang B, Tello-Ruiz MK, Piri RD, Wang N, Kim DW, Zeng Y, O’Connor CH, Li X, Gilbert AM, Baggs E, Krasileva KV, Portwood JL 2nd, Cannon EKS, Andorf CM, Manchanda N, Snodgrass SJ, Hufnagel DE, Jiang Q, Pedersen S, Syring ML, Kudrna DA, Llaca V, Fengler K, Schmitz RJ, Ross-Ibarra J, Yu J, Gent JI, Hirsch CN, Ware D, Dawe RK (2021) De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 373(6555):655–662. https://doi.org/10.1126/science.abg5289
Ionita-Laza I, Rogers AJ, Lange C, Raby BA, Lee C (2009) Genetic association analysis of copy-number variation (CNV) in human disease pathogenesis Genomics. Elsevier, Amsterdam
Jonsson E, Yamada M, Vale RD, Goshima G (2015) Clustering of a kinesin-14 motor enables processive retrograde microtubule-based transport in plants. Nat Plants 1(7):15087. https://doi.org/10.1038/NPLANTS.2015.87
Jumper J, Evans R, Pritzel A et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. https://doi.org/10.1038/s41586-021-03819-2
Kato Y, Miyakawa T, Tanokura M (2018) Overview of the mechanism of cytoskeletal motors based on structure. Biophys Rev 10(2):571–581. https://doi.org/10.1007/s12551-017-0368-1
Kollmar M (2016) Fine-tuning motile cilia and flagella: evolution of the dynein motor proteins from plants to humans at high resolution. Mol Biol Evol 33(12):3249–3267. https://doi.org/10.1093/molbev/msw213
Kumar S, Suleski M, Craig JM, Kasprowicz AE, Sanderford M, Li M, Stecher G, Hedges SB (2022) TimeTree 5: an expanded resource for species divergence times. Mol Biol Evol 39(8):msac174. https://doi.org/10.1093/molbev/msac174
Lan M, Kang E, Liu X, Fu Y, Zhu L (2023) Stable ARMADILLO REPEAT KINESIN 2 in light inhibits hypocotyl elongation and facilitates light-induced cortical microtubule reorientation in Arabidopsis. J Exp Bot 74(3):800–816. https://doi.org/10.1093/jxb/erac473
Lang D, Ullrich KK, Murat F, Fuchs J, Jenkins J, Haas FB, Piednoel M, Gundlach H, Van Bel M, Meyberg R, Vives C, Morata J, Symeonidi A, Hiss M, Muchero W, Kamisugi Y, Saleh O, Blanc G, Decker EL, van Gessel N, Grimwood J, Hayes RD, Graham SW, Gunter LE, McDaniel SF, Hoernstein SNW, Larsson A, Li FW, Perroud PF, Phillips J, Ranjan P, Rokshar DS, Rothfels CJ, Schneider L, Shu S, Stevenson DW, Thümmler F, Tillich M, Villarreal Aguilar JC, Widiez T, Wong GK, Wymore A, Zhang Y, Zimmer AD, Quatrano RS, Mayer KFX, Goodstein D, Casacuberta JM, Vandepoele K, Reski R, Cuming AC, Tuskan GA, Maumus F, Salse J, Schmutz J, Rensing SA (2018) The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J 93(3):515–533. https://doi.org/10.1111/tpj.13801
Lawrence CJ, Dawe RK, Christie KR, Cleveland DW, Dawson SC, Endow SA, Goldstein LS, Goodson HV, Hirokawa N, Howard J, Malmberg RL, McIntosh JR, Miki H, Mitchison TJ, Okada Y, Reddy AS, Saxton WM, Schliwa M, Scholey JM, Vale RD, Walczak CE, Wordeman L (2004) A standardized kinesin nomenclature. J Cell Biol 167(1):19–22. https://doi.org/10.1083/jcb.200408113
Lee YR, Liu B (2004) Cytoskeletal motors in Arabidopsis. Sixty-one kinesins and seventeen myosins. Plant Physiol. https://doi.org/10.1104/pp.104.052621
Lee YR, Li Y, Liu B (2007) Two Arabidopsis phragmoplast-associated kinesins play a critical role in cytokinesis during male gametogenesis. Plant Cell 19(8):2595–2605. https://doi.org/10.1105/tpc.107.050716
Lee YR, Qiu W, Liu B (2015) Kinesin motors in plants: from subcellular dynamics to motility regulation. Curr Opin Plant Biol 28:120–126. https://doi.org/10.1016/j.pbi.2015.10.003
Li Y, Deng Z, Kamisugi Y, Chen Z, Wang J, Han X, Wei Y, He H, Terzaghi W, Cove DJ, Cuming AC, Chen H (2021) A minus-end directed kinesin motor directs gravitropism in Physcomitrella patens. Nat Commun 12:4470. https://doi.org/10.1038/s41467-021-24546-2
Li J, Xu Y, Chong K (2012) The novel functions of kinesin motor proteins in plants. Protoplasma. https://doi.org/10.1007/s00709-011-0357-3
Li S, Sun T, Ren H (2015) The functions of the cytoskeleton and associated proteins during mitosis and cytokinesis in plant cells. Front Plant Sci 27(6):282. https://doi.org/10.3389/fpls.2015.00282
Liu B, Lee YJ (2022) Spindle assembly and mitosis in plants. Annu Rev Plant Biol 20(73):227–254. https://doi.org/10.1146/annurev-arplant-070721-084258
Liu Y, Wang J, Ge W, Wang Z, Li Y, Yang N, Sun S, Zhang L, Wang X (2017) Two highly similar poplar paleo-subgenomes suggest an autotetraploid ancestor of Salicaceae Plants. Front Plant Sci 12(8):571. https://doi.org/10.3389/fpls.2017.00571
Lucas J, Geisler M (2022) Sequential loss of dynein sequences precedes complete loss in land plants. Plant Physiol 189(3):1237–1240. https://doi.org/10.1093/plphys/kiac151
Malcos JL, Cyr RJ (2011) An ungrouped plant kinesin accumulates at the preprophase band in a cell cycle-dependent manner. Cytoskeleton (hoboken) 68(4):247–258. https://doi.org/10.1002/cm.20508
Miki H, Okada Y, Hirokawa N (2005) Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol 15(9):467–476. https://doi.org/10.1016/j.tcb.2005.07.006
Nebenführ A, Dixit R (2018) Kinesins and myosins: molecular motors that coordinate cellular functions in plants. Annu Rev Plant Biol 29(69):329–361. https://doi.org/10.1146/annurev-arplant-042817-040024
Oh SA, Bourdon V, Das’ Pal M, Dickinson H, Twell D (2008) Arabidopsis kinesins HINKEL and TETRASPORE act redundantly to control cell plate expansion during cytokinesis in the male gametophyte. Mol Plant 1(5):794–799
Oh SA, Allen T, Kim GJ, Sidorova A, Borg M, Park SK, Twell D (2012) Arabidopsis fused kinase and the Kinesin-12 subfamily constitute a signalling module required for phragmoplast expansion. Plant J 72(2):308–319. https://doi.org/10.1111/j.1365-313X.2012.05077.x
Oppenheimer DG, Pollock MA, Vacik J, Szymanski DB, Ericson B, Feldmann K, Marks MD (1997) Essential role of a kinesin-like protein in Arabidopsis trichome morphogenesis. Proc Natl Acad Sci USA 94(12):6261–6266. https://doi.org/10.1073/pnas.94.12.6261
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20(2):289–290. https://doi.org/10.1093/bioinformatics/btg412
Pellicer J, Leitch IJ (2020) The Plant DNA C-values database (release 7.1): an updated online repository of plant genome size data for comparative studies. New Phytol 226:301–305. https://doi.org/10.1111/nph.16261
Reddy AS, Day IS (2001) Kinesins in the Arabidopsis genome: a comparative analysis among eukaryotes. BMC Genomics 2:2. https://doi.org/10.1186/1471-2164-2-2
Reddy VS, Reddy AS (1999) A plant calmodulin-binding motor is part kinesin and part myosin. Bioinformatics 15(12):1055–1057. https://doi.org/10.1093/bioinformatics/15.12.1055
Richardson DN, Simmons MP, Reddy AS (2006) Comprehensive comparative analysis of kinesins in photosynthetic eukaryotes. BMC Genomics 31(7):18. https://doi.org/10.1186/1471-2164-7-18
Robinson D, Vanacloig-Pedros E, Cai R, Place M, Hose J, Gasch AP (2023) Gene by enviroment interactions influence the fitness cost of gene copynumber variation in yeast. G3: Genes Genomes Genet. https://doi.org/10.1093/g3journal/jkad159
Schneider R, Persson S (2015) Connecting two arrays: the emerging role of actin-microtubule cross-linking motor proteins. Front Plant Sci 2(6):415. https://doi.org/10.3389/fpls.2015.00415
Scholey JM (2003) Intraflagellar transport. Annu Rev Cell Dev Biol 19:423–443. https://doi.org/10.1146/annurev.cellbio.19.111401.091318
She ZY, Yang WX (2017) Molecular mechanisms of kinesin-14 motors in spindle assembly and chromosome segregation. J Cell Sci 130(13):2097–2110. https://doi.org/10.1242/jcs.200261
Shen Z, Collatos AR, Bibeau JP, Furt F, Vidali L (2012) Phylogenetic analysis of the Kinesin superfamily from Physcomitrella. Front Plant Sci 16(3):230. https://doi.org/10.3389/fpls.2012.00230
Shen Z, Liu YC, Bibeau JP, Lemoi KP, Tüzel E, Vidali L (2015) The kinesin-like proteins, KAC1/2, regulate actin dynamics underlying chloroplast light-avoidance in Physcomitrella patens. J Integr Plant Biol 57(1):106–119. https://doi.org/10.1111/jipb.12303
Song H, Golovkin M, Reddy AS, Endow SA (1997) In vitro motility of AtKCBP, a calmodulin-binding kinesin protein of Arabidopsis. Proc Natl Acad Sci USA 94(1):322–327. https://doi.org/10.1073/pnas.94.1.322
Su D, Yang L, Shi X, Ma X, Zhou X, Hedges SB, Zhong B (2021) Large-scale phylogenomic analyses reveal the monophyly of bryophytes and neoproterozoic origin of land plants. Mol Biol Evol 38(8):3332–3344. https://doi.org/10.1093/molbev/msab106
Suetsugu N, Yamada N, Kagawa T, Yonekura H, Uyeda TQ, Kadota A, Wada M (2010) Two kinesin-like proteins mediate actin-based chloroplast movement in Arabidopsis thaliana. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.0912773107
Sun J, Zhang M, Qi X et al (2020) Armadillo-repeat kinesin1 interacts with Arabidopsis atlastin RHD3 to move ER with plus-end of microtubules. Nat Commun 11:5510. https://doi.org/10.1038/s41467-020-19343-2
Tanz SK, Castleden I, Hooper CM, Vacher M, Small I, Millar HA (2013) SUBA3: a database for integrating experimentation and prediction to define the SUBcellular location of proteins in Arabidopsis. Nucleic Acids Res. https://doi.org/10.1093/nar/gks1151
Tseng KF, Wang P, Lee YJ, Bowen J, Gicking AM, Guo L, Liu B, Qiu W (2018) The preprophase band-associated kinesin-14 OsKCH2 is a processive minus-end-directed microtubule motor. Nat Commun 9(1):1067. https://doi.org/10.1038/s41467-018-03480-w
Vale RD (2003) The molecular motor toolbox for intracellular transport. Cell 112(4):467–480. https://doi.org/10.1016/s0092-8674(03)00111-9
Vanstraelen M, Van Damme D, De Rycke R, Mylle E, Inzé D, Geelen D (2006) Cell cycle-dependent targeting of a kinesin at the plasma membrane demarcates the division site in plant cells. Curr Biol 16(3):308–314. https://doi.org/10.1016/j.cub.2005.12.035
Vicente JJ, Wordeman L (2015) Mitosis, microtubule dynamics and the evolution of kinesins. Exp Cell Res. https://doi.org/10.1016/j.yexcr.2015.02.010
Vinogradova MV, Malanina GG, Reddy AS, Fletterick RJ (2009) Structure of the complex of a mitotic kinesin with its calcium binding regulator. Proc Natl Acad Sci USA 106(20):8175–8179. https://doi.org/10.1073/pnas.0811131106
Wang W, Cao L, Wang C, Gigant B, Knossow M (2015) Kinesin, 30 years later: recent insights from structural studies. Prot Sci 24:1047–1056. https://doi.org/10.1002/pro.2697
Wang J, Yu J, Sun P, Li C, Song X, Lei T, Li Y, Yuan J, Sun S, Ding H, Duan X, Shen S, Shen Y, Li J, Meng F, Xie Y, Wang J, Hou Y, Zhang J, Zhang X, Li XQ, Paterson AH, Wang X (2020) Paleo-polyploidization in Lycophytes. Genom Proteom Bioinform. https://doi.org/10.1016/j.gpb.2020.10.002
Wang QH, Zhang J, Liu Y, Jia Y, Jiao YN, Xu B, Chen ZD (2022a) Diversity, phylogeny, and adaptation of bryophytes: insights from genomic and transcriptomic data. J Exp Bot 73(13):4306–4322. https://doi.org/10.1093/jxb/erac127
Wang J, Zhang L, Wang J, Hao Y, Xiao Q, Teng J, Shen S, Zhang Y, Feng Y, Bao S, Li Y, Yan Z, Wei C, Wang L, Wang J (2022b) Conversion between duplicated genes generated by polyploidization contributes to the divergence of poplar and willow. BMC Plant Biol 22(1):298. https://doi.org/10.1186/s12870-022-03684-9
Welburn JP (2013) The molecular basis for kinesin functional specificity during mitosis. Cytoskeleton (hoboken) 70(9):476–493. https://doi.org/10.1002/cm.21135
Wickstead B, Gull K (2006) A “holistic” kinesin phylogeny reveals new kinesin families and predicts protein functions. Mol Biol Cell 17(4):1734–1743. https://doi.org/10.1091/mbc.e05-11-1090
Wickstead B, Gull K (2007) Dyneins across eukaryotes: a comparative genomic analysis. Traffic 8(12):1708–1721. https://doi.org/10.1111/j.1600-0854.2007.00646.x
Wickstead B, Gull K, Richards TA (2010) Patterns of kinesin evolution reveal a complex ancestral eukaryote with a multifunctional cytoskeleton. BMC Evol Biol 27(10):110. https://doi.org/10.1186/1471-2148-10-110
Yamada M, Goshima G (2017) Mitotic spindle assembly in land plants: molecules and mechanisms. Biology (Basel) 6(1):6. https://doi.org/10.3390/biology6010006
Yamada M, Tanaka-Takiguchi Y, Hayashi M, Nishina M, Goshima G (2017) Multiple kinesin-14 family members drive microtubule minus end-directed transport in plant cells. J Cell Biol 216(6):1705–1714. https://doi.org/10.1083/jcb.201610065
Ye Z, Yuan Z, Xu H, Pan L, Chen J, Gatera A, Uzair M, Xu D (2022) Genome-wide identification and expression analysis of kinesin family in Barley (Hordeum vulgare). Genes (Basel) 13(12):2376. https://doi.org/10.3390/genes13122376
Yokoyama R, O’toole E, Ghosh S, Mitchell DR (2004) Regulation of flagellar dynein activity by a central pair kinesin. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.0406817101
Zhu C, Dixit R (2012) Functions of the Arabidopsis kinesin superfamily of microtubule-based motor proteins. Protoplasma 249(4):887–899. https://doi.org/10.1007/s00709-011-0343-9
Acknowledgements
Funding and release time for this project was provided to Lucas by the University of Wisconsin—Oshkosh Faculty Development Program, PERC Program, and College of Letters and Science.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Kerry Geiler-Samerotte.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Lucas, J., Geisler, M. Plant Kinesin Repertoires Expand with New Domain Architecture and Contract with the Loss of Flagella. J Mol Evol 92, 381–401 (2024). https://doi.org/10.1007/s00239-024-10178-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-024-10178-9