
Abstract
14-3-3 proteins are signal moderators in sensing various stresses and play essential functions in plant growth and development. Although, 14-3-3 gene families have been identified and characterized in many plant species, its evolution has not been studied systematically. In this study, the plant 14-3-3 family was comprehensively analyzed from green algae to angiosperm. Our result indicated that plant 14-3-3 originated during the early evolutionary history of green algae and expanded in terricolous plants. Twenty-six 14-3-3 genes were identified in the tea genome. RNA-seq analysis showed that tea 14-3-3 genes display different expression patterns in different organs. Moreover, the expression of most tea 14-3-3 genes displayed variable expression patterns under different abiotic and biotic stresses. In conclusion, our results elucidate the evolutionary origin of plant 14-3-3 genes, and beneficial for understanding their biological functions and improving tea agricultural traits in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The sessile nature of plants requires the plants to continuously develop complex mechanism to adjust physiological processes response to changing environmental stress. 14-3-3 proteins are characterized as phosphothreonine /phosphoserine binding proteins, act as scaffolds to bind and regulate other proteins, thus to regulate complex environmental signaling pathways (Denison et al. 2011; Ferl et al. 2002). Therefore, 14-3-3 proteins play major functional roles in most processes, including signal transduction, primary metabolism, protein trafficking, and stress reactions.
Plant 14-3-3 proteins comprise multiple isoforms and they were named as GF14 or GRF for they are a part of protein/G-box complex (de Vetten and Ferl 1994; Rosenquist et al. 2001). With the completion of the genome sequence in a variety of plants, many members of 14-3-3 proteins have been annotated. 14-3-3 protein was originally identified in the model plant Arabidopsis thaliana. To date, many 14-3-3 s have been identified in plant genomes, including Arabidopsis, rice, maize, soybean, grape, and so on (Chen et al. 2006; Cheng et al. 2018; Denison et al. 2011; Wang et al. 2019). Previous studies have demonstrated that plant 14-3-3 proteins have numerous roles in multiple physiological processes. For example, three barley 14-3-3 isoforms were expressed in embryo (Testerink et al. 1999). Most banana 14-3-3 genes displayed high expression during fruit development and postharvest ripening (Li et al. 2016). In Arabidopsis, reduction of GRF10 and GRF9 with antisense technology increased the leaf starch accumulation, indicating that 14-3-3 proteins regulate starch synthesis (Sehnke et al. 2001). Overexpression of cotton 14-3-3L promoted fiber elongation and maturation, while downregulation of 14-3-3L slowed fiber initiation and elongation (Zhou et al. 2015). Most Arabidopsis and rice GRFs displayed variable expression in response to heat, cold and salt stresses (Yashvardhini et al. 2018). Overexpressed AtGRF6 in cotton improved tolerance to drought stress by regulate stomatal conductance (Yan et al. 2004). Due to their broad roles, the physiological functions of 14-3-3 genes are of great interest in plant science.
Tea is the world’s most popular drink, rich in various helpful secondary metabolites (theanine, caffeine, flavonoids) and more than two billion cups are consumed every day (Brody 2019). As a type of evergreen woody plant, tea plant (Camellia sinensis) is primarily cultivated in tropical and subtropical regions. During growth, tea plant is often affected by various environment stresses (e.g., drought, heat, cold, salt, pest), which significantly constrain the yield and quality of tea products (Zhou et al. 2014). Therefore, analyzing the molecular mechanisms of tea plant response to environment stresses is of great significance, and many genes related to stress resistance have been discovered (Zhang et al. 2019). Tea plants underwent two whole-genome duplication (WGD) events by whole genome analysis (Wei et al. 2018; Xia et al. 2017). Many genes associated with secondary metabolisms (e.g., serine carboxypeptidase-like acyltransferase gene) and disease resistance (nucleotide-binding sites with leucine-rich repeats gene, pattern-recognition receptors gene) were significantly amplified in the tea plant genome (Wei et al. 2018; Xia et al. 2017). However, the 14-3-3 family in tea plant has not been characterized to date and the biological functions of this family remain unknown.
In this study, we performed a genome-wide identification and analysis of 14-3-3 proteins in tea and other plant species. Phylogenetic analyses were performed to delineate the evolutionary history of the 14-3-3 family in major angiosperm lineages. The expression patterns in tissues and in response to abiotic and biotic stresses were characterized by examining publicly available RNA-seq data and qRT-PCR. The results explore the evolutionary relationship of 14-3-3 gene family in plant species and provide a theoretical basis for future studies of the biological functions of 14-3-3 gene family members in tea.
Materials and Methods
Genome-Wide Identification of Plant 14-3-3 Proteins
The protein sequences of Arabidopsis thaliana, Coffea canephora, Theobroma cacao, Oryza sativa, Glycine max, Zea mays, Amborella trichopoda, Selaginella moellendorffii, Physcomitrella patens, Chlamydomonas reinhardtii were downloaded from Phytozome (http://www.phytozome.net/) and TPIA (http://tpia.teaplant.org/index.html), respectively. HMMER search (E-value = 1e − 10) and NCBI Basic Local Alignment Search Tool algorithms (BLASTP, E-value = 1e − 10) were employed to search 14-3-3 proteins. All obtained protein sequences were examined for the presence of the 14-3-3 domain (PF00244) using the Hidden Markov Model (HMM) of Pfam (http://pfam.sanger.ac.uk/search) (Finn et al. 2016) and SMART (http://smart.emblheidelberg.de) (Letunic et al. 2015) (Table S1).
Multiple Sequence Alignment and Phylogenetic Analysis
Because the phylogenetic tree based on amino acid sequences was poor statistical support, we performed the Bayesian tree based on protein-coding nucleotide sequences (Table S2). Nucleotide sequences were aligned by MEGA and the hypervariable 5’and 3’ ends were trimmed. The Bayesian tree was constructed with MrBayes 3.2.1 software with the fixed Whelan and Goldman model, four Markov chains and an average SD of 0.01 (Ronquist and Huelsenbeck 2003).
Gene Structure, Motif Analysis, Chromosomal Location and Synteny Analysis
Gene structure analysis was conducted using Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/) (Hu et al. 2015). The motifs of tea 14-3-3 proteins were analyzed using the online MEME program (http://meme-suite.org/tools/meme) (Bailey et al. 2009). The motif distribution type was zero or one occurrence per sequence, maximum number of motifs:7, and only motifs with E-value > 0.05 were present. The location data of tea 14-3-3 genes were obtained from the genome annotation files and the chromosomal location was drawn and visualized using CIRCOS (Krzywinski et al. 2009). The gene duplication landscape was obtained using the MCScanX with the default parameters (Wang et al. 2012) and the syntonic map was generated using CIRCOS with the putative duplicated genes were linked by the connection lines.
Expression Analysis of Tea 14-3-3 Genes
To determine the expression patterns of tea 14-3-3 genes, the publicly available RNA-seq data were used as a resource (transcriptome data were downloaded from the TPIA database (http://tpia.teaplant.org/) (Xia et al. 2019). These transcriptome data include 8 tissues including apical bud, flower, fruit, young leaf, mature leaf, old leaf, root and stem. The fragments per kilobase per million reads (FPKM) value representing the expression level of CsGRF genes. The heatmap with k-means clustering was generated using R software.
RNA Extraction and qRT-PCR Analysis
The tea plants (Camellia sinensis cv. Xinyanghong 10) were grown in a green-house maintained at 23 ± 3 °C with 65 ± 5% room humidity and a 16/8 h (day/night) at Xinyang Normal University, Henan, China. The potted plants were watered and fertilized by the same standards. 2-year-old tea plants of uniform growth without signs of insects or disease were chosen for experiments.
For cold and salt stresses, plants were exposed to 4◦C for 3 days and 100 mmol/L NaCl for 2 days. For drought stress, plants were exposed to drought by withholding water for 7 days, followed by a 24-h recovery period. For insect feeding treatment, fifteen larvae from 3rd or 4th instar E. oblique were placed on each of 3 totally unfed tea plants for obtaining geometrid-damaged tea plant. A group of plants that did not experience any stress were used as control (CK).
The Total RNA was isolated from the leaf for each treatment and RNA quality and quantity were determined using 1% agarose gel electrophoresis. One microgram of RNA was reverse transcribed to cDNA using the PrimeScript RT reagent Kit. Quantitative RT-PCR (qRT-PCR) was performed using SYBR Premix EX Taq on an ABI StepOnePlus machine. Relative expression was calculated by the 2−ΔΔCt method. CsPTB was used as an internal control gene. The expression ratio was represented relative to the control value observed for the gene and fold change was calculated by the formula 2−ΔΔCt, where ΔΔCt = (Ctgene—Ctptb) treatment − (Ctgene – Ctptb) control. The experiments were repeated in triplicate. Specific primers for CsGRF genes were designed using Primer 5 software (Table S3).
Results
Identification of GRF Genes in major Plant Lineages
The complete set of GRF genes were identified from tea and other major plant species based on Hidden Markov model (HMM) algorithm and BLASTP. 147 sequences, each containing a 14-3-3 domain, were identified from 11 different plant species including green algae Chlamydomonas reinhardtii, moss Physcomitrella patens, pteridophyta Selaginella moellendorffii, basal angiosperm Amborella trichopoda, monocots (Oryza sativa, Zea mays), and dicots (Arabidopsis thaliana, Theobroma cacao, Glycine max, Camellia sinensis, Coffea canephora) (Fig. 1). The tea and other 10 plants encode different numbers of GRF isoforms, ranging from two in the green algae C.reinhardtii to 11 in P.patens, 8 in O.sativa, 15 in A.thaliana, with the highest copy number being 26 in C. sinensis. These results indicate that GRFs were originated during the early evolutionary history of algae. Land plants contained higher copy numbers than algae, indicating that expansions of GRF genes occurred after land plants diverged from green algae (Fig. 1).

Summary of the GRF gene among 11 species
26 GRF proteins were identified in tea, with CsGRF21 displayed the shortest coding sequence (363 bp), amino acid length (120 aa), and the smallest molecular weight (13.57 kDa), while CsGRF14 showed the longest coding sequence (867 bp), amino acid length (288 aa), and the biggest molecular weight (32.85 kDa) (Table 1). Their isoelectric point (pI) values ranged from 4.67 (CsGRF16 and CsGRF18) to 8.78 (CsGRF21) (Table 1).
Phylogenetic Classification of GRF Genes into Two Groups
The GRF protein sequence can be divided into three sections: the conserved core region, and a more divergent amino and carboxyl terminus, respectively. The core region contains nine antiparallel a-helices which are function in protein dimerization. The amino acid sequences of tea GRF were highly conserved in the core region with nine a-helices were identified, while its N-terminal and C-terminal regions were more divergent (Fig. 2). This characterization was same as the GRF proteins in other plants, such as Arabidopsis, rice and soybean (Chen et al. 2006; Wang et al. 2019; Wu et al. 1997).

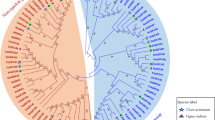
Sequence alignment of tea GRF proteins. Nine α-helices were marked as α1-α9
Although many plant GRF proteins have been identified, the evolutionary origin was still not clear. In previous studies, many neighbour-joining (NJ) phylogenetic trees were constructed based on GRF amino acid sequences, however, these NJ trees were not good in statistical support. To explore the evolutionary origin of plant GRF gene family members, we used the core protein-coding nucleotide sequences and phylogenetic tree was conducted with Bayesian method (Fig. 3). Based on the phylogenetic analyses, plant GRF genes can be divided into two major groups (ε group and non-ε group) with non-ε group contains more isoforms (Fig. 3), consistent with the former classification in other plants, including Arabidopsis (Ferl et al. 2002), rice (Yashvardhini et al. 2018), grape (Cheng et al. 2018), and soybean (Wang et al. 2019). Both groups contain genes from moss, indicating an early origin of GRF genes before the divergence of land plants. The green alga C.reinhardtii has two GRF genes and they were included in the non-ε group for them containing four and one introns, respectively.

Phylogenetic tree of GRF in plant species based on Bayesian analysis of coding nucleotide sequences. Section colours indicate epsilon and non-epsilon types of isoforms. Cs: Camellia sinensis, Ca: Coffea arabica, Thecc: Theobroma cacao, At: Arabidopsis thaliana, Loc Os: Oryza sativa, Gm: Glycine max, Pp: Physcomitrella patens, GRMZM: Zea mays, AmTr: Amborella trichopoda, Sm: Selaginella moellendorffii, Cre: Chlamydomonas reinhardtii (Color figure online)
In A.trichopoda, O.sativa, Z.mays, C.arabica and C.sinensis, the majority of GRF isoforms were included in the non-ε group, while in lower plants and Eudicots, the number of GRF isoforms in ε group was equal to that in non-ε group (Figs. 1, 3). These results indicate different patterns of expansion of GRF proteins in monocots and eudicots, respectively. 26 GRF isoforms were identified in tea (Fig. 3). While only 8 and 11 GRF isoforms were identified in cacao and coffee, respectively (Fig. 4). The tea genome experiences two rounds of whole-genome duplications (WGD) that occurred ~ 30 to 40 and ~ 90 to 100 Mya since the γ-event ∼140 Mya (Wei et al. 2018; Xia et al. 2017). However, the coffee and cacao genomes display no sign of WGD in its lineages since the γ triplication at the origin of the core eudicots (Argout et al. 2011; Denoeud et al. 2014) (Fig. 4). The different expansion patterns of GRF isoforms among these three economical plants suggesting functional variations GRF among tea, coffee and cacao.

Evolution of GRF genes in tea, coffee and cacao. A Distribution of genes encoding GRFs among tea tree, coffee and cacao genomes. B Phylogenetic tree of GRF genes from tea, coffee, cacao and Arabidopsis based on Bayesian analysis
Structure Analysis of CsGRF Genes and CsGRF Proteins
To butter understand the gene structural evolution, the exon–intron organization of CsGRF genes were analyzed with the online service GSDS based on their genome and coding sequences (Fig. 5). The ε group has six to seven introns, while the majority of the non-ε group has tree introns, except for CsGRF1, CsGRF3, CsGRF4, CsGRF18, CsGRF20 genes have no introns (Fig. 5). Exon–intron gene structure correlates with division into epsilon and non-epsilon groups. The exon/intron pattern in terms of the number of introns and exon length were obviously different in the two groups of GsGRF genes, suggesting the diversity of CsGRF genes during the evolution. To investigate the protein sequence features of the CsGRFs, 7 motifs were predicted by the MEME tool (Fig. 5). Majority of CsGRF (18/26, 69%) contained all seven motifs, while the others members contained variable numbers of motifs with CsGRF21, CsGRF23, CsGRF26 only had motifs 4 and 5. The differences in the type and number of motifs in CsGRFs indicate the structural basis for the diversity in protein function.

Conserved motifs and gene structure in CsGRF14s. A Exon/intron structures of CsGRF14 genes. B Conserved motifs of the CsGRF14s. Each motif is represented by a number in colored box
Chromosome Distribution and Synteny Analysis of CsGRFs
Twenty-three CsGRF genes were distributed unevenly on nine chromosomes, and we named the CsGRF genes according to their positions on chromosomes (Fig. 6). The chr1 contained six CsGRF genes, and chr2, 6, 8 only contained one CsGRF gene. Segmental duplication played an essential role in CsGRF expansion in Arabidopsis, rice, Vitis, Soybean and Populus (Cheng et al. 2018; Tian et al. 2015; Wang et al. 2019; Wu et al. 1997; Yashvardhini et al. 2018). To better understand the evolution of CsGRF genes, we conducted a synteny analysis (Fig. 7A). 14 CsGRF isoforms exhibiting segmental duplication events between different chromosomes and contigs (CsGRF5/CsGRF7, CsGRF6/CsGRF7, CsGRF8/CsGRF16, CsGRF9/CsGRF15, CsGRF10/CsGRF17, CsGRF12/CsGRF13, CsGRF22/CsGRF15, CsGRF23/CsGRF26) (Fig. 7A). 8 isoforms were located on one block and lacked duplicates on their corresponding blocks (CsGRF1, CsGRF11, CsGRF14, CsGRF18, CsGRF19, CsGRF20, CsGRF21, CsGRF24). These results suggested that CsGRF genes were also evolved from segment duplication. Similar results were also identified in GRF genes in T.cacao and C.canephora.

Genomic localization of CsGRF14 genes. Twenty-two GRF14 genes were located on 9 out of 15 chromosomes

Synteny analysis of GRF genes in Camellia sinensis, Theobroma cacao, and Coffee canephora. A Synteny analysis of the GRF genes in C.sinensis, T.cacao, and C.canephora, respectively. B Synteny relationship of GRF genes among Arabidopsis, C.sinensis, T.cacao, and C.canephora. The syntenic pairs were prepared according to the homology between them. The positions of all the GRF genes are depicted in the chromosomes. Vertical lines highlight orthologs
To compare the origin and evolutionary relatedness among tea, Arabidopsis, T.cacao and C.canephora, the shared synteny was analyzed (Fig. 7B). In tea, 10 GRF genes (38%) were found to have syntenic pairing with 11 Arabidopsis GRF genes (73%). In C.canephora, 9 GRF genes (81%) were found to have syntenic pairing with 6 T.cacao GRF genes (75%). These results furthered indicated that segmental duplication contributed predominantly to expansion of GRF genes.
Expression Pattern of CsGRF Genes in Different Tissues and Under Abiotic and Biotic Stresses
The expression patterns of CsGRF genes in different tea tissues (e.g., apical bud, flower, fruit, young leaf, mature leaf, old leaf, root and stem) were performed based on the previous RNA-seq data generated by Wei and co-workers (Wei et al. 2018) (Fig. 8). Only two GRF genes (CsGRF18 and CsGRF19) did not expressed in that dataset, and the other 24 CsGRF genes were expressed in more than one but not all tissues. In addition, most CsGRF genes showed a distinct tissue-specific expression pattern, suggesting the diversity of their roles (Fig. 8). For instance, six genes (CsGRF4/7/9/11/16/24) displayed high expression in the flower. Two genes (CsGRF5/22) were specifically expressed in old leaf. CsGRF1 and CsGRF12 had a significantly transcript accumulation in fruit and root, respectively. These tissue-specific expression patterns were consistent with the previous research in AtGRFs, OsGRFs, GmGRFs, VvGRFs and PtGRFs (Cheng et al. 2018; Tian et al. 2015; Wang et al. 2019; Yashvardhini et al. 2018), indicating that CsGRF genes had extensive functional divergence.

Expression patterns of GRF genes in different tissues of tea plant
Different abiotic and biotic stresses such as cold, drought, salinity, and pest adversely affect plant growth and development. Considerable evidence has shown that GRF proteins can regulate the expression of many other stress-related genes and play vital roles in response to both abiotic and biotic stresses (Denison et al. 2011; Yang et al. 2019). To explore the roles of CsGRF genes in tea plant under diverse environmental conditions, the expression patterns of CsGRF genes in different stresses (cold, PEG, NaCl, Ectropis oblique damage) were examined using qRT-PCR (Fig. 9). Ten genes showed no or very low expression in leaves were not further analyzed. The expression levels of most CsGRF genes were changed, and all of them showed significantly different expression patterns in response to different stresses (Fig. 9). The expression of CsGRF14 and CsGRF17 genes were specially upregulated under cold stress, while the expression of CsGRF10 and CsGRF16 were specially downregulated under cold and E.oblique stresses, respectively (Fig. 9). Expression levels of CsGRF21 were increased under drought, salt and E.oblique stresses, while expression levels of CsGRF5 were decreased under these stresses. CsGRF9 was significantly upregulated under abiotic stresses (cold, salt, drought), while CsGRF9 was upregulated under cold stress and downregulated under drought and salt stresses. These results indicate the different roles of CsGRFs in different stress conditions and reveal that the response mechanism is complex and diverse.

Expression analysis of CsGRF genes in response to cold, drought, salt and E. oblique treatments by qRT-PCR. Expression data were normalized to CsPTB gene expression level and every CsGRF genes under control was normalized as ‘1’. The mean expression value was calculated from three replications. The standard deviations of three biological replicates are represented by the error bars. Asterisks on top of the bars indicating statistically significant differences (*p < 0.05)
Discussion
GRFs Genes Have Been Conserved During Speciation
Plant GRFs play important roles in the developmental regulation and in response to environmental stresses. Although, many plant GRFs have been identified, the evolutionary origin and phylogenetic relationship of GRFs are not yet fully explored. In this study, a comprehensive phylogenetic analysis of the plant GRFs from algae, Bryophyta, Pteridophyta and angiosperms was performed (Fig. 3). All the GRFs genes were divided into two groups (ε group and non-ε group), which is in accordance with previous studies in Arabidopsis, rice, grape and soybean (Cheng et al. 2018; Wang et al. 2019; Yashvardhini et al. 2018). In the ancient terricolous plants, there are 11 and 6 GRF isoforms in Physomitrella patens and Selaginella moellendorffii, respectively (Figs. 1, 3). In neonatal terricolous plants, there are 17, 8, 25, 11, 26, 18, 8 and 15 GRF isoforms in Amborella trichopoda, Oryza sativa, Zea mays, Coffea canephora, Camellia sinensis, Glycine max, Theobroma cacao and Arabidopsis thaliana, respectively. These results indicate that the number of GRF was expanded in land plants. Based on the phylogenetic analysis, plant GRFs were originated during the early evolutionary history of algae (Fig. 3).
26 GRF isoforms were identified from tea genome (Figs. 1, 3), and this number was substantially more than those identified in other plants, including coffee and cacao (Figs. 1, 3, 4). In previous studies, the tea genome was shown to experience two WGD events, one occurred ~ 30 to 40 million years ago (Mya) and the second occurred ~ 90 to 100 Mya (Wei et al. 2018; Xia et al. 2017). The high GRFs numbers in tea genome, indicating that the WGD duplication might be the main mechanism of GRF gene family expansion during the evolution of tea. Previous studies have revealed that segmental duplication events contributed to GRF gene duplication in rice, grape and soybean (Cheng et al. 2018; Wang et al. 2019; Yashvardhini et al. 2018). The synteny analysis also showed that segment duplication was the major gene duplication for GRF expansion in tea. Regulatory genes and signaling genes were showed to be retained after duplication comparted to the genome-wide average, and many genes associated with secondary metabolisms and disease resistance were significantly amplified in the tea genome (Wei et al. 2018; Xia et al. 2017). GRFs play important roles in regulating complex environmental signaling pathways and networks, and allowing crosstalk between different pathways. Therefore, the GRFs have been conserved throughout evolution.
Functional Divergence in CsGRF Genes
Many researches have suggested that plant GRF gens are signal moderators, its transcription were regulated by various environmental signals, consistent with their diverse roles in growth, development and stress responses (Denison et al. 2011). Most tea GRF genes were differentially expressed in the different tissues of tea plant (Fig. 5), indicating that CsGRFs play different roles in different organs or tissues. This phenomenon is also reported in other species, including Arabidopsis, rice, grape, maize and soybean (Cheng et al. 2018; Denison et al. 2011; Wang et al. 2019; Yashvardhini et al. 2018).
The transcription of the GRFs genes was either induced or inhibited by cold, drought, salt and E.oblique (Fig. 9), indicating that CsGRFs play important roles in plant response to various abiotic and biotic stresses. In addition, we found that many close CsGRF genes exhibit the opposite expression trend under different stress. For example, CsGRF9 was upregulated under cold, drought and salt stresses, while CsGRF10 was only down-regulated under cold stress. CsGRF24 was upregulated under cold, drought and E.oblique stresses, while CsGRF16 was only down-regulated under E.oblique stress. CsGRF2 and CsGRF21 were upregulated under salt stress, while its expressions display an opposite trend under E.oblique stress with CsGRF2 was down-regulated while CsGRF21 was up-regulated. Therefore, GRFs have different expression patterns in different tissues, various abiotic and biotic stresses, suggesting that the CsGRF genes are involved in the tea plant growth, development and responses to stresses, and different isoforms may play specific roles in specific processes. The complex and diverse expression patterns of CsGRF genes suggested that they potentially related to environmental adaptation and phytochemical properties within the tea lineage.
References
Argout X, Salse J, Aury JM, Guiltinan MJ, Droc G, Gouzy J, Allegre M, Chaparro C, Legavre T, Maximova SN, Abrouk M, Murat F, Fouet O, Poulain J, Ruiz M, Roguet Y, Rodier-Goud M, Barbosa-Neto JF, Sabot F, Kudrna D, Ammiraju JS, Schuster SC, Carlson JE, Sallet E, Schiex T, Dievart A, Kramer M, Gelley L, Shi Z, Bérard A, Viot C, Boccara M, Risterucci AM, Guignon V, Sabau X, Axtell MJ, Ma Z, Zhang Y, Brown S, Bourge M, Golser W, Song X, Clement D, Rivallan R, Tahi M, Akaza JM, Pitollat B, Gramacho K, D’Hont A, Brunel D, Infante D, Kebe I, Costet P, Wing R, McCombie WR, Guiderdoni E, Quetier F, Panaud O, Wincker P, Bocs S, Lanaud C (2011) The genome of Theobroma cacao. Nat Genet 43:101
Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:202
Brody H (2019) Tea Nature 566:1
Chen F, Li Q, Sun L, He Z (2006) The rice 14-3-3 gene family and its involvement in responses to biotic and abiotic stress. DNA Res 13:53
Cheng C, Wang Y, Chai F, Li S, Xin H, Liang Z (2018) Genome-wide identification and characterization of the 14-3-3 family in Vitis vinifera L during berry development and cold- and heat-stress response. BMC Genomics 19:579
de Vetten NC, Ferl RJ (1994) Two Genes Encoding GF14 (14-3-3) Proteins in Zea mays (structure, expression, and potential regulation by the G-box-binding complex). Plant Physiol 106:1593
Denison FC, Paul AL, Zupanska AK, Ferl RJ (2011) 14-3-3 proteins in plant physiology. Semin Cell Dev Biol 22:720
Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, Aury JM, Bento P, Bernard M, Bocs S, Campa C, Cenci A, Combes MC, Crouzillat D, Da Silva C, Daddiego L, De Bellis F, Dussert S, Garsmeur O, Gayraud T, Guignon V, Jahn K, Jamilloux V, Joët T, Labadie K, Lan T, Leclercq J, Lepelley M, Leroy T, Li LT, Librado P, Lopez L, Muñoz A, Noel B, Pallavicini A, Perrotta G, Poncet V, Pot D, Priyono RM, Rouard M, Rozas J, Tranchant-Dubreuil C, VanBuren R, Zhang Q, Andrade AC, Argout X, Bertrand B, de Kochko A, Graziosi G, Henry RJ, Jayarama MR, Nagai C, Rounsley S, Sankoff D, Giuliano G, Albert VA, Wincker P, Lashermes P (2014) The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345:1181
Ferl RJ, Manak MS, Reyes MF (2002) The 14-3-3s. Genome Biol 3:3010
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44:D279
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G (2015) GSDS 20: an upgraded gene feature visualization server. Bioinformatics 31:1296
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639
Letunic I, Doerks T, Bork P (2015) SMART: recent updates, new developments and status in 2015. Nucleic Acids Res 43:D257
Li M, Ren L, Xu B, Yang X, Xia Q, He P, Xiao S, Guo A, Hu W, Jin Z (2016) Genome-wide identification, phylogeny, and expression analyses of the 14-3-3 family reveal their involvement in the development, ripening, and abiotic stress response in banana. Front Plant Sci 7:1442
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572
Rosenquist M, Alsterfjord M, Larsson C, Sommarin M (2001) Data mining the arabidopsis genome reveals fifteen 14-3-3 genes expression is demonstrated for two out of five novel genes. Plant Physiol 127:142
Sehnke PC, Chung HJ, Wu K, Ferl RJ (2001) Regulation of starch accumulation by granule-associated plant 14-3-3 proteins. Proc Natl Acad Sci USA 98:765
Testerink C, van der Meulen RM, Oppedijk BJ, de Boer AH, Heimovaara-Dijkstra S, Kijne JW, Wang M (1999) Differences in spatial expression between 14-3-3 isoforms in germinating barley embryos. Plant Physiol 121:81
Tian F, Wang T, Xie Y, Zhang J, Hu J (2015) Genome-wide identification, classification, and expression analysis of 14-3-3 gene family in Populus. PLoS ONE 10:e0123225
Wang Y, Ling L, Jiang Z, Tan W, Liu Z, Wu L, Zhao Y, Xia S, Ma J, Wang G, Li W (2019) Genome-wide identification and expression analysis of the 14-3-3 gene family in soybean (Glycine max). PeerJ 7:e7950
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, Lee TH, Jin H, Marler B, Guo H, Kissinger JC, Paterson AH (2012) MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40:e49
Wei C, Yang H, Wang S, Zhao J, Liu C, Gao L, Xia E, Lu Y, Tai Y, She G, Sun J, Cao H, Tong W, Gao Q, Li Y, Deng W, Jiang X, Wang W, Chen Q, Zhang S, Li H, Wu J, Wang P, Li P, Shi C, Zheng F, Jian J, Huang B, Shan D, Shi M, Fang C, Yue Y, Li F, Li D, Wei S, Han B, Jiang C, Yin Y, Xia T, Zhang Z, Bennetzen JL, Zhao S, Wan X (2018) Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc Natl Acad Sci USA 115:4151
Wu K, Rooney MF, Ferl RJ (1997) The Arabidopsis 14-3-3 multigene family. Plant Physiol 114:1421
Xia EH, Li FD, Tong W, Li PH, Wu Q, Zhao HJ, Ge RH, Li RP, Li YY, Zhang ZZ, Wei CL, Wan XC (2019) Tea Plant Information Archive: a comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol J 17:1938
Xia EH, Zhang HB, Sheng J, Li K, Zhang QJ, Kim C, Zhang Y, Liu Y, Zhu T, Li W, Huang H, Tong Y, Nan H, Shi C, Shi C, Jiang JJ, Mao SY, Jiao JY, Zhang D, Zhao Y, Zhao YJ, Zhang LP, Liu YL, Liu BY, Yu Y, Shao SF, Ni DJ, Eichler EE, Gao LZ (2017) The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol Plant 10:866
Yan J, He C, Wang J, Mao Z, Holaday SA, Allen RD, Zhang H (2004) Overexpression of the Arabidopsis 14-3-3 protein GF14 lambda in cotton leads to a “stay-green” phenotype and improves stress tolerance under moderate drought conditions. Plant Cell Physiol 45:1007
Yang Z, Wang C, Xue Y, Liu X, Chen S, Song C, Yang Y, Guo Y (2019) Calcium-activated 14-3-3 proteins as a molecular switch in salt stress tolerance. Nat Commun 10:1199
Yashvardhini N, Bhattacharya S, Chaudhuri S, Sengupta DN (2018) Molecular characterization of the 14-3-3 gene family in rice and its expression studies under abiotic stress. Planta 247:229
Zhang Z, Feng X, Wang Y, Xu W, Huang K, Hu M, Zhang C, Yuan H (2019) Advances in research on functional genes of tea plant. Gene 711:143940
Zhou L, Xu H, Mischke S, Meinhardt LW, Zhang D, Zhu X, Li X, Fang W (2014) Exogenous abscisic acid significantly affects proteome in tea plant (Camellia sinensis) exposed to drought stress. Hortic Res 1:14029
Zhou Y, Zhang ZT, Li M, Wei XZ, Li XJ, Li BY, Li XB (2015) Cotton (Gossypium hirsutum) 14-3-3 proteins participate in regulation of fibre initiation and elongation by modulating brassinosteroid signalling. Plant Biotechnol J 13:269
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (32170351), Natural Science Foundation of Henan (No. 212102110154) and Nanhu Scholars Program for Young Scholars of XYNU. The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Contributions
Z-BZ and Z-GF designed the research. Z-BZ, X-KW and SW wrote the manuscript. X-KW, SW, QG performed the identification of GRF genes, protein structure, evolution analysis, and expression analysis. WZ participated in manuscript preparation and revision.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Handling editor: Ananias Escalante.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, ZB., Wang, XK., Wang, S. et al. Expansion and Diversification of the 14-3-3 Gene Family in Camellia sinensis. J Mol Evol 90, 296–306 (2022). https://doi.org/10.1007/s00239-022-10060-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-022-10060-6