
Abstract
Green plants and algae have plastids of which chloroplasts are most common. Chloroplasts contain their own genomes, which are relatively stable. Approximately 120 genes are in plastomes and mostly encode proteins used in photosynthesis, protein synthesis, and DNA replication. They provide valuable resources to study the plastogenomics, which helps in understanding the process of evolution in one of the oldest living plants and their interrelationship with angiosperms. Comparing the components of plastomes with those of whole genomes or proteome sequences provides important insights into plant phylogeny and evolution. In the recent years, phylogenomics based on chloroplast genomes has demonstrated numerous advantages in plant phylogenetics. Chloroplast phylogenomics can provide a framework for assessing the impact of reticulate evolution in the early evolution of ferns, especially now the more nuclear data is becoming available. In this review, an attempt is made to study plastid genomes and comparisons of gene content, gene arrangement, gene expression, and gene sequences between extant among various fern species.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Ferns are the closest sister group to all seed plants, yet little is known about their genomes other than that they are generally colossal. Tree ferns first occurred in the Late Triassic. Ferns as a whole include lineages that diverged from one another prior to the divergence of the major seed plant clades. In a broad sense, ferns include four main clades such as psilotoids (whisk ferns) + ophioglossoids, equisetoids (horsetails), marattioids, and leptosporangiates (Sessa et al. 2014). As our ability to infer evolutionary trees has improved, classifications aimed at recognizing natural groups have become increasingly predictive and stable (IPPG 2016). Most broad analyses of green plant relationships based on nuclear gene sequence data have relied largely on 18S/26S rDNA sequences (Soltis et al. 1999) although recent analyses have employed numerous nuclear genes (review Kumar et al., this volume). Most studies that have used nuclear markers were based on only a single locus and/or were at relatively shallow phylogenetic depths, with the goal of understanding reticulate patterns of evolution caused by hybridization and allopolyploidy (Chen et al. 2014). Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae) (Ruhfel et al. 2014).
2 Methodology
Chloroplasts have their own DNA and ribosomes because of which they are able to synthesize some of their proteins and replicate independent of the nucleus. Plastids also contain 70s ribosomes that translate the mRNA produced from these genes.
Chloroplast genome sequences are most valuable for understanding plant evolution and phylogeny (Daniell et al. 2016). The utilities of whole chloroplast genomes, or plastomes, in fern phylogenetics have been documented in earlier studies (Wolf et al. 2003; Der 2010; Lu et al. 2015).
2.1 Plastid Genome Sequencing
The availability of next-generation sequencing (NGS) platforms (Alanazi et al. 2021) and bioinformatic tools (Langmead and Nellore 2018) has a great impact on understanding of plastogenomics or chloroplast (cp) genomics and its application in biotechnology (Daniell et al. 2016). In studies conducted before the availability of high-throughput methods, isolated chloroplasts were used for the amplification of the entire chloroplast genome by rolling circle amplification (Bausher et al. 2006). Eleven chloroplast genes encode ndh subunits, which are involved in photosynthesis. The ndh proteins assemble into the photosystem I complex to mediate cyclic electron transport in chloroplasts (Munekage et al. 2004) and facilitate chlororespiration (Peltier and Cournac 2002).
Plastomes of pteridophytes (spore-bearing vascular plants) are mined from NCBI organelle genome database. Plant plastomеs possess a quadripartite structure composed of large single-copy (LSC) and small single-copy (SSC) regions divided by two parts of inverted repeat (IR) (Olejniczak et al. 2016). Olejniczak et al. (2016) reported that plastome size of higher plants is usually around 150,000 bp in length and comprises approximately 120–130 genes, among which about 75 genes encode proteins of photosystems I and II, as well as for other proteins, involved in photosynthesis (Daniell et al. 2016), while other genes encode ribosomal RNA and proteins and transfer RNA. IRs are usually regarded as the most stable part of the plastome (Olejniczak et al. 2016). Logacheva et al. (2017) suggested that IRs typically range in size from 15 to 30 kbp and contain a core set of genes consisting of four rRNA genes (4.5S, 5S, 16S, and 23S rRNA) and five tRNA genes (trnAUG, trnI-GAU, trnN-GUU, trnR-ACG, and trnV-GAC). Plastome structure, gene contents, and GC contents are analyzed by the in-house developed Python code (see Kwon et al. 2020). Intronic features including presence/absence, length, and intron phases are analyzed manually in the annotated information in NCBI. Peng et al. (2020) sequenced complete chloroplast genome of the fern Asplenium tenerum (Aspleniaceae). The complete plastid genome of A. tenerum (GenBank accession no. MT700551) is 154,628 bp in length with an overall GC content of 40.82%. The genome displays a typical quadripartite structure consisting a small single-copy region (SSC; 21,374 b p), a large single-copy region (LSC; 81,205 bp), and a pair of identical inverted repeats (IR; 26,117 bp). Peng et al. (2020) suggested that the genome encoded a non-redundant gene set similar to that of the other Aspleniaceae plastomes, including 84 protein-coding genes, 8 rRNA genes, and 34 tRNA genes. Nine protein-coding genes (ndhA, rpl2, rpl16, petD, petB, rpoC1, atpF, rps16, and ndhB) were disrupted by one intron, and three genes (clpP, rps12, and ycf3) by two, including the trans-spliced rps12 gene. Logacheva et al. (2017) characterized plastid genomes of three species of Dryopteris, using sequencing of chloroplast DNA-enriched samples, and performed comparative analysis with available plastomes of Polypodiales, the most species-rich group of ferns. Logacheva et al. (2017) determined the marked conservation of gene content and relative evolution rate of genes and intergenic spacers in the IRs of Polypodiales. Faster evolution of the four intergenic regions had been demonstrated (trnA-orf42, rrn16-rps12, rps7-psbA, and ycf2-trnN).
The chloroplast translation initiation factor 1 (infA) is a homolog of the essential gene infA in Escherichia coli (Millen et al. 2001). This gene initiates translation in collaboration with two nuclear-encoded initiation factors to mediate interactions between mRNA, ribosomes, and initiator tRNA-Met (Millen et al. 2001).
Another evolutionary event is horizontal gene transfer (HGT) affecting the plastome structure (Logacheva et al. 2017). HGT between nucleus, mitochondria, and plastids has been shown to occur at a high rate and contributed significantly to the plant genome evolution by relocating and refashioning of the genes and consequently contributing to genetic diversity.
2.2 Simple Sequence Repeats (SSRs)
Microsatellites, or simple sequence repeats (SSRs), are ubiquitous throughout both the coding and non-coding regions of all eukaryotic genomes (Powell et al. 1996). Recently, they have been found and characterized within protein-coding genes and their untranslated regions (UTRs). Several studies on possible SSR functions have been undertaken (Li et al. 2004).
SSRs derived from EST (expressed sequence tag) libraries (EST-SSRs) show a higher rate of interspecies transferability than genomic SSRs, which reside in the non-coding region of the genome (Zwenger et al. 2010). Most of the SSR variation are functionally neutral, but the variations in coding regions of SSR may have functional significance, including chromosomal organization, DNA structure, protein binding, gene transcription, and translation (Li et al. 2002, 2004) which provides the basis for rapid evolution (Parisod et al. 2010).
Li et al. (2004) reviewed the SSR distributions within expressed sequence tags (ESTs) and genes including protein coding, 3′-UTRs and 5′-UTRs, and introns and discussed the consequences of SSR repeat-number changes in those regions of both prokaryotes and eukaryotes. Li et al. (2004) reported that substantial data indicates that SSR expansions and/or contractions in protein-coding regions can lead to a gain or loss of gene function via frameshift mutation or expanded toxic mRNA. They further said that SSR variations in 5′-UTRs could regulate gene expression by affecting transcription and translation and the SSR expansions in the 3′-UTRs cause transcription slippage and produce expanded mRNA, which can be accumulated as nuclear foci and which can disrupt splicing and, possibly, disrupt other cellular function.
2.3 Transcriptional Regulation
The regulation of transcription, that is, the synthesis of messenger RNA from a genomic DNA template, plays a crucial role in plant development. According to Lang et al. (2010), transcriptional regulation is primarily achieved by transcription-associated proteins (TAPs, comprising transcription factors [TFs] and other transcriptional regulators [TRs]), which control gene regulatory networks. Evolutionary retention of duplicated genes encoding transcription-associated proteins (TAPs, comprising transcription factors and other transcriptional regulators) has been hypothesized to be positively correlated with increasing morphological complexity and paleopolyploidizations, especially within the plant kingdom.
2.4 Posttranscriptional RNA Processing
Over the past 23 years, it has been well documented that RNAs transcribed from most eukaryotic genes can undergo a variety of posttranscriptional RNA processing events (splicing, capping, polyadenylation) that are required to convert RNA precursors into mature RNA species (Yoshinaga et al. 1996, see review Gott and Emeson (2000). Gott and Emeson (2000) suggested that the term RNA editing describes numerous cellular processes that result in the modification of RNA sequences differing from that designated by their DNA (or RNA) templates. The RNA sequence revisions, which include both the insertion and deletion of nucleotides and the conversion of one base to another, involve a wide range of largely unrelated mechanisms (Gott and Emeson 2000). Such mechanisms affect mRNAs, tRNAs, rRNAs, and 7 SLRNA (Ben-Shlomo et al. 1999) which in turn can alter the function or coding potential of the modified transcripts (see review Du et al. 2020).
The majority of the RNA-editing events that have been identified thus far involve changes in mRNA sequences and result in the production of altered protein products. Creation of new start and stop codons by uridine insertion and cytidine to uridine (C-to-U) conversions has been observed in plant organelles (Gott and Emeson 2000). Stop codons are also subject to removal by U-to-C changes in plants, most frequently in hornworts (Yoshinaga et al. 1996). “Silent” codon changes are also observed, but more often editing creates codons for highly conserved or functionally essential amino acids (Gott and Emeson 2000).
Labiak and Karol (2017) used next-generation sequencing methods to study changes in gene composition, plastome architecture, and putative RNA-editing sites. Although the rapid development of high-throughput sequencing technology has led to an explosion of plastome sequences, annotation remains a significant bottleneck for plastomes. In the absence of cDNA, the annotation of RNA editing in plastomes must be done manually. However, as compared to manual annotation, Robison and Wolf (2019) developed a tool ReFernment which offers a greater speed and accuracy for annotating RNA-editing sites. This software should be especially useful for researchers generating large numbers of plastome sequences for taxa with high levels of RNA editing. Likewise, Qu et al. (2019) introduced Plastid Genome Annotator (PGA), a standalone command line tool that can perform rapid, accurate, and flexible batch annotation of newly generated target plastomes based on well-annotated reference plastomes. PGA accurately identifies gene and intron boundaries as well as intron loss. PGA uses reference plastomes as the query and unannotated target plastomes as the subject to locate genes, which Qu et al. (2019) referred to as the reverse query-subject basic local alignment search tool (BLAST) (Altschul et al. 1990) search approach. BLAST was proposed by Altschul et al. (1990) as a new approach to rapid sequence comparison, and it directly approximates alignments that optimize a measure of local similarity, the maximal segment pair (MSP) score.
2.5 Plastid RNA Editing
The term RNA editing was first coined by Benne and colleagues (Benne et al. 1986) to describe the insertion of uridines into the cytochrome oxidase subunit II mRNA in kinetoplasts of Trypanosoma brucei and Crithidia fasciculata. Gott and Emeson (2000) said that RNA editing can be broadly defined as any site-specific alteration in an RNA sequence that could have been copied from the template, excluding changes due to processes such as RNA splicing and polyadenylation. They further reported that changes in gene expression attributed to editing have been described in organisms from unicellular protozoa to man and can affect the mRNAs, tRNAs, and rRNAs present in all cellular compartments. However, very little is known about intrageneric variation in frequency of plant RNA editing, and almost no study has been conducted in ferns as reported by Fauskee et al. (2021).
Recent studies of plant RNA editing have demonstrated that the number of editing sites can vary widely among large taxonomic groups. Grosche et al. (2012) suggested that RNA editing is a posttranscriptional process that results in modifications of ribonucleotides at specific locations. Thus, RNA editing acts upon transcripts from mitochondrial, nuclear, and chloroplast genomes.
Replacing Cytidine-to-Uridine (C-to-U) during RNA editing, is a process for converting a specific nucleotide of RNA in organellar genomes e.g. mitochondria, and plastids, throughout land plants but U to C is used less frequently. (Shikanai 2006; Du et al. 2020). In most cases, RNA editing alters translated amino acids or creates new start codons. Spike moss genus Selaginella (lycophytes), has the highest frequency of RNA editing. It has been used as a model to test the effects of extreme RNA editing on phylogenetic reconstruction (Du et al. 2020). They predicted the C-to-U RNA-editing sites in coding regions of 18 Selaginella plastomes and reconstructed the phylogenetic relationships within Selaginella based on 3 data set pairs consisting of plastome or RNA-edited coding sequences, first and second codon positions, and translated amino acid sequences, respectively. Du et al. (2020) reported that the numbers of RNA-editing sites in plastomes were highly correlated with the GC content of first and second codon positions, but not correlated with the GC content of plastomes as a whole.
Contrast phylogenetic analyses showed that there were substantial differences (e.g., the placement of clade B in Selaginella) between the phylogenies generated by the plastome and RNA-edited data sets. This empirical study provides evidence that extreme C-to-U RNA editing in the coding regions of organellar genomes alters the sequences used for phylogenetic reconstruction and might even confound phylogenetic reconstruction.
Shikanai (2006) wrote further that to specify the site of editing, the cis-element adjacent to the editing site functions as a binding site for the trans-acting factor. Genetic approaches using Arabidopsis thaliana have clarified that a member of the protein family with pentatricopeptide repeat (PPR) motifs is essential for RNA editing to generate a translational initiation codon of the chloroplast ndhD gene. The PPR motif is a highly degenerate unit of 35 amino acids and appears as tandem repeats in proteins that are involved in RNA maturation steps in mitochondria and plastids. The Arabidopsis genome encodes approximately 450 members of the PPR family, some of which possibly function as trans-acting factors binding the cis-elements of the RNA-editing sites to facilitate access of an unidentified RNA-editing enzyme (Shikanai 2006).
RNA editing can alter individual nucleotides in primary transcripts, which can cause the amino acids encoded by edited RNA to deviate from the ones predicted from the DNA template (see Jiang et al. 2012). Technique of bioinformatics is used to analyze the effect of editing events on protein secondary and three-dimensional structures. Example of cotton is given here to show comparison of RNA posttranscriptional editing in vascular plants and seed plants. Jiang et al. (2012) found that 21 editing sites in cotton chloroplast transcripts can affect protein secondary structures and 7 editing sites can alter three-dimensional protein structures. These results imply that 24 editing sites in seed plant cotton (all these editing sites were C-to-U conversion) may play an important role in their protein structures and functions (Jiang et al. 2012). As reported by Chen et al. (2011), C-to-U changes are most common in seed plants.
Grosche et al. (2012) further reported that in chloroplasts, single-nucleotide conversions in mRNAs via RNA editing occur at different frequencies across the plant kingdom. These range from several hundred edited sites in some mosses and ferns to lower frequencies in seed plants and the complete lack of RNA editing in the liverwort Marchantia polymorpha. RNA-editing sites have imbalanced distribution in genes, and most of them may function by changing protein structure or interaction (Chen et al. 2011). Grosche et al. (2012) said that analyses of the C-to-U conversions and the genomic context in which the editing sites are embedded provide evidence in favor of the hypothesis that chloroplast RNA editing evolved to compensate mutations in the first land plants. It was concluded that RNA-editing sites can be rapidly gained or lost throughout evolution but start or stop codons are relatively stable.
Ruiz-Ruano et al. (2019) reported that gene content of Vandenboschia speciosa (Hymenophyllaceae) of fern order Hymenophyllales plastome was similar to that in most ferns but an important number of genes required U-to-C RNA editing for proper protein translation and two genes showed start codons alternative to the canonical AUG (AUA).
3 Plastome
In addition to photosynthesis, chloroplast play vital roles in other aspects of plant physiology and development, including the synthesis of nucleotides, fatty acids, amino acids, vitamins, phytohormones, metabolites, and the assimilation of sulfur and nitrogen (Neuhaus and Emes 2000; Daniell et al. 2016).
Plastids (chloroplasts) possess their own genetic information and, consequently, express heritable traits (Bock 2007). Wicke et al. (2011) as well as few reported instances of gene duplication or horizontal gene transfer. The plastid genome provides a wealth of phylogenetically informative data that are relatively easy to obtain and use (Soltis and Soltis 1998).
Plastid genomes are structurally highly conserved coding and considerable diverse non-coding spacer regions. Because of their very low level of recombination, they are valuable sources of genetic markers for phylogenetic analyses (Daniell et al. 2006; Gao et al. 2009; Xu et al. 2019). Wolf et al. (2010) examined for the first time the structure of the plastome across fern phylogeny. According to Grewe et al. (2013), the order of these plastid genes has remained consistent for most species, such that large syntenic tracks can be easily identified between genomes (Grewe et al. 2013).
The size of photosynthetic land plant plastid chromosomes ranges from 120 to 160 kb. The plastome in photosynthetic plants comprises 70 (gymnosperms) to 88 (liverworts) protein-coding genes and 33 (most eudicots) to 35 (liverworts) structural RNA genes (Bock 2007), totaling 100–120 unique genes. An almost universal feature of the circular chloroplast genome is a large inverted repeat sequence, some 10–25 kilobase pairs (kb) in size, which separates the remainder of the molecule into single-copy regions of 80 kb and 20 kb (Shim et al. 2021). The chloroplast genome includes 120–130 genes, primarily participating in photosynthesis, transcription, and translation.
Wicke et al. (2011) grouped plastid genes into four groups such as those involved in primary and secondary photosynthesis pathways, genes involved in sulfate transport and lipid acid synthesis, genes involved in transcription and translation, and a number of structural RNA genes. Wicke et al. (2011) demonstrated only a few functional gene gains, and more frequent gene losses have been inferred for land plants; the plastid Ndh complex is one example of multiple independent gene losses. Zhu et al. (2016) reported that the plastid genome (plastome) of nearly all land plants has a highly conserved quadripartite structure composed of two copies of an inverted repeat (IR) and two single-copy (SC) regions, termed the large single-copy (LSC) and small single-copy (SSC) regions. Intronic SSRs can affect gene transcription, mRNA splicing, or export to cytoplasm (Li et al. 2004).
4 Plastogenomics
Plastomes have now been extensively used for exploring phylogenetic relationships and understanding evolutionary processes of plants (Lehtonen 2011). Sequence data from the plastid genome have transformed plant systematics and contributed greatly to the current view of plant relationships (Ruhfel et al. 2014). Phylogenetic and biotechnological investigations are allowing novel insights and expanding the scope of plastome research (Ruhlman and Jansen 2014). Structural changes in the cp genome, such as gene rearrangements (Tangphatsornruang et al. 2010), gene/intron losses or duplications (Guisinger et al. 2011), and small inversions (Yi and Kim 2012), are well known at the genus, family, or ordinal levels of seed plants. However, the cp genome studies in ferns are limited to just a few lineages.
Liu et al. (2021) reported that most fern plastomes consist of four parts, including a pair of large inverted repeats (IRs), a large single-copy (LSC) region, and a small single-copy (SSC) region. Almost all fern IRs contains a core gene set of four ribosomal RNAs (16S, 23S, 4.5S, and 5S) and several tRNA genes (trnA-UGC, trnI-GAU, trnN-GUU, and trnR-ACG). IR regions are responsible for variations in chloroplast genome size and rearrangement and thus promoting genomic evolution (Raubeson et al. 2007; Gao et al. 2013; Grewe et al. 2013; Daniell et al. 2016; Logacheva et al. 2017). In chloroplast genome variables are present in LCS and SSC regions, while expansion and contraction were noted in the IR region (Li et al. 2016; Asaf et al. 2017). Logacheva et al. (2017) reported that IRs of Polypodiales plastomes are dynamic and are regulated by gene loss, duplication, and putative lateral transfer from mitochondria.
Molecular phylogenetic investigations have revolutionized our understanding of fern phylogeny (Rothfels et al. 2015). According to Rothfels et al. (2015), deep divergences in fern phylogeny have been recorded mainly in 12 studies almost exclusively on plastid data (Table 4.1). These studies relied almost exclusively on data from a single linkage group, the plastid genome, which is maternally inherited in ferns (Gastony and Yatskievych 1992; Guillon and Raquin 2000).
Lu et al. (2015) reported phylogeny of ferns based on plastome sequence data (Fig. 4.1). All the 132 complete plastome sequences in the NCBI RefSeq collection from GenBank as of 10 Feb 2019 were downloaded and analyzed by Liu et al. (2020). Sequenced species mainly included tree ferns (Cyatheales) and polypod ferns (Polypodiales), which contain most of the extant fern diversity. These include the inversion of a 3 kb region that is shared by Equisetum L. and other ferns (Gao et al. 2009) and the loss of chlB, chlL, and chlN in Psilotum Sw. and Tmesipteris Bernh (Grewe et al. 2013; Zhong et al. 2014).

Lu et al. (2015) reported phylogeny of ferns based on plastome sequence data. Source: Lu, J.M. et al. (2015). Chloroplast phylogenomics resolves key relationships in ferns. Jnl of Systematics Evolution, 53: 448–457. doi: 10.1111/jse.12180. Reproduced with license number 5085221071437 dated 10 June 2021
5 Phylogeny of Ferns Based on Plastome Sequence Data
Studies on chloroplast genomes of ferns and lycophytes are relatively few in comparison with those on seed plants. Lu et al. (2015) suggested a basic phylogenetic framework of extant ferns (Fig. 4.1). However, the relationships among a few key nodes remain unresolved or poorly supported.
Pteridophytes are free-sporing vascular plants comprising two classes Lycopodiopsida (lycophytes) and Polypodiopsida (ferns) which form distinct evolutionary lineages in the tracheophyte phylogenetic tree (Shmakov 2016).
5.1 Lycopodiopsida (Lycophytes)
Only three orders are currently recognized within Lycopodiopsida, including Lycopodiales, Isoetales, and Selaginellales (Shmakov 2016). Order Lycopodiales includes 1 family and 16 genera, whereas orders Isoetales and Selaginellales each contain a single genus Selaginella (Zhou and Zhang 2015; Weststrand and Korall 2016a, b). Complete plastome sequences have been made available for all the three important orders: (1) Lycopodiales, Huperzia species; (Guo et al. 2016); (2) Isoetales, Isoetes flaccida (Karol et al. 2010); and (3) Selaginellales, Selaginella species (Smith 2009).
Huperzia serrata (Lycopodiaceae) chloroplast genome is reported to contain 120 unique genes, including 86 coding genes, 4 rRNA genes, and 30 tRNA genes (Guo et al. 2016). Wolf et al. (2005) studied completed genome of Huperzia lucidula (Lycopodiaceae) which is 154,373 bp, containing inverted repeats of 15,314 bp each, a large single-copy region of 104,088 bp, and a small single-copy region of 19,657 bp. Tsuji et al. (2007) determined complete nucleotide sequence of the chloroplast genome of Selaginella uncinata, a lycophyte belonging to the basal lineage of the vascular plants. The circular double-stranded DNA is 144,170 bp, with an inverted repeat of 25,578 bp separated by a large single-copy region (LSC) of 77,706 bp and a small single-copy region (SSC) of 40,886 bp. Tsuji et al. (2007) showed that the gene order and arrangement are almost identical between the plastomes of Huperzia lucidula (Lycopodiaceae) and bryophytes, but the plastome of S. uncinata is considerably different from those of bryophytes. Several new pieces of evidence for monilophyte monophyly of Isoetes flaccida, a heterosporous lycophyte, have been recorded.
Selaginella with 700 species is distributed in a diverse range of habitats which include deserts, tropical rainforests, and alpine and arctic regions. Selaginella plastomes have the highest GC content and fewest genes and introns of any photosynthetic land plant. Uniquely, the canonical inverted repeat was converted into a direct repeat (DR) via large-scale inversion in some Selaginella species. Ancestral reconstruction identified additional putative transitions between an inverted and DR orientation in Selaginella and Isoetes plastomes. ADR orientation does not disrupt the activity of copy-dependent repair to suppress substitution rates within repeats. Thus, gene relocation in lycophyte plastomes occurs via overlapping inversions rather than transposase/recombinase-mediated processes (Shim et al. 2021). Shim et al. (2021) studied two Selaginella spp. Selaginella stauntoniana and Selaginella involvens and reported that unlike the inverted repeat (IR) structures typically found in plant plastomes, Selaginella species had direct repeat (DR) structures (Shim et al. 2021). A genus-wide comparison of genomic features, including GC contents, structural changes in the genome, and gene losses, revealed that Selaginella have reduced LSC regions and longer SSCs than LSCs, except for the three species of Selaginella lepidophylla, Selaginella hainanensis, and S. uncinata (Shim et al. 2021 Fig. 4.2). Most Selaginella species shared a unique plastome structure consisting of a set of direct repeats (DRs) instead of the inverted repeats (IRs) found in most plastomes. Shim et al. (2021) confirmed the unusual DR structure through the assembly of the S. tamariscina plastome (Fig.4.2). Shim et al. (2021) concluded that the plastome sequences of Selaginella species were smaller than those of non-Selaginella and typical land plants.

Shim et al. (2021) presented map of complete plastid genomes of the Selaginella tamariscina, Selaginella stauntoniana, and Selaginella involvens. Shaded areas indicate regions involved in the inversion event. Source: Shim, Hyeonah et al. (2021) “Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers” Int. J. Mol. Sci. 22, no. 2: 641. doi: 10.3390/ijms22020641. An open-access article distributed under the terms of the Creative Commons CC BY license
Low guanine and cytosine (GC) content is one of the more conspicuous features of plastid DNA (ptDNA). Smith (2009) reported that as of February 2009, all completely sequenced plastid genomes have GC content below 43% except for the ptDNA of the lycophyte Selaginella uncinata, which is 55% GC. Thus, there is genus-wide GC bias in Selaginella ptDNA, within the Lycopsida class (and among plants in general). Shim et al. (2021) concluded that these findings provide convincing support for the earlier proposed theory that the GC content of land-plant organelle DNA is positively correlated and directly connected to levels of organelle RNA editing.
5.2 Polypodiopsida (Ferns)
Recently, the complete cp genome sequences of four orders of eusporangiate ferns were analyzed, and the data aided in understanding the evolutionary history of eusporangiate ferns (Grewe et al. 2013; Karol et al. 2010). Seed plant lineages usually show small ranges of variation in both GC contents and effective numbers of codons (ENCs). Kim et al. (2014) reported that GC contents and the effective numbers of codons (ENCs) values of the early diverged leptosporangiate ferns showed intermediate levels between eusporangiate and core leptosporangiate ferns. The core leptosporangiate ferns show higher ENCs than early diverged leptosporangiate ferns. The cp gene sequences clearly indicated that the cp genome similarity between O. cinnamomea (Osmundales) and eusporangiate ferns is symplesiomorphies, rather than synapomorphies. Therefore, Kim et al. (2014) are in agreement with the view that Osmundales is a distinct early diverged lineage in the leptosporangiate ferns. In Pteridaceae, all subfamilies accepted by Christenhusz et al. (2011) were found to be monophyletic, although the monophyly of Cheilanthoideae had poor support. By contrast, numerous pteridoid genera, including Adiantum, were not monophyletic.
5.2.1 Eusporangiate
Grewe et al. (2013) sequenced the plastid genomes from three early diverging species: Equisetum hyemale (Equisetales), Ophioglossum californicum (Ophioglossales), and Psilotum nudum (Psilotales). A comparison of fern plastid genomes showed that some lineages have retained inverted repeat (IR) boundaries originating from the common ancestor of land plants, while other lineages have experienced multiple IR changes including expansions and inversions (Grewe et al. 2013; Fig. 4.3).

Grewe et al. (2013) presented plastome maps for newly sequenced monilophytes. Boxes on the inside and outside of the outer circle represent genes transcribed clockwise and anti-clockwise, respectively. The inner circle displays the GC content represented by dark gray bars. The location of the IRs is marked on the inner circle and represented by a thicker black line in the outer circle. The large euphyllophyte LSC inversion and the small monilophyte LSC inversion are highlighted on the outer circle by blue and purple bars, respectively. Source: Grewe et al. (2013). Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol Biol 13, 8. doi: 10.1186/1471-2148-13-8. This is an open-access article distributed under the terms of the Creative Commons CC BY license
5.2.2 Polypodiales
5.2.2.1 Leptosporangiate Ferns
Leptosporangiate ferns account for 80% of nonflowering vascular plants (including gymnosperms and lycophytes) (Schneider et al. 2004; Schuettpelz and Pryer 2009; Rai and Graham 2010), and the leptosporangiate order Polypodiales is by far the largest fern order, with more than 7000 extant species. The unique chloroplast genomic rearrangement of core leptosporangiate ferns (Salviniales, Cyatheales, and Polypodiales) and Schizaeales can be explained by an expansion of the IRs and “two inversions” (Wolf et al. 2003) which mainly affect the orientation and gene content of the IRs (Hasebe and Iwatsuki 1992).
Fan et al. (2021) studied complete chloroplast genomes of Athyrium brevifrons Nakai ex Kitagawa, D. crassirhizoma Nakai, Dryopteris goeringiana (Kunze) Koidz, and Polystichum tripteron (Kunze) Presl. Simple sequence repeats (SSRs), nucleotide diversity analysis, and RNA editing were investigated in all four species (Fan et al. 2021). Genome comparison analysis revealed that single-copy regions were more highly conserved than IR regions. IR boundary expansion and contraction varied among the four ferns (Fan et al. 2021; Figs.4.4 and 4.5). The genome size ranged from 149,468 (D. crassirhizoma) to 151,341 bp (A. brevifrons). The chloroplast genomes had a circular assembly and exhibited a typical quadripartite structure, including one LSC region (82,384–82,799 bp), one SSC region (21,600–21,708 bp), and two IR regions (22,040–22,682 bp).

Fan et al. (2021) presented morphological characteristics of D. crassirhizoma, D. goeringiana, P. tripteron, and A. brevifrons. (Source: Fan, R, Ma, W, Liu, S, Huang, Q. Integrated analysis of three newly sequenced fern chloroplast genomes: Genome structure and comparative analysis. Ecol Evol. 2021; 11: 4550–4563. doi: https://doi.org/10.1002/ece3.7350). This is an open-access article distributed under the terms of the Creative Commons CC BY license

Fan et al. (2021) depicted the chloroplast genome maps of D. crassirhizoma, D. goeringiana, A. brevifrons, and P. tripteron. Genes drawn inside the circle are transcribed clockwise, and those outside the circle are transcribed counterclockwise. The light gray inner circle corresponds to the A + T content and the dark gray to the G + C content. Genes belonging to different functional groups are shown in different colors. (Source: Fan R, Ma W, Liu S, Huang Q. Integrated analysis of three newly sequenced fern chloroplast genomes: Genome structure and comparative analysis. Ecol Evol. 2021 Mar 18;11(9):4550–4563. doi: https://doi.org/10.1002/ece3.7350). This is an open-access article distributed under the terms of the Creative Commons CC BY license
The complete chloroplast genomes of Dryopteris goeringiana (Kunze) Koidz, D. crassirhizoma Nakai, Athyrium brevifrons Nakai ex Kitagawa, and Polystichum tripteron (Kunze) Presl were sequenced by Fan et al. (2020). Simple sequence repeats (SSRs), nucleotide diversity analysis, and RNA editing were investigated in all four species. They also demonstrated that ferns have a higher G + C content and a higher number of C-to-U RNA editing events than other plants. Fan et al. (2021), speculated that D. crassirhizoma, D. goeringiana, D. decipiens, P. tripteron, and C. devexiscapulae are closely related. D. crassirhizoma and D. goeringiana are closely related to D. decipiens. P. tripteron was identified as a sister species of C. devexiscapulae. Interestingly, D. decipiens and C. devexiscapulae (Fig. 4.5) were found to be clustered into one branch in a study by Wei et al. (2017). The genomes of Azolla and Salvinia offer a new opportunity to examine the evolution of plant genes and gene families across all Viridiplantae (land plants plus green algae) (Li et al. 2018).
Li et al. (2018) reported on the genomes of Azolla filiculoides and Salvinia cucullata (Salviniales) and presented evidence for episodic whole-genome duplication in ferns – one at the base of “core leptosporangiates” and one specific to Azolla. One fern-specific gene seems to have been derived from bacteria through horizontal gene transfer (Fig. 4.6). This gene provides insect resistance. The relatively small genome (0.75 Gb; Obermayer et al. 2002) of Azolla is exceptional among ferns, a group that is notorious for genomes as large as 148 Gb (Hidalgo et al. 2017) and averaging 12 Gb (Sessa and Der 2016). Azolla is one of the fastest-growing plants on the planet, with demonstrated potential to be a significant carbon sink (Li et al. 2018).

Genome size evolution in Salviniales (Li et al. 2018). (a) Members of Salviniales have smaller genome sizes than other ferns (averaging 1C = 12 Gb). Two whole-genome duplication (WGD) events identified in this study were mapped onto the phylogeny, with divergence time estimates obtained from Testo and Sundue. (b, c) Whole genomes were assembled from A. filiculoides (b) and S. cucullata (c). (d, e) The genome of S. cucullata has substantially reduced levels of RNA (d) and DNA (e) transposons compared to A. filiculoides. Image in panel c courtesy of P.-F. Source: Li, FW., Brouwer, P., Carretero-Paulet, L. et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nature Plants 4, 460–472 (2018). doi: 10.1038/s41477-018-0188-8. This is an open-access article distributed under the terms of the Creative Commons CC BY license
Azolla is also remarkable in harboring an obligate, N2-fixing cyanobacterium, Nostoc azollae, within specialized leaf cavities. Furthermore, the Azolla genome lacks genes that are common to arbuscular mycorrhizal and root nodule symbioses. Li et al. (2018) identified several putative transporter genes specific to Azolla-cyanobacterial symbiosis. These genomic resources will help in exploring the biotechnological potential of Azolla and address fundamental questions in the evolution of plant life (Li et al. 2018).
5.2.2.1.1 Polypodiaceae
Polypods are the lineage of most derived ferns that diversified in the Cretaceous period, displaying an ecologically opportunistic response to the diversification of angiosperms. (Schneider et al. 2004). They suggested that plastomes of polypods have undergone multiple complex genomic reconfigurations during fern evolution, and thus, their plastomes differ substantially from the plastomes of basal ferns (Psilotales, Ophioglossales, Marattiales, and Equisetales). Earlier plastome evolution among Polypodiaceae is considered relatively static compared with that in lineages other than polypods (Wolf et al. 2010), but Liu et al. (2021) suggested that the plastomes of Polypodiaceae are dynamic molecules, rather than constituting static genomes as previously thought. They further indicated that dispersed repeats flanking insertion sequences contribute to the repair mechanism induced by double-strand breaks and are probably a major driver of structural evolution in the plastomes of Polypodiaceae, e.g., Neolepisorus fortunei, Neolepisorus ovatus, and Phymatosorus cuspidatus (Liu et al. 2021; Fig. 4.7).

Liu et al. (2021) presented plastome gene maps of Neolepisorus fortunei, Neolepisorus ovatus, and Phymatosorus cuspidatus. The plastome map represents all three species since their gene numbers, orders, and names are the same, except that N. fortunei has lost the trnR-UCG gene. Genes located outside and within the black circle are transcribed in the clockwise and counterclockwise directions, respectively. Different colors represent genes belonging to different functional groups Source: Liu et al. (2021). Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol. 2021 Jan 7;21(1):31. doi: https://doi.org/10.1186/s12870-020-02800-x. This article is licensed under a Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain
6 Analysis of SSRs in Plastomes
Genomic microsatellites (simple sequence repeats; SSRs), iterations of 1–6 bp nucleotide motifs, have been detected in the genomes of every organism (Guichoux et al. 2011). Nevertheless, SSRs are usually just considered as evolutionarily neutral DNA markers (e.g. Schlötterer and Wiehe 1999). SSR genetic and evolutionary mechanisms remain controversial. Li et al. (2002) presented SSR putative functions/effects. Xu et al. (2019) investigated the phylogenetic relatedness among the plastid genomes of 30 species. The ferns of Leptosporangiatidae, Psilophytinae, and Equisetinae were grouped into three separate clades, respectively. The two Eusporangiate ferns were not grouped in one clade, with Mankyua chejuensis B.Y. Sun was closer to Psilophytinae and the other one Angiopteris evecta (G. Forst.) Hoffm. (Marattiaceae) was identified as a sister genus to Leptosporangiatidae. The Leptosporangiatidae ferns formed two clades: Osmuda and the other clade contained the other species. This tree also indicated that the moss Physcomitrella patens (Hedw.) Bruch & Schimp was grouped in one clade with Leptosporangiatidae, Psilophytinae, Equisetinae, and Eusporangiate (Xu et al. 2019;Fig 4.8).

Xu et al. (2019) analyzed plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Source: Xu, L., Xing, Y., Wang, B. et al. Plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Chin Med 14, 9 (2019). doi: 10.1186/s13020-019-0230-4. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
6.1 SSR Sequence Analysis
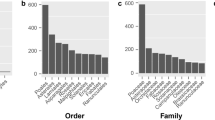
A total of 74 (1024) bp and 82 SSR (1191) bp loci that were 1024 bp and 1191 bp long, respectively, were detected in the D. crassirhizoma and O. japonica plastid genomes. The number of mono-repeats was dominant in the plastid genomes of both species. Compared with D. fragrans, D. decipiens, and O. cinnamomeum (Xu et al. 2019; Fig. 4.8), they found that the three species from Dryopteris (Dryopteridaceae) had more SSR mono-repeats than the two species from Osmunda.
Liu et al. (2021) recorded the total number of SSRs in 12 Polypodiaceae species, which ranged from 38 to 51. Four kinds of SSRs were detected: mononucleotides (62.8–88.3%), dinucleotides (8.7–20.9%), trinucleotides (6.6–22.5%), and tetranucleotides (0–4.4%). However, tetranucleotide repeats were discovered in only the plastomes of L. clathratus, L. hemionitideus, L. hederaceum, S. yakushimensis, and D. roosii (Fig. 4.9). SSRs were much more frequently located in the LSC region (48.0–71.1%) than in IR (10.5–36.0%) and SSC regions (9.3–22.0%).

Source: Liu et al. (2021) Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol 21, 31 (2021). The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data
The “Monilophyte” clade comprising ferns, horsetails, and whisk ferns receives unequivocal support from molecular data as the sister clade to seed plants. However, the branching order of its earliest emerging lineages, the Equisetales (horsetails), the Marattiales, the Ophioglossales/Psilotales, and the large group of leptosporangiate ferns, has remained dubious (Knie et al. 2015). However, Knie et al. (2015) ultimately obtained a well-supported molecular phylogeny is placing Marattiales as sister to leptosporangiate ferns and horsetails as sister to all remaining monilophytes.
Gao et al. (2009) studied complete chloroplast genome sequence of a tree fern Alsophila spinulosa. This provided insights into evolutionary changes in fern chloroplast genomes. Gao et al. (2009) reported that the Alsophila cp genome is 156,661 base pairs (bp) in size and has a typical quadripartite structure with the large single-copy (LSC, 86,308 bp) and small single-copy (SSC, 21,623 bp) regions separated by two copies of an inverted repeat (IRs, 24,365 bp each). Thick black lines on the inner cycle indicate the inverted repeats (IRA and IRB) which separate the genome into the large single-copy (LSC) and small single-copy (SSC) regions. Gao et al. (2013) determined the complete chloroplast genome sequences of Lygodium japonicum (Lygodiaceae), a member of schizaeoid ferns (Schizaeales), and Marsilea crenata (Marsileaceae), a representative of heterosporous ferns (Salviniales). Wolf et al. (2003) determined the complete nucleotide sequence of the chloroplast genome of the leptosporangiate fern, Adiantum capillus-veneris L. (Pteridaceae).
Xu et al. (2019) carried out a SSR sequence analysis. A total of 74 and 82 SSR loci that were 1024 bp and 1191 bp long, respectively, were detected in the D. crassirhizoma and O. japonica plastid genomes. The number of mono-repeats was dominant in the plastid genomes of both species. There were 54 and 62 SSRs located in the LSC, 14 and 12 located in the IR, and 8 and 6 located in the SSC in the D. crassirhizoma and O. japonica plastid genome, respectively. Compared with D. fragrans, D. decipiens, and O. cinnamomeum, Xu et al. (2019) found that the three species from Dryopteris (Dryopteridaceae) had more SSR mono-repeats than the two species from Osmunda. The position of Osmunda may indicate that Osmunda diverged early in the lineage of leptosporangiate ferns (Kim et al. 2014).
Liu et al. (2020) have sequenced the complete plastid genome of a scaly tree fern Alsophila spinulosa (ab. Alsophila) (Cyatheaceae). In addition to tree ferns, heterosporous and polypod ferns are the other two main lineages within the “core leptosporangiates” (Li et al. 2016). They confirmed that two major rearrangements distinguish higher leptosporangiate ferns from basal fern lineages. The Alsophila cp genome is very similar to that of the polypod fern Adiantum in terms of gene content, gene order, and GC content (Wolf et al. 2003). Sun et al. (2017) studied the complete chloroplast genome of the medical fern Drynaria roosii in order to understand the evolution of the genome of the fern. In D. roosii, the circular double-stranded cpDNA sequence of 154,305 bp consists of two inverted repeat (IRA and IRB) regions of 23,416 bp each, a large single-copy (LSC) region of 86,040 bp, and a small single-copy (SSC) region of 21,433 bp. The phylogenetic position of D. roosii was closely clustered with Adiantum capillus-veneris, Cheilanthes lindheimeri, and Pteridium aquilinum subsp. Aquilinum as sister species and then clustered with Alsophila spinulosa, Lygodium japonicum, Diplopterygium glaucum, and Osmundastrum cinnamomeum. D. roosii belongs to Polypodiales. The complete chloroplast genome of D. roosii provides utility information for ferns’ evolutionary and genomic studies.
7 Genes Translocated into the Plastid Inverted Repeat
Li et al. (2016) reported that plant chloroplast genomes (plastomes) are characterized by an inverted repeat (IR) region and two larger single-copy (SC) regions, and they further suggested that patterns of molecular evolution in the IR and SC regions differ, most notably by a reduced rate of nucleotide substitution in the IR compared to the SC region. Rates of molecular evolution vary dramatically among organismal lineages and across genomes (Bromham and Penny 2003), and understanding what causes this rate variation is a fundamental topic in evolutionary biology (Lanfear et al. 2010). Li et al. (2016) demonstrated that when genes are translocated into the IR, their nucleotide substitution rates dropped significantly (two- to threefold). They further reported that this deceleration is not shared with other nontranslocated chloroplast genes. They concluded that in addition to rate deceleration, GC content increases following translocation, indicating that the IR affects both substitution rates and GC content (Fig. 4.10). It may be finally concluded that without the knowledge of genome structure, or modeling for possible hidden rate shifts, the evolutionary inferences could be grossly misleading (Rothfels and Schuettpelz 2014).

Li et al. (2016) showed chloroplast genome structure. (a) The typical plant chloroplast genome (plastome) comprises a pair of inverted repeat (IR) regions separating a single-copy (SC) region. The red arrowheads indicate the direction in which the rRNA genes in the IR are transcribed. (b) In some fern chloroplast genomes, the direction of transcription in the IR is inverted. (c) Genome rearrangements have resulted in changes in IR gene content. The phylogeny on the left shows the relationships among the sampled fern chloroplast genomes; a part of their genome organization is shown on the right. The tree topology and divergence times are derived from Rothfels et al. (2015). Gene lengths are not to scale, gene arrow tips indicate the direction of transcription, and a few genes are omitted for clarity. “*” indicates that chlN is not always present. Note that rps12 in Ophioglossales, Psilotales, and Equisetales lacks the second intron, and therefore there is no exon 3. Source: Li, F. W., Kuo, L. Y., Pryer, K. M., & Rothfels, C. J. (2016). Genes Translocated into the Plastid Inverted Repeat Show Decelerated Substitution Rates and Elevated GC Content. Genome biology and evolution, 8(8), 2452–2458. doi: 10.1093/gbe/evw167. Reproduced with license number 5101790146234 dated 4 July 2021
Plastid ribosomes are ubiquitous organelles in plant cells and play a vital role in the biosynthesis of proteins. In higher plants, plastid ribosomes contain approximately 60 ribosomal proteins that are encoded in both the plastid and the nuclear genetic compartments (Eneas-Filho et al. 1981). The plastid ribosomal protein S12 encoded by the rps12 gene is a highly conserved protein located in the functional center of the 30S subunit of the ribosome (Yamaguchi and Subramanian 2003).
The plastomes of polypods have undergone multiple complex genomic reconfigurations during fern evolution, and thus, their plastomes differ substantially from the plastomes of basal ferns (Psilotales, Ophioglossales, Marattiales, and Equisetales).
The variation in the exon location and intron content of the rps12 gene in fern plastomes provides a unique opportunity to explore the effect of gene structure on sequence evolution. Liu et al. (2020) reconstructed the phylogeny of ferns and inferred the patterns and rates of plastid rps12 gene evolution in a phylogenetic context (Figs. 4.11 and 4.12). In most ferns, the first exon of rps12 is located in the LSC, whereas the second and third exons reside in the IRs. The plastomes of polypods have undergone multiple complex genomic reconfigurations during fern evolution, and thus, their plastomes differ substantially from the plastomes of basal ferns (Psilotales, Ophioglossales, Marattiales, and Equisetales).

Liu et al. (2020) depicted sizes of each part of 16 fern complete plastome sequences. Liu, S., Wang, Z., Wang, H. et al. Patterns and Rates of Plastid rps12 Gene Evolution Inferred in a Phylogenetic Context using Plastomic Data of Ferns. Sci Rep 10, 9394 (2020). doi: https://doi.org/10.1038/s41598-020-66219-y. This is an open-access article distributed under the terms of the Creative Commons CC BY license

Liu et al. (2020) depicted phylogram showing intron losses and the distribution of the rps12 gene in fern plastomes. The absence of the rps12 intron is indicated with a red line; the dashed line denotes that all the exons of rps12 were present in only one copy. Source: Liu, S., Wang, Z., Wang, H. et al. (2020). Patterns and Rates of Plastid rps12 Gene Evolution Inferred in a Phylogenetic Context using Plastomic Data of Ferns. Sci Rep 10, 9394. doi: https://doi.org/10.1038/s41598-020-66219-y. This is an open-access article distributed under the terms of the Creative Commons CC BY license
8 Plant Transcriptome Evolution
Plant genomes encode many lineage-specific, unique transcription factors (Wilhelmsson et al. 2017). Wilhelmsson et al. (2017) further stated that expansion of transcription-associated proteins (TAPs) (total number of TAP genes per genome) comprising transcription factors and transcriptional regulators has been found to coincide with the evolution of morphological complexity (Lang et al. 2010). Lang et al. (2010) further reiterated that evolutionary retention of duplicated genes encoding transcription-associated proteins (TAPs) has been hypothesized to be positively correlated with increasing morphological complexity and paleopolyploidizations, especially within the plant kingdom. Both, the emergence and expansion of TAP families during land plant evolution, suggest a clear trend of increasing transcriptional complexity along with morphological complexity (Lang et al. 2010). Using phylogenetic comparative (PC) analyses, Lang et al. (2010) defined the timeline of TAP loss, gain, and expansion among Viridiplantae.
The evolution of plant transcription-associated proteins, including transcription factors (TFs, binding in sequence-specific manner to cis-regulatory elements to enhance or repress transcription) and transcriptional regulators (TRs, acting as part of the transcription core complex, via unspecific binding, protein-protein interaction, or chromatin modification) of ferns were exclusively represented by the Pteridium aquilinum transcriptome (Wilhelmsson et al. 2017). Szövényi et al. (2021) compared patterns of various levels of genome and epigenomic organization found in seed-free plants to those of seed plants, e.g., some genomic features appear to be fundamentally different. For instance, hornworts, Selaginella, and most liverworts are devoid of whole-genome duplication, in stark contrast to other land plants. However, model systems are crucial to further our understanding about how changes in genes translate into evolutionary novelties (Szövényi et al. 2021).
The finding that the transcriptional regulator Polycomb group EZ (PcG_EZ) was lost in ferns is corroborated here by whole-genome data (Li et al. 2018). Conversely, the transcription factor ULTRAPETALA, which originated at the base of euphyllophytes and is present in P. aquilinum, was apparently secondarily lost in Salviniales. Conversely, the transcription factor ULTRAPETALA, which originated at the base of euphyllophytes and is present in P. aquilinum, was apparently secondarily lost in Salviniales (Li et al. 2018).
9 Discussion
Plastid genome data are beneficial in resolving species definitions because organelle-based “barcodes” can be established for a species and then used to unmask interspecies phylogenetic relationships (Yang et al. 2013). Sequence data from the plastid genome have transformed plant systematics and contributed greatly to the current view of plant relationships. The plastid genome provides a wealth of phylogenetically informative data that are relatively easy to obtain and use (Olmstead and Palmer 1994; Soltis and Soltis 1998). Next-generation sequencing has provided a wealth of plastid genome sequence data from an increasingly diverse set of green plants (Viridiplantae) (Rothfels et al. 2015).
Grewe et al. (2013) reported that phylogenetic affinities were revealed by mapping rare genomic structural changes in a phylogenetic context. Chloroplast genomes have stable maternal heredity which excludes recombination and are remarkably conserved in land plants. This has made them a valuable and ideal resource for species identification, plant phylogenetics, population genetics, and genetic engineering taxon-rich phylogenetic analyses (Nock et al. 2014). Rai and Graham (2010) also examined the utility of a large plastid-based data set in inferring backbone relationships for monilophytes and found it highly congruent with earlier multigene studies, corroborating clades in common across studies (Knie et al. 2015). However, the relationships among major fern lineages, especially the placement of Equisetales, remain enigmatic (Grewe et al. 2013; see also Wickett et al. 2014).
The ferns of Psilophytinae formed a sister clade to Equisetinae with strong support, which was different from a previous study (Borgstrom et al. 2011). Pryer et al. (2004) based on phylogenetic analyses confirmed that (1) Osmundaceae are sister to the rest of the leptosporangiates, (2) resolved a diverse set of ferns formerly thought to be a subsequent grade as possibly monophyletic (Dipteridaceae, Matoniaceae, Gleicheniaceae, Hymenophyllaceae), and (3) placed schizaeoid ferns as sister to a large clade of “core leptosporangiates” that includes heterosporous ferns, tree ferns, and polypods. Whole plastome sequences have been used to study evolution (Kim et al. 2014; Labiak and Karol 2017). Szövényi et al. (2021) reported that during the past few years, several high-quality genomes have been published from Charophyte algae, bryophytes, lycophytes, and ferns. Szövényi et al. (2021) compared patterns of various levels of genome and epigenomic organization found in seed-free plants to those of seed plants. They reported that in stark contrast to other land plants, Selaginella and most liverworts are devoid of whole-genome duplication.
The phylogenetic relationships among these four basal fern orders are the most debated topics in fern phylogeny. Pryer et al. (2001) suggested that maximum likelihood analysis showed unambiguously that horsetails and ferns together are the closest relatives to seed plants. However, this refutes the prevailing view that horsetails and ferns are transitional evolutionary grades between bryophytes and seed plants and has important implications for our understanding of the development and evolution of plants. Although lycophytes were abundant and dominant in land flora during the Carboniferous era (Kenrick and Crane 1997), the class Lycopodiopsida diverged shortly after land plants evolved to acquire vascular tissues (Banks et al. 2011). Lycopods are similar to ferns in this regard, but ferns are the sister group of the seed plants (gymnosperms plus angiosperms), whereas the lycopods are sister to all other vascular plants (ferns plus the seed plants) (Christenhusz and Chase 2014). Psilotales (which include Psilotum and Tmesipteris) are closer to ferns (Hendy and Penny 1989; Pryer et al. 2001; Qiu et al. 2006, 2007; Korall et al. 2010; Zhong et al. 2011, 2014).
Clark et al. (2016) suggested that genome size was correlated with chromosome number across all ferns despite some substantial variation in both traits. Marchant et al. (2019) reported a single ancient polyploidy event and spread of repeat elements in the evolutionary history of C-Fern (Ceratopteris richardii) based on both genomic and cytogenetic data. According to Shim et al. (2021), plastid genomes (plastomes) are typically 120–160 kb long, with a quadripartite architecture comprising one long single-copy (LSC) region and a short single-copy (SSC) region separated by two inverted repeats (IRA and IRB). Shim et al. (2021) reported that the early vascular plants in the genus Selaginella are valuable resources for deciphering plant evolution. Comparative analyses of 19 lycophytes by Shim et al. (2021) revealed unique phylogenetic relationships between Selaginella species and related lycophytes. The changes were reflected by structural rearrangements involving two rounds of large inversions that resulted in dynamic changes between IR and DR blocks in the plastome sequence. Furthermore, lycopods present other uncommon characteristics, e.g., a small genome size, drastic reductions in gene and intron numbers, a high GC content, and extensive RNA editing. Their findings suggest that Selaginella plastomes have undergone unique evolutionary events yielding genomic features unparalleled in other lycophytes, ferns, or seed plants. Banks et al. (2011) reported that the transition from a gametophyte to a sporophyte dominated life cycle required far fewer new genes than the transition from a non-seed vascular to a flowering plant.
Fan et al. (2021) observed vast differences in SSRs among D. fragrans, D. crassirhizoma, and D. goeringiana, although they belonged to the same genus. They speculated that the types of SSRs are associated more with the surrounding environment than with the genus. This could explain the considerable differences among SSRs within the same species (Fan et al. 2021). Evolutionary significance has been shown in comparative studies between ferns and angiosperms. Palmer and Stein (1982) reported that Osmunda chloroplast genome was found to be remarkably similar in size, conformation, physical organization, and map positions of known genes, to chloroplast DNA from a number of angiosperms. Gene probes from tobacco, corn, and spinach were used to map the positions of six genes on the Osmunda cinnamomea chloroplast chromosome. Palmer and Stein (1982) made comparative studies on gene probes from tobacco, corn, and spinach to map the positions of six genes on the chloroplast chromosome. The 16S and 23S ribosomal RNAs are encoded by duplicate genes which lie within the inverted repeat. Genes for the large subunit of ribulose-1,5-bisphosphate carboxylase, a photosystem II polypeptide, and the alpha and beta subunits of chloroplast coupling factor are located in three different segments of the large single-copy region. The major difference between chloroplast DNA from this fern and angiosperms is that the inverted repeat is smaller in Osmunda (8–13 kb) than in angiosperms (22–25 kb).
Zhong et al. (2014) studied chloroplast genomes of a tree fern (Dicksonia squarrosa) (Cyatheaceae that includes the Alsophila genus) and a “fern ally” (Tmesipteris elongata). Gao et al. (2009) reported that Alsophila cp genome shows a high degree of synteny with that of Adiantum, but differs considerably from two basal ferns (Angiopteris evecta and Psilotum nudum). Availability of cp genome sequence from other tree ferns will facilitate interpretation of the evolutionary changes of fern cp genomes. Complete cp genome sequences of Angiopteris yunnanensis Hieron. (Marattiaceae) (Liu et al. 2019), Angiopteris evecta (G. Forst.) Hoffm. (Roper et al. 2007), and Angiopteris angustifolia C. Presl (Zhu et al. 2016) have been published. Comparative analyses confirmed the conservatism of plastid genome sequences among the species of Angiopteris and the distant related marattioid genus Christensenia (Jiang et al. 2019; Liu et al. 2019).
Haufler (1987, 2002 and 2014) suggested that ferns underwent multiple cycles of polyploidy. They reported whole-genome duplications (WGDs) accompanied by subsequent diploidization involving gene silencing, but without apparent chromosome loss, hence high chromosome numbers were retained leading to polyploidy. The high chromosome count of horsetails could then indeed be caused by a few paleopolyploidies of which a large fraction of genetic material has been retained (Haufler 1987). Vanneste et al. (2015) demonstrated that horsetails underwent an independent paleopolyploidy during the Late Cretaceous prior to the diversification of the genus but did not experience any recent polyploidizations that could account for their high chromosome number. This hypothesis has been provided by observations that polyploidy contributes to c. 31% of speciation events in ferns compared with c. 15% in angiosperms (Wood et al. 2009). This could also explain the high chromosome counts of homosporous ferns. Alternatively, the ancestor of all vascular plants could have exhibited a relatively high chromosome number (Soltis and Soltis 1987) so that a single paleopolyploidy could have resulted in the very high chromosome number in horsetails. It is emphasized that both theories are not mutually exclusive. The evolution of fern genomes has been considered paradoxical owing to the conservation of high chromosome numbers in taxa with demonstrated diploid gene expression (Haufler 2014).
WGD is often proposed as a driver of species diversification (Landis et al. 2018). Nakazato et al. (2006) suggested that abundance of gene duplicates is a potential mechanism for the past polyploidization in Ceratopteris richardii. They constructed a high-resolution genetic linkage map of the homosporous fern model species, C. richardii (n = 39). Single genome duplication isolates an individual from its parental species and forces the nascent polyploid to overcome numerical inferiority and parental competition if it is to survive (Levin 1975). The concurrent duplication of all nuclear genes is accompanied by widespread changes in gene expression (Adams and Wendel 2005) and often chromosomal rearrangements (Levin 2002; Gaeta et al. 2007). Yet despite the potential for ecological and genomic havoc, polyploidy is remarkably frequent, especially among plants. By some accounts, 20–40% (Stebbins 1971) of extant flowering plant species are neopolyploids, and as many as 70% are thought to have some polyploid ancestry (De Bodt et al. 2005; Cui et al. 2006).
Clark et al. (2016) reported that genome size was correlated with chromosome number across all ferns despite some substantial variation in both traits. They observed a trend toward conservation of the amount of DNA per chromosome, although Osmundaceae and Psilotaceae have substantially larger chromosomes.
Plastid genomes display remarkable organizational stability over evolutionary time (Robison et al. 2018). The chloroplast (cp) (plastid genomes – plastomes) has small size, high copy number, conservation, and extensive characterization at the molecular level (Raubeson and Jansen 2005). Plastomes contain high proportions of protein-coding genes compared with plant nuclear genomes, with many of these genes being essential to photosynthesis (Wicke et al. 2011). Chloroplast genomes have a typically circular structure with one large single-copy (LSC) region, one short single-copy (SSC) region, and two inverted repeat (IR) regions, ranging from 120 to 170 kb in length (Downie and Palmer 1992). Bromham and Penny (2003) reported that plant chloroplast genomes (plastomes) are characterized by an inverted repeat (IR) region and two larger single-copy (SC) regions. Patterns of molecular evolution in the IR and SC regions differ, most notably by a reduced rate of nucleotide substitution in the IR compared to the SC region. Xu et al. (2015) suggested that gain and loss of genes, gene content duplication, and gene order rearrangements appear to be phylogenetically and species informative.
Transcriptome sequencing or gene sequence resources as the 1000 Plants Project (The 1000 Plants Project http://www.onekp.com) are available, but genes alone are insufficient to answer the most pressing questions in fern and land plant genome evolution. In contrast, other phyloplastomic studies tend to support grouping Equisetales and Ophioglossales + Psilotales together as a monophyletic group and sister to the remaining ferns. Given that plastid genes generally evolve more slowly than the nuclear genes, these topological differences may be due to different numbers of phylogenetically informative sites contained within the diverse molecular data.
The relationships between the different horsetail species are now well resolved with Equisetum bogotense basal to both the subgenera Hippochaete and Equisetum that each contain seven species (Guillon 2004, 2007), while the Equisetopsida are most likely sister to both the whisk ferns (Psilotales) and ophioglossoid ferns (Ophioglossales; Grewe et al. 2013). Both molecular dating studies (Des Marais et al. 2003; Pryer et al. 2004) and fossil evidence (Stewart and Rothwell 1993) indicate that extant horsetails diverged in the Early Cenozoic, not long after the Cretaceous-Paleogene boundary ∼66 Mya. The phylogenetic relationships among many ferns have been studied through different methods, and at the broadest level, Borgstrom et al. (2011) results were congruent with previous studies (Des Marais et al. 2003; Grewe et al. 2013); however, the phylogenetic evolution of ferns poses several unanswered answers. Liu et al. (2021) carried out whole-chloroplast genome comparison among Polypodiaceae. The three newly obtained plastomes of Polypodiaceae (N. ovatus, N. fortunei, and P. cuspidatus) were compared with nine previously published plastomes representing three subfamilies of Polypodiaceae, i.e., Microsoroideae, Platycerioideae, and Drynarioideae. The Polypodiaceae plastomes appeared to be structurally similar to each other, showing a typical quadripartite structure consisting of two IRs separated by LSC and SSC. Gao et al. (2018) have shown that repetitive structures with a higher GC content contribute to increasing the thermal stability of the Dryopteris fragrans plastome and maintaining its structure in the face of thermal changes during millions of years of evolution including Cretaceous period when Angiosperms made their appearance (Schneider et al. 2004). Thus, speculate that these repeating structures with a high GC content may be one of the molecular foundations of the adaptation of Polypodiaceae to the environment, which also provides new insights for understanding the environmental adaptation mechanism of plants.
10 Conclusion
Almost all recognized extant fern families and nearly all monilophyte genera at the early diverging nodes have now been sampled in published molecular phylogenetic studies, with few exceptions. Further analyses of the fern chloroplast genomes should provide new insights into the plastid genome evolution. Phylogenomics based on chloroplast genomes has shown many advantages in plant phylogenetics in recent years. With more nuclear data becoming available recently, chloroplast phylogenomics can provide a framework for testing the impact of reticulate evolution in the early evolution of ferns. The examination of plastid genomic features, such as gene content, gene order, intron gain/loss, genome size, nucleotide composition, and codon usage, may also offer independent tests of hypotheses derived from analysis of DNA sequence data. Further plastome sequencing of marattioid ferns and early diverging leptosporangiate ferns will likely be necessary to solidify the sister relationship between these two lineages, but the position of Equisetum is unlikely to be resolvable with more plastome data. However, Grewe et al. (2013) concluded that Equisetopsida is sister to Psilotopsida.
11 Prospects
There are structural variations, gene contents, and GC contents of the chloroplast genomes from green algae to flowering plants (Kwon et al. 2020). The diversity of plastome structures in ferns is insufficiently explored (Logacheva et al. 2016). They further suggested that the ancient HGT DNA transfer from mitochondrial to plastid genome occurred in a common ancestor of ferns which is an evolutionary event that can affect plastome structure. Wilhelmsson et al. (2017) updated rule sets for domain-based classification of transcription-associated proteins (TAPs), comprising transcription factors and transcriptional regulators. Kwon et al. (2020) also presented and corrected some false annotations on the introns in protein-coding and tRNA genes in the genome database, which might be confirmed by the chloroplast transcriptome analysis in the future.
RNA-editing sites should be corrected when plastid or mitochondrial genes and can be used for phylogenetic studies, particularly in those lineages with abundant organellar RNA-editing sites, e.g., hornworts, quillworts, spike mosses, and some seed plants (Du et al. 2020). Expansion or reduction or deletion of IRs resulted in the length variation of the plastomes. Ribosomal RNA genes, rrn, were located in the IRs so that they were present in a duplicate except of the species that had lost one of the IRs. The plastid introns are long compared with the nuclear introns, which might be related with the spliceosome nuclear introns and self-splicing group II plastid introns (Kwon et al. 2020). There were many annotation artifacts in the intron positions in the NCBI database. Fauskee et al. (2021) reported that RNA-editing sites among the three species Adiantum (Pteridaceae), A. shastense, A. aleuticum, and A. capillus-veneris showed a higher degree of conservation, with reverse (U-to-C) editing sites than forward (C-to-U) sites and sites involving start and stop codons were highly conserved. In contrast to this, in seed plants, RNA editing most commonly involves C-to-U changes (Chen et al. 2011). Further studies are needed to study the role of RNA editing in plant evolution.
References
Adams KL, Wendel JF (2005) Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8(2):135–141
Alanazi KM, Ali MA, Kim SY, Rahman MO, Farah MA, Alhemaid F, Lee J (2021) The cp genome characterization of Adenium obesum: gene content, repeat organization and phylogeny. Saudi J Biol Sci 28(7):3768-3775. https://doi.org/10.1016/j.sjbs.2021.03.048
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410. https://doi.org/10.1016/S0022-2836(05)80360-2
Asaf S, Khan AL, Khan MA, Waqas M, Kang SM, Yun BW, Lee IJJSR (2017) Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Sci Rep 7(1):7556
Banks JA, Nishiyama T, Hasebe M, Bowman JL, Gribskov M, Depamphilis CW, Albert VA, Aono N, Aoyama T, Ambrose BA et al (2011) The Selaginella genome identifies genetic changes associated with the evolution of vascular plants. Science 332:960–963
Bausher MG, Singh ND, Lee SB, Jansen RK, Daniell H (2006) The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple’: organization and phylogenetic relationships to other angiosperms. BMC Plant Biol 6:21
Benne R, Burg JVD, Brakenhoff JPJ, Sloof P, Boom JHV et al (1986) Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46:819–826
Ben-Shlomo H, Levitan A, Shay NE, Goncharov I, Michaeli S (1999) RNA editing associated with the generation of two distinct conformations of the trypanosomatid Leptomonas collosoma 7SL RNA. J Biol Chem 274(36):25642–25650. https://doi.org/10.1074/jbc.274.36.25642
Bock R (2007) Structure, function, and inheritance of plastid genomes. In: Bock R (ed) Cell and molecular biology of plastids. Springer, Berlin, pp 29–63
Borgstrom E, Lundin S, Lundeberg J (2011) Large scale library generation for high throughput sequencing. PLoS One 6(4):e19119
Bromham L, Penny D (2003) The modern molecular clock. Nat Rev Genet 4:216–224
Chen H, Deng L, Jiang Y, Lu P, Yu J (2011) RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J Integr Plant Biol 53(12):961–970. https://doi.org/10.1111/j.1744-7909.2011.01082.x
Chen CW, Nagan LT, Hidayat A (2014) Biodiversity, evolution and biogeography of plants. Blumea 59(1):49–58
Christenhusz MJ, Chase MW (2014) Trends and concepts in fern classification. Ann Bot 113(4):571–594. https://doi.org/10.1093/aob/mct299
Christenhusz MJM, Zhang X, Schneider H (2011) A linear sequence of extant lycophytes and ferns. Phytotaxa 19:7–54
Clark J, Hidalgo O, Pellicer J, Liu H, Marquardt J, Robert Y, Christenhusz M, Zhang S, Gibby M, Leitch IJ, Schneider H (2016) Genome evolution of ferns: evidence for relative stasis of genome size across the fern phylogeny. New Phytol 210(3):1072–1082. https://doi.org/10.1111/nph.13833
Cui L, Wall PK, Leebens-Mack JH et al (2006) Widespread genome duplications throughout the history of flowering plants. Genome Res 16:738–749
Daniell H, Lin CS, Yu M et al (2016) Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol 17:134. https://doi.org/10.1186/s13059-016-1004-2
De Bodt S, Maere S, Van de Peer Y (2005) Genome duplication and the origin of angiosperms. Trends Ecol Evol 20:591–597
Der JP (2010) Genome perspectives on evolution in bracken fern. PhD thesis, Utah: Utah State University
Des Marais DL, Smith AR, Britton DM, Pryer KM (2003) Phylogenetic relationships and evolution of extant horsetails, equisetum, based on cp DNA sequence data (rbcL and trnL-F). Int J Plant Sci 164:737–751
Downie SR, Palmer JD (1992) The use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In: Soltis PS, Soltis DE, Doyle JJ (eds) Molecular systematics of plants. Chapman and Hall, New York, NY, pp 14–35
Du XY, Lu JM, Li DZ (2020) Extreme plastid RNA editing may confound phylogenetic reconstruction: a case study of Selaginella (lycophytes). Plant Divers 42(5):356–361. https://doi.org/10.1016/j.pld.2020.06.009
Eneas-Filho J, Hartley MR, Mache R (1981) Pea chloroplast ribosomal proteins: characterization and site of synthesis. Mol Gen Genet 184:484–488. https://doi.org/10.1007/BF00352527
Fan R, Ma W, Liu S, Huang Q (2021) Integrated analysis of three newly sequenced fern chloroplast genomes: genome structure and comparative analysis. Ecol Evol 11(9):4550–4563. https://doi.org/10.1002/ece3.7350
Fauskee BD, Sigel EM, Pryer KM, Grusz AL (2021) Variation in frequency of plastid RNA editing within Adiantum implies rapid evolution in fern plastomes. Am J Bot 108(5):820–827
Gaeta RT, Pires JC, Iniguez-Luy F, Leon E, Osborn TC (2007) Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19:3403–3417
Gao L, Yi X, Yang Y, Su Y, Wang T (2009) Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern cp genomes. BMC Evol Biol 9:130
Gao L, Wang B, Wang ZW, Zhou Y, Su YJ, Wang T (2013) Plastome sequences of Lygodium japonicum and Marsilea crenata reveal the genome organization transformation from basal ferns to core leptosporangiates. Genome Biol Evol 5:1403–1407. https://doi.org/10.1093/gbe/evt099
Gastony GJ, Yatskievych G (1992) Maternal inheritance of the chloroplast and mitochondrial genomes in cheilanthoid ferns. Am J Bot 79:716–722
Gott JM, Emeson RB (2000) Functions and mechanisms of RNA editing. Annum Rev Genet 34:499–531
Grewe F, Guo W, Gubbels EA, Hansen AK, Mower JP (2013) Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol Biol 13:8. https://doi.org/10.1186/1471-2148-13-8
Grosche C, Funk HT, Maier UG, Zauner S (2012) The chloroplast genome of Pellia endiviifolia: gene content, RNA-editing pattern, and the origin of chloroplast editing. Genome Biol Evol 4(12):1349–1357. https://doi.org/10.1093/gbe/evs114
Guichoux E, Lagache L, Wagner S, Chaumeil P, Léger P, Lepais O et al (2011) Current trends in microsatellite genotyping. Mol Ecol Resour 11:591–611
Guillon JM (2004) Phylogeny of horsetails (equisetum) based on the chloroplast rps4 gene and adjacent noncoding sequences. Syst Bot 29:251–259
Guillon JM (2007) Molecular phylogeny of horsetails (equisetum) including chloroplast atpB sequences. J Plant Res 120:569–574
Guillon JM, Raquin C (2000) Maternal inheritance of chloroplasts in the horsetail Equisetum variegatum (Schleich.). Curr Genet 37:53–56
Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2011) Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol 28:583–600
Guo ZY, Zhang HR, Shrestha N, Zhang XC (2016) Complete chloroplast genome of a valuable medicinal plant, Huperzia serrata (Lycopodiaceae), and comparison with its congener. Appl in plant Sci 4(11):apps.1600071. https://doi.org/10.3732/apps.1600071
Gao R, Wang W, Huang Q, Fan R, Wang X, Feng P et al (2018) Complete chloroplast genome sequence of Dryopteris fragrans (L.) Schott and the repeat structures against the thermal environment. Sci Rep 8:1–11
Hasebe M, Iwatsuki K (1992) Gene localization on the chloroplast DNA of the maiden hair fern; Adiantum capillus-veneris. Bot Mag (Tokyo) 105:413–419
Hasebe M, Wolf PG, Pryer KM, Ueda K, Ito M, Sano R, Gastony GJ et al (1995) Fern phylogeny based on rbcL nucleotide sequences. Am Fern J 85:134–181
Haufler CH (1987) Electrophoresis is modifying our concepts of evolution in homosporous pteridophytes. Am J Bot 74:953–966
Haufler CH (2002) Homospory 2002: an odyssey of progress in pteridophyte genetics and evolutionary biology. Bio Sci 52:1081–1093
Haufler CH (2014) Ever since Klekowski: testing a set of radical hypotheses revives the genetics of ferns and lycophytes. Am J Bot 101:2036–2042
Hendy MD, Penny DA (1989) Framework for the quantitative study of evolutionary trees. Syst Zool 38:297–309
Hidalgo O, Pellicer J, Christenhusz M, Schneider H, Leitch AR, Leitch IJ (2017) Is there an upper limit to genome size. Trends Plant Sci 22:567–573
IPPG (2016) A community-derived classification for extant lycophytes and ferns. J Syst and Evol 54(6):563–603. https://doi.org/10.1111/jse.12229
Jiang Y, Fan SL, Song MZ, Yu JN, Yu SX (2012) Identification of RNA editing sites in cotton (Gossypium hirsutum) chloroplasts and editing events that affect secondary and three-dimensional protein structures. Genet Mol Res 11(2):987–1001. https://doi.org/10.4238/2012
Jiang Q, Schneider H, Liu H (2019) Complete chloroplast genome of Angiopteris yunnanensis (Marattiaceae). Mitochondrial DNA B Resour 4(2):3912–3913. https://doi.org/10.1080/23802359.2019.1688111
Karol KG, Arumuganathan K, Boore JL, Duffy AM, Everett KD, Hall JD, Hansen SK, Kuehl JV, Mandoli DF, Mishler BD, Olmstead RG, Renzaglia KS, Wolf PG (2010) Complete plastome sequences of Equisetum arvense and Isoetes flaccida: implications for phylogeny and plastid genome evolution of early land plant lineages. BMC Evol Biol 10:321. https://doi.org/10.1186/1471-2148-10-321
Kenrick P, Crane PR (1997) The origin and early evolution of plants on land. Nature 389:33–39
Kim HT, Chung MG, Kim KJ (2014) Chloroplast genome evolution in early diverged leptosporangiate ferns. Mol Cells 37(5):372–382. https://doi.org/10.14348/molcells.2014.2296
Knie N, Fischer S, Grewe F, Polsakiewicz M, Knoop V (2015) Horsetails are the sister group to all other monilophytes and Marattiales are sister to leptosporangiate ferns. Mol Phylogenet Evol 90:140–149. https://doi.org/10.1016/j.ympev.2015.05.008
Korall P, Schuettpelz E, Pryer KM (2010) Abrupt deceleration of molecular evolution linked to the origin of arborescence in ferns. Evolution 64:2786–2792
Kuo L-Y, Li F-W, Chiou W-L, Wang C-N (2011) First insights into fern matK phylogeny. Mol Phylogenet Evol 59:556–566
Kwon EC, Kim JH, Kim NS (2020) Comprehensive genomic analyses with 115 plastomes from algae to seed plants: structure, gene contents, GC contents, and introns. Genes Genomics 42(5):553–570. https://doi.org/10.1007/s13258-020-00923-x
Labiak PH, Karol KG (2017) Plastome sequences of an ancient fern lineage reveal remarkable changes in gene content and architecture. Am J Bot 104:1008–1018. https://doi.org/10.3732/ajb.1700135
Landis JB, Soltis DE, Li Z, Marx HE, Barker MS, Tank DC, Soltis PS (2018) Impact of whole-genome duplication events on diversification rates in angiosperms. Am J Bot 105:348–363. https://doi.org/10.1002/ajb2.1060
Lanfear R, Welch JJ, Bromham L (2010) Watching the clock: studying variation in rates of molecular evolution between species. Trends Ecol Evol 25:495–503
Lang D, Weiche B, Timmerhaus G, Richardt S, Riaño-Pachón DM, Corrêa LG, Reski R, Mueller-Roeber B, Rensing SA (2010) Genome-wide phylogenetic comparative analysis of plant transcriptional regulation: a timeline of loss, gain, expansion, and correlation with complexity. Genome Biol Evol 2:488–503. https://doi.org/10.1093/gbe/evq032
Langmead B, Nellore A (2018) Cloud computing for genomic data analysis and collaboration. Nat Rev Genet 19:208–219. https://doi.org/10.1038/nrg.2017.113
Lehtonen S (2011) Towards resolving the complete Fern tree of life. PLoS One 6(10):e24851. https://doi.org/10.1371/journal.pone.0024851
Levin DA (1975) Minority cytotype exclusion in local plant populations. Taxon 24:35–43
Levin DA (2002) The role of chromosomal change in plant evolution. Oxford University Press, New York
Li YC, Korol AB, Fahima T, Beiles A, Nevo E (2002) Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Mol Ecol 11:2453–2465
Li YC, Korol AB, Fahima T, Nevo E (2004) Microsatellites within genes: structure, function, and evolution. Mol Biol Evol 6:991–1007. https://doi.org/10.1093/molbev/msh073
Li FW, Kuo LY, Pryer KM, Rothfels CJ (2016) Genes translocated into the plastid inverted repeat show decelerated substitution rates and elevated GC content. Genome Biol Evol 8:2452–2458. https://doi.org/10.1093/gbe/evw167
Li FW, Brouwer P, Carretero-Paulet L et al (2018) Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat Plants 4:460–472. https://doi.org/10.1038/s41477-018-0188-8
Liu HM, Schneider H, Yu Y, Fujiwara T, Khine PK (2019) Towards the conservation of the Mesozoic relict fern Christensenia—a fern species with extremely small populations in China. J Plant Res 132(5):601–616
Liu S, Wang Z, Wang H et al (2020) Patterns and rates of plastid rps12 gene evolution inferred in a phylogenetic context using Plastomic data of ferns. Sci Rep 10:9394. https://doi.org/10.1038/s41598-020-66219-y
Liu S, Wang Z, Su Y et al (2021) Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol 21:31. https://doi.org/10.1186/s12870-020-02800-x
Logacheva MD, Krinitsina AA, Belenikin MS et al (2017) Comparative analysis of inverted repeats of polypod fern (Polypodiales) plastomes reveals two hypervariable regions. BMC Plant Biol 17:255. https://doi.org/10.1186/s12870-017-1195-z
Lu J-M, Zhang N, Du X-Y, Wen J, Li D-Z (2015) Chloroplast phylogenomics resolves key relationships in ferns. J Syst Evol 53:448–457
Marchant DB, Sessa EB, Wolf PG, Heo K, Barbazuk WB, Soltis PS, Soltis DE (2019) The C-Fern (Ceratopteris richardii) genome: insights into plant genome evolution with the first partial homosporous fern genome assembly. Sci Rep 9(1):18181. https://doi.org/10.1038/s41598-019-53968-8
Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L et al (2001) Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13:645–658
Munekage Y, Hashimoto M, Miyake C, Tomizawa K-I, Endo T, Tasaka M, Shikanai T (2004) Cyclic electron flow around photosystem I is essential for photosynthesis. Nature 429:579–582
Nakazato T, Jung MK, Housworth EA, Rieseberg LH, Gastony GJ (2006) Genetic map-based analysis of genome structure in the homosporous fern Ceratopteris richardii. Genetics 173(3):1585–1597. https://doi.org/10.1534/genetics.106.055624
Neuhaus HE, Emes MJ (2000) Nonphoto synthetic metabolism in plastids. Annu Rev Plant Physiol Plant Mol Biol 51:111–140
Nock CJ, Baten A, King GJ (2014) Complete chloroplast genome of Macadamia integrifolia confirms the position of the Gondwanan early-diverging eudicot family Proteaceae. BMC Genomics 15(S9):S13. https://doi.org/10.1186/1471-2164-15-S9-S13
Obermayer R, Leitch IJ, Hanson L, Bennett MD (2002) Nuclear DNA C-values in 30 species double the familial representation in pteridophytes. Ann Bot 90:209–217
Olejniczak SA, Lojewska E, Kowalczyk T, Sakowicz T (2016) Chloroplasts: state of research and practical applications of plastome sequencing. Planta 244(3):517–527
Olmstead RG, Palmer JD (1994) Chloroplast DNA systematics – a review of methods and data analysis. Am J Bot 81:1205–1224. https://doi.org/10.2307/2445483
Palmer JD, Stein DB (1982) Chloroplast DNA from the fern Osmunda cinnamomea: physical organization, gene localization and comparison to angiosperm. Curr Genet 5:165–170. https://doi.org/10.1007/BF00391801
Parisod C, Holderegger R, Brochmann C (2010) Evolutionary consequences of autopolyploidy. New Phytol 186:5–17
Peltier G, Cournac L (2002) Chlororespiration. Annu Rev Plant Biol 53:523–550
Peng Y, Zhang S, Shanshan D, Peng T (2020) The complete chloroplast genome of the fern Asplenium tenerum (Aspleniaceae). Mitochondrial DNA B 5(3):3743–3374. https://doi.org/10.1080/23802359.2020.1831985
Powell W, Michele M, Chaz A, Michael H, Julie V, Scott T et al (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed 2:225–238. https://doi.org/10.1007/BF00564200
Pryer KM, Smith AR, Skog JE (1995) Phylogenetic relationships of extant pteridophytes based on evidence from morphology and rbcL sequences. Am Fern J 85:205–282
Pryer KM, Schneider H, Smith AR, Cranfill R, Wolf PG, Hunt JS, Sipes SD (2001) Horsetails and ferns are a monophyletic group and the closest living relatives to seed plants. Nature 409:618–622
Pryer KM, Schuettpelz E, Wolf PG, Schneider H, Smith AR, Cranfill R (2004) Phylogeny and evolution of ferns (Monilophytes) with a focus on the early leptosporangiate divergences. Am J Bot 91:1582–1598. https://doi.org/10.3732/ajb.91.10.1582
Qiu YL et al (2006) The deepest divergences in land plants inferred from phylogenomic evidence. Proc Natl Acad Sci U S A 103:15511–15516
Qiu YL et al (2007) A non-flowering land plant phylogeny inferred from nucleotide sequences of seven chloroplast, mitochondrial and nuclear genes. Int J Plant Sci 168:691–708
Qu XJ, Moore MJ, Li DZ, Yi TS (2019) PGA: a software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods. 15:50. https://doi.org/10.1186/s13007-019-0435-7
Rai HS, Graham SW (2010) Utility of a large, multigene plastid data set in inferring higher-order relationships in ferns and relatives (monilophytes). Am J Bot 97:1444–1456. https://doi.org/10.3732/ajb.0900305
Raubeson LA, Jansen RK (2005) Chloroplast genomes of plants. In: Henry RJ (ed) Plant diversity and evolution: genotypic and phenotypic variation in higher plants. CABI Publishing, London, pp 45–68
Raubeson LA, Peery R, Chumley TW, Dziubek C, Fourcade HM, Boore JL, Jansen RK (2007) Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8(1):174
Robison TA, Wolf PG (2019) Refernment: an R package for annotating RNA editing in plastid genomes. Appl Plant Sci. 7(2):e01216. https://doi.org/10.1002/aps3.1216
Robison TA, Grusz AL, Wolf PG, Mower JP, Fauskee BD, Sosa K, Schuettpelz E (2018) Mobile elements shape Plastome evolution in ferns. Genome Biol and Evol 10(10):2558–2571. https://doi.org/10.1093/gbe/evy189
Roper JM, Kellon Hansen S, Wolf PG, Karol KG, Mandoli DF, Everett KDE, Kuehl J, Boore JL (2007) The complete plastid genome sequence of Angiopteris evecta (G. Forst.) Hoffm. (Marattiaceae). Am Fern J 97(2):95–106
Rothfels CJ, Schuettpelz E (2014) Accelerated rate of molecular evolution for vittarioid ferns is strong and not driven by selection. Syst Biol 63:31–54
Rothfels CJ, Sundue MA, Kuo LY, Larsson A, Kato M, Schuettpelz E, Pryer KM (2012) Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Syst Biol 61:490–509
Rothfels CJ, Li F-W, Sigel EM, Huiet L, Larsson A, Burge DO, Ruhsam M, Deyholos M, Soltis DE, Stewart CN, Jr Shaw SW, Pokorny L, Chen T, de Pamphilis C, DeGironimo L, Chen L, Wei X, Sun X, Korall P, Stevenson DW, Graham SW, Wong GK-S, Pryer KM (2015) The evolutionary history of ferns inferred from 25 low-copy nuclear genes. Am J Bot 102:1089–1107. https://doi.org/10.3732/ajb.1500089
Ruhfel BR, Gitzendanner MA, Soltis PS et al (2014) From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol Biol 14:23. https://doi.org/10.1186/1471-2148-14-23
Ruhlman TA, Jansen RK (2014) The plastid genomes of flowering plants. Methods Mol Biol 1132:3–38. https://doi.org/10.1007/978-1-62703-995-6-1
Ruiz-Ruano FJ, Navarro-Domínguez B, Camacho JPM, Garrido-Ramos MA (2019) Full plastome sequence of the fern Vandenboschia speciosa (Hymenophyllales): structural singularities and evolutionary insights. J Plant Res 132(1):3–17. https://doi.org/10.1007/s10265-018-1077-y
Sessa EB, Banks JA, Barker MS et al (2014) Between Two Fern Genomes. GigaSci 3:15. https://doi.org/10.1186/2047-217X-3-15
Schlötterer C, Wiehe T (1999) Microsatellites, a neutral marker to infer selective sweeps. In: Goldstein DB, Schlötterer C (eds) Microsatellites: evolution and applications. Oxford University Press, Oxford, pp 238–247
Schneider H, Schuettpelz E, Pryer KM, Cranfill R, Magallón S, Lupia R (2004) Ferns diversified in the shadow of angiosperms. Nature 428:553–557
Schuettpelz E, Pryer KM (2007) Fern phylogeny inferred from 400 leptosporangiate species and three plastid genes. Taxon 56:1037–1050
Schuettpelz E, Pryer KM (2009) Evidence for a Cenozoic radiation of ferns in an angiosperm-dominated canopy. Proc Natl Acad Sci U S A 106:11200–11205
Schuettpelz E, Korall P, Pryer KM (2006) Plastid atpA data provide improved support for deep relationships among ferns. Taxon 55:897–906
Sessa EB, Der JP (2016) Evolutionary genomics of ferns and lycophytes. Adv Bot Res 78:215–254
Shikanai T (2006) RNA editing in plant organelles: machinery, physiological function and evolution. Cell Mol Life Sci 63(6):698–708. https://doi.org/10.1007/s00018-005-5449-9
Shim H, Lee HJ, Lee J, Lee HO, Kim JH, Yang TJ, Kim NS (2021) Plastid genomes of the early vascular plant genus Selaginella have unusual direct repeat structures and drastically reduced gene numbers. Int J Mol Sci 22(2):641. https://doi.org/10.3390/ijms22020641
Shmakov AA (2016) Community-derived classification for extant lycophytes and ferns. J Syst Evol 54:563–603
Smith DR (2009) Unparalleled GC content in the plastid DNA of Selaginella. Plant Mol Biol 71:627–639
Soltis DE, Soltis PS (1987) Polyploidy and breeding systems in homosporous pteridophyta - a reevaluation. Am Nat 130:219–232
Soltis DE, Soltis PM (1998) Choosing an approach and an appropriate gene for phylogenetic analysis. In: Soltis DE, Soltis PM, Doyle J (eds) Molecular systematics of plants II. Kluwer, Boston, pp 1–42
Soltis PS, Soltis DE, Chase MW (1999) Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402:402–404
Stebbins GL (1971) Chromosomal evolution in higher plants. Edward Arnold, London
Stewart WN, Rothwell GW (1993) Paleobotany and the evolution of plants. Cambridge University Press, Cambridge
Sun M, Li J, Li D, Shi L (2017) Complete chloroplast genome sequence of the medical fern Drynaria roosii and its phylogenetic analysis. Mitochondrial DNA B Resour 2(1):7–8. https://doi.org/10.1080/23802359.2016.1275835
Szövényi P, Gunadi A, Li FW (2021) Charting the genomic landscape of seed-free plants. Nat Plants 7:554–565. https://doi.org/10.1038/s41477-021-00888-z
Tangphatsornruang S, Sangsrakru D, Chanprasert J, Uthaipaisanwong P, Yoocha T, Jomchai N, Tragoonrung S (2010) The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Res 17:11–22
Tsuji S, Ueda K, Nishiyama T, Hasebe M, Yoshikawa S, Konagaya A, Nishiuchi T, Yamaguchi K (2007) The chloroplast genome from a lycophyte (microphyllophyte), Selaginella uncinata, has a unique inversion, transpositions and many gene losses. J Plant Res 120:281–290
Vanneste K, Sterck L, Myburg AA, Van de Peer Y, Mizrachi E (2015) Horsetails are ancient polyploids: evidence from Equisetum giganteum. Plant Cell 27(6):1567–1578. https://doi.org/10.1105/tpc.15.00157
Wei R, Yan YH, Harris AJ, Kang JS, Shen H, Xiang QP, Zhang XC (2017) Plastid Phylogenomics resolve deep relationships among Eupolypod II ferns with rapid radiation and rate heterogeneity. Genome Biol Evol 9(6):1646. https://doi.org/10.1093/gbe/evx107
Weststrand S, Korall P (2016a) Phylogeny of Selaginellaceae: there is value in morphology after all! Am J Bot 103:2136–2159
Weststrand S, Korall P (2016b) A subgeneric classification of Selaginella (Selaginellaceae). Am J Bot 103:2160–2169
Wicke S, Schneeweiss GM, dePamphilis CW et al (2011) The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol 76:273–297. https://doi.org/10.1007/s11103-011-9762-4
Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA, Ruhfel BR, Wafula E, Der JP, Graham SW, Mathews S, Melkonian M, Soltis DE, Soltis PS, Miles NW, Rothfels CJ, Pokorny L, Shaw AJ, DeGironimo L, Stevenson DW, Surek B, Villarreal JC, Roure B, Philippe H, dePamphilis CW, Chen T, Deyholos MK, Baucom RS, Kutchan TM, Augustin MM, Wang J, Zhang Y, Tian Z, Yan Z, Wu X, Sun X, Wong GK, Leebens-Mack J (2014) Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc Natl Acad Sci U S A 111(45):E4859–E4868. https://doi.org/10.1073/pnas.1323926111
Wikström N, Pryer KM (2005) Incongruence between primary sequence data and the distribution of a mitochondrial atp1 group II intron among ferns and horsetails. Mol Phylogenet Evol 36:484–493
Wilhelmsson P, Mühlich C, Ullrich KK, Rensing SA (2017) Comprehensive genome-wide classification reveals that many plant-specific transcription factors evolved in Streptophyte algae. Genome Biol Evol 9(12):3384–3397. https://doi.org/10.1093/gbe/evx258
Wolf PG, Rowe CA, Sinclair RB, Hasebe M (2003) Complete nucleotide sequence of the chloroplast genome from a Leptosporangiate Fern, Adiantum capillus-veneris L. DNA Res 10(2):59–65. https://doi.org/10.1093/dnares/10.2.59
Wolf PG, Karol KG, Mandoli DF, Kuehl J, Arumuganathan K, Ellis MW, Mishler BD et al (2005) The first complete chloroplast genome sequence of a lycophyte, Huperzia lucidula (Lycopodiaceae). Gene 350:117–128
Wolf PG, Roper JM, Duffy AM (2010) The evolution of chloroplast genome structure in ferns. Genome 53:731–738. https://doi.org/10.1139/G10-061
Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH (2009) The frequency of polyploid speciation in vascular plants. Proc Natl Acad Sci U S A 106:13875–13879
Xu JH, Liu Q, Hu W, Wang T, Xue Q, Messing J (2015) Dynamics of chloroplast genomes in green plants. Genomics 106(4):221–231. https://doi.org/10.1016/j.ygeno.2015.07.004
Xu L, Xing Y, Wang B et al (2019) Plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Chin Med 14:9. https://doi.org/10.1186/s13020-019-0230-4
Yamaguchi K, Subramanian AR (2003) Proteomic identification of all plastid-specific ribosomal proteins in higher plant chloroplast 30S ribosomal subunit. Eur J Biochem 270:190–205. https://doi.org/10.1046/j.1432-1033.2003.03359.x
Yang JB, Yang SX, Li HT, Yang J, Li DZ (2013) Comparative chloroplast genomes of camellia species. PLoS One 8(8):e73053. https://doi.org/10.1371/journal.pone.0073053
Yi DK, Kim KJ (2012) Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS One 7:e35872
Yoshinaga K, Iinuma H, Masuzawa T, Uedal K (1996) Extensive RNA editing of U to C in addition to C to U substitution in the rbcL transcripts of hornwort chloroplasts and the origin of RNA editing in green plants. Nucleic Acids Res 24:1008–1014
Zhong B et al (2011) Systematic error in seed plant phylogenomics. Genome Biol Evol 3:1340–1348
Zhong B, Fong R, Collins LJ, McLenachan PA, Penny D (2014) Two new fern chloroplasts and decelerated evolution linked to the long generation time in tree ferns. Genome Biol Evol 6:1166–1173
Zhou X-M, Zhang L (2015) A classification of Selaginella (Selaginellaceae) based on molecular (chloroplast and nuclear), macromorphological, and spore features. Taxon 64:1117–1140
Zhu A, Guo W, Gupta S, Fan W, Mower JP (2016) Evolutionary dynamics of the plastid inverted repeat: the effects of expansion, contraction, and loss on substitution rates. New Phytol 209:1747–1756. https://doi.org/10.1111/nph.13743
Zwenger SR, Reinsvold RE, Basu C (2010) Analysis and functional annotation of expressed sequence tags from the diesel tree (Copaifera officinalis). Plant Biotechnol 27:385–391
Acknowledgments
This chapter is dedicated to my (AK) teacher Dr. K. K. Sharma for introducing the subject of pteridophytes now “monilophytes” during master’s study at the University of Rajasthan, Jaipur. We are also thankful to Professor S.K. Verma and Professor S.P. Khullar noted pteridologists for the support and during the preparation of this MS. Authors acknowledge with thanks the authors of publications quoted here:
-
Table 4.1: Rothfels, C.J., et al. (2015). The evolutionary history of ferns inferred from 25 low-copy nuclear genes. American Journal of Botany, 102: 1089-1107. https://doi.org/10.3732/ajb.1500089. Reproduced with license number 5087470694801 dated 14 June 2021.
-
Figure 4.1: Lu, J.-M. et al. (2015). Chloroplast phylogenomics resolves key relationships in ferns. Jnl of Systematics Evolution, 53: 448–457. https://doi.org/10.1111/jse.12180. Reproduced with license number 5085221071437 dated 10 June 2021
-
Figure 4.2: Shim, Hyeonah et al. (2021) “Plastid Genomes of the Early Vascular Plant Genus Selaginella Have Unusual Direct Repeat Structures and Drastically Reduced Gene Numbers” Int. J. Mol. Sci. 22, no. 2: 641. https://doi.org/10.3390/ijms22020641. An open-access article distributed under the terms of the Creative Commons CC BY license
-
Figure 4.3: Grewe et al. (2013). Complete plastid genomes from Ophioglossum californicum, Psilotum nudum, and Equisetum hyemale reveal an ancestral land plant genome structure and resolve the position of Equisetales among monilophytes. BMC Evol Biol 13, 8. https://doi.org/10.1186/1471-2148-13-8
-
Figure 4.4: Fan R, Ma W, Liu S, Huang Q. Integrated analysis of three newly sequenced fern chloroplast genomes: Genome structure and comparative analysis. Ecol Evol. 2021 Mar 18;11(9):4550–4563. doi: https://doi.org/10.1002/ece3.7350. PMID: 33976830; PMCID: PMC8093657
-
Figure 4.5: Fan R, Ma W, Liu S, Huang Q. Integrated analysis of three newly sequenced fern chloroplast genomes: Genome structure and comparative analysis. Ecol Evol. 2021 Mar 18;11(9):4550–4563. doi: https://doi.org/10.1002/ece3.7350. This is an open-access article distributed under the terms of the Creative Commons CC BY license
-
Figure 4.6: Li, FW., Brouwer, P., Carretero-Paulet, L. et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nature Plants 4, 460–472 (2018). https://doi.org/10.1038/s41477-018-0188-8. This is an open-access article distributed under the terms of the Creative Commons CC BY license
-
Figure 4.7: Liu S, et al. (2021). Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol. 2021 Jan 7;21(1):31. doi: https://doi.org/10.1186/s12870-020-02800-x. This article is licensed under a Creative Commons Attribution 4.0 International License. http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain
-
Figure 4.8: Xu, L., Xing, Y., Wang, B. et al. Plastid genome and composition analysis of two medical ferns: Dryopteris crassirhizoma Nakai and Osmunda japonica Thunb. Chin Med 14, 9 (2019). https://doi.org/10.1186/s13020-019-0230-4. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
-
Figure 4.9: Liu et al. (2021). Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol 21, 31 (2021). The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/)
-
Figure 4.10: Li, F. W., Kuo, L. Y., Pryer, K. M., & Rothfels, C. J. (2016). Genes Translocated into the Plastid Inverted Repeat Show Decelerated Substitution Rates and Elevated GC Content. Genome biology and evolution, 8(8), 2452–2458. https://doi.org/10.1093/gbe/evw167. Reproduced with license number 5101790146234 dated 4 July 2021
-
Figure 4.11: Liu et al. (2020) depicted sizes of each part of 16 fern complete plastome sequences. Liu, S., Wang, Z., Wang, H. et al. Patterns and Rates of Plastid rps12 Gene Evolution Inferred in a Phylogenetic Context using Plastomic Data of Ferns. Sci Rep 10, 9394 (2020). https://doi.org/10.1038/s41598-020-66219-y. This is an open-access article distributed under the terms of the Creative Commons CC BY license
-
Figure 4.12: Liu, S., Wang, Z., Wang, H. et al. (2020). Patterns and Rates of Plastid rps12 Gene Evolution Inferred in a Phylogenetic Context using Plastomic Data of Ferns. Sci Rep 10, 9394. https://doi.org/10.1038/s41598-020-66219-y. This is an open-access article distributed under the terms of the Creative Commons CC BY license
AK is thankful to his wife Professor Dr. Vijay Rani Kumar Retd Professor of Botany for critically examining the MS and valuable suggestions.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kumar, A., Giri, P., Uniyal, P.L. (2022). Plastogenomics Provides a Tool to Study Phylogenetic Interrelationships of Monilophytes: A Review. In: Marimuthu, J., Fernández, H., Kumar, A., Thangaiah, S. (eds) Ferns. Springer, Singapore. https://doi.org/10.1007/978-981-16-6170-9_4
Download citation
DOI: https://doi.org/10.1007/978-981-16-6170-9_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-6169-3
Online ISBN: 978-981-16-6170-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)