
Abstract
We examine the impact of likelihood surface characteristics on phylogenetic inference. Amino acid data sets simulated from topologies with branch length features chosen to represent varying degrees of difficulty for likelihood maximization are analyzed. We present situations where the tree found to achieve the global maximum in likelihood is often not equal to the true tree. We use the program covSEARCH to demonstrate how the use of adaptively sized pools of candidate trees that are updated using confidence tests results in solution sets that are highly likely to contain the true tree. This approach requires more computation than traditional maximum likelihood methods, hence covSEARCH is best suited to small to medium-sized alignments or large alignments with some constrained nodes. The majority rule consensus tree computed from the confidence sets also proves to be different from the generating topology. Although low phylogenetic signal in the input alignment can result in large confidence sets of trees, some biological information can still be obtained based on nodes that exhibit high support within the confidence set. Two real data examples are analyzed: mammal mitochondrial proteins and a small tubulin alignment. We conclude that the technique of confidence set optimization can significantly improve the robustness of phylogenetic inference at a reasonable computational cost. Additionally, when either very short internal branches or very long terminal branches are present, confident resolution of specific bipartitions or subtrees, rather than whole-tree phylogenies, may be the most realistic goal for phylogenetic methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The maximum likelihood (ML) method of phylogenetic inference puts the determination of evolutionary relationships among taxa within a well understood statistical framework. ML estimators have the lowest variance of any estimation method. Also, the ML estimated tree has been shown to converge on the true tree in the limit of infinite data for some substitution models (Chang 1996; Rogers 2001). For these reasons the ML estimated (ML*) tree found for a given sequence alignment is sometimes presented as the sole candidate phylogeny. However, the given tree may in fact be an unreliable estimate for at least two reasons: the purported ML tree may be only a local, rather than a global, maximum in tree space that was found by a particular search algorithm and the amount of information in the sequence alignment may not be sufficient to unambiguously determine the ML tree (Mossel and Steel 2005; Rokas et al. 2003; Pollock et al. 2002; Bininda-Emonds et al. 2001; Cummings et al. 1995).
The problem of how to search effectively in tree space is a formidable one, as the number of possible trees grows factorially with the number of taxa. Typically, searching is performed via systematic rearrangement of the current best tree. Commonly available topological rearrangement algorithms include nearest-neighbor interchange (NNI), subtree pruning and regrafting (SPR), and tree bisection and reconnection (TBR). These are implemented in the popular programs PAUP* (Swofford 2002) and Phylip (Felsenstein 2004). NNI-based search explores a relatively small neighborhood of the current best tree and thus tends to converge to local maxima. SPR and TBR will search tree space more broadly, though it is a challenge to design algorithms that are practical for large trees. Recent algorithms (see, e.g., Hordijk and Gascuel 2005; Stamatakis et al. 2004) use limited SPR moves and can sometimes “escape” local maxima and still converge rapidly on an estimated ML tree. It is a fact, however, that for the problem of phylogenetic inference, a local maximum that a program “escaped” from may be the true solution. For sequence alignments of finite length, stochastic deviations from the asymptotic likelihood may be greater than the difference in values for two or more trees. This leads to the wrong topology exhibiting the ML.
This problem can be handled in a hypothesis testing context by considering confidence sets of trees. A (1 – α) × 100% confidence interval for topologies specifies a set of trees for which the calculated likelihood differences are insignificant at the given α level. This means that the false rejection rate when evaluating hypotheses is α × 100%. To construct such a set, one usually begins with a set of potential ML trees and uses their site likelihood differences to pare the set down to those topologies that cannot be rejected as a possible best tree. A bootstrap method for doing so was first suggested by Felsenstein (1985). Subsequent methodologies have been put forth by various authors; for an overview, see Felsenstein (2003). In general, one endeavors to construct a set that is as small as possible yet still contains the true tree most of the time. Confidence tests are sometimes applied to the results of a number of independent tree searches begun from random initial starting trees. Clearly, the better an algorithm is at finding a global likelihood maximum, the less likely it is that alternative hypotheses will be present in the set of output topologies. For this reason, it is desirable to retain information on local maxima encountered during the tree search.
The program TRExML (Wolf et al. 2000), written with this goal in mind, retains the k most likely trees during its search, then applies a confidence region criterion to the candidate set after the search is completed. The parameter k is ad hoc and fixed ahead of time. Furthermore, TRExML uses a modified version of the stepwise sequence addition algorithm of fastDNAML which explores tree space relatively narrowly and also is significantly biased by sequence addition order (Felsenstein 1981; Olsen et al. 1994).
The recently implemented program covSEARCH (Blouin et al. 2005a) is explicitly designed both to find the ML* tree and to explore the likelihood surface for alternative hypotheses that cannot be rejected from a statistical point of view. Like TRExML, covSEARCH retains pools of candidate ML trees during the search but combines exhaustive SPR rearrangements of the current pool with confidence tests to dynamically determine the size and content of the pool. The phylogenetic confidence tests of Kishino and Hasegawa (1989) (KH and RELL-ws), Shimodaira and Hasegawa (SH; 1999), and Strimmer and Rambaut (2002) (ELW) and a variant proposed here (ECDF-site) are available. The use of confidence set heuristics automates the selection of pool size and adapts it to the statistics of the likelihood surface for the alignment under consideration. This can improve accuracy in a computationally efficient way. covSEARCH can take advantage of a parallel computing environment, improving the feasibility of searches requiring large pools of trees to be returned. Nonetheless, the exhaustive SPR rearrangements make the current software most appropriate for either relatively small trees (tens of taxa) or large trees with some constrained nodes.
In this work we apply covSEARCH to simulated amino acid sequences in order to examine in some detail under what circumstances it is advisable to look beyond the ML* tree for inference. We present situations where the ML* tree found after many trial searches is most often not the true tree. For most criteria, the final confidence set does usually contain the true topology. We consider the suitability of using a simple consensus method as a way to summarize the content of the covSEARCH output set. Like the ML* tree, however, the consensus tree can be an unreliable estimate of the true tree. We suggest examining individual node frequencies within the confidence set to determine support for specific bipartitions. More importantly, it is clear from our results that interpreting the ML* tree as the true tree is incorrect. This interpretation assumes that the user provides infinite data and that the model of substitution is not misspecified.
Methods
Heuristic Search Algorithms
The covSEARCH topology optimization algorithm is seeded with a single tree. The default is the BIONJ tree (Gascuel 1997); alternatively a user may specify a starting tree. All possible SPR rearrangements are performed on the initial tree to generate the first candidate tree pool. Likelihoods are estimated for all of the trees in the pool. For the confidence set heuristic algorithm, a confidence test is then applied to the candidate pool and trees that fall below the preselected significance level for the test are deleted from the pool. All possible SPRs are performed on all of the remaining trees in the candidate pool to generate a new pool. This process is iterated until there is no longer any change to the candidate pool.
Confidence set heuristic topology optimization algorithm
-
1.
Seed pool P with BIONJ or user-specified tree, \( {\rm P}_0 = \tau _{init} \)
-
2.
Perform search iteration step i.
-
a.
Perform all possible SPR rearrangements on all \( t\,\epsilon\,{\rm P}_i \) and add newly generated trees temporarily to pool: \( {\rm P}^\prime_i = {\rm P}_i \cup SPR({\rm P}_i) \)
-
b.
Evaluate likelihoods for all \( t \in P^{\prime}_i \) and determine current best tree \( \tau _{ML}* \)
-
c.
Apply ML phylogeny confidence test to \( {\rm P}^{\prime}_{i} \), evaluating p-values \( p_{t_j} \) for each candidate topology tj.
-
d.
Delete from the pool all t j whose p-value falls below the designated significance level: \( {\rm P}_{i + 1} \leftarrow CS({\rm P}^{\prime}_{i}) = \{t_j :p_{t_j} > \alpha\}. \)
-
a.
-
3.
If \( ({\rm P}_{i + 1} \ne {\rm P}_i) i \leftarrow i + 1 \) and return to step 2;
otherwise
output pool contents and exit.
The implemented confidence test procedures are outlined below. The reader is encouraged to see the references (Kishino et al. 1990; Shimodaira and Hasegawa 1999; Strimmer and Rambaut 2002; Goldman et al. 2000) for further details.
In the covSEARCH implementation of the KH test (Kishino et al. 1990), a tree τ is rejected if L ML* – L τ lies outside the 1 – α confidence interval for N(0,S·Var(δ)), where S is the number of sites and Var(δ) is the variance of the distribution of site log likelihood differences. For the RELL-ws (resampling estimated log likelihood-winning sites) variation, the number of positive site likelihood differences is tabulated from a number of resampled sites and the alternative tree is said to be rejected at level α if more than (1 – α) × 100% of the paired site likelihood differences favor the ML* tree. These tests are defined for the comparison of a single topology against the ML* topology and their use in contexts that require multiple comparisons is not technically correct (Goldman et al. 2000).
The procedure of Shimodaira and Hasegawa (1999) explicitly accounts for the possibility that any of the candidate topologies may be the ML tree, performing a single all-against-all comparison rather than multiple tests of pairs of topologies. Briefly, one calculates the test statistic T = L ML* – L for each topology τ. One then constructs replicates of the test statistic for each topology using bootstrap resampling (RELL was used in this work). For each bootstrap sample b∈1..B the test statistic replicate \( T_{\tau b} = max_t \{ \tilde L^{\prime}_{tb} - \tilde L^{\prime}_{\tau b}\} \) is calculated, where \( \tilde L_{\tau b} \) signifies that the log likelihood has been centered by subtracting the corresponding row average. One then computes \( p_\tau = card\{ b:T_{\tau b} > T_\tau \} /B \) and the confidence set includes those topologies for which \( p_\tau \ge \alpha \), where α is the specified significance level.
The empirical cumulative distribution function (ECDF-site) method is based on the raw distribution of site log likelihood differences, \( cdf(\delta \equiv l_{i,ML}* - l_{i,\tau }) \). A candidate tree is rejected if δ=0 lies beyond 1 – p, i.e., the probability that site log likelihoods in the ML* tree are greater than those for the test tree exceeds 1 × α.
The expected likelihood weight (ELW) method of Strimmer and Rambaut (2002) uses an average over bootstrap samples to compute the expectation of the likelihood weight: \( w_\tau \approx {1 \over b}\sum\nolimits_{b = 1}^B w_{\tau b} \) with \( w_\tau = {L_\tau \over \Sigma L_t} \). The confidence set is computed as the smallest set such that the probability of one of the included candidates being the correct model for the data exceeds a specified confidence value. That is, if one orders the topologies according to likelihood weight such that \( w_\tau > w_{\tau + 1} \), the probability of the set of the first m topologies containing the true topology is \( \sum\nolimits_{t = 1}^m {\omega _t} \). Thus the confidence set at significance level α consists of the first \( t_{min} \) topologies in the ordered list, where \( t_{min} \) is the smallest index for which \( P(\tau _{true} \in C) \ge 1 - \alpha \).
The windowed likelihood algorithm used to examine convergence is identical to the confidence set heuristic algorithm given above except that steps 2c and 2d are replaced by:
Windowed likelihood heuristic search
2c’. Delete from the pool all trees t j such that \( L_{ML^* } - L_{t_j } > W_L \).
Unlike the confidence set algorithm, the windowed likelihood algorithm does not allow for a predefined significance level; the window parameter W L must be selected by the user at the start of each run. W L = 0 means the search is single threaded and the only SPR search path pursued for each iteration is the one that modifies the current best topology. Accuracy with respect to the ML tree is determined relative to the ML tree found among all searches and confidence sets that were actually run, i.e., a global maximum cannot be guaranteed.
Software Tools and Simulation Details
The covSEARCH program used for inference was developed at Dalhousie University (Blouin et al. 2005a). ML and confidence tests are implemented in the package libcov (Butt et al. 2005). Some source code has been incorporated from freely available programs including branch length optimization routines from TREE-PUZZLE (Schmidt et al. 2002) and a random number generator from PAML (Yang 2005). Consensus tree calculations are based on the Phylip Consense program (Felsenstein 2004). Simulated protein sequences were generated using the program covTREE (Blouin et al. 2005b), with the exception of Fig. 3b, for which a modified version of covTREE that implements random selection of various model parameters was used. Source code for covTREE, libcov, and covSEARCH is available for download at the following URL: http://www.cs.dal.ca/∼cblouin.
For the results shown in Figs. 1 and 2, each topology is characterized by two parameters: internal branch length, L i , and terminal branch length, L E . Alignments were simulated using α=0.5 with four site rate categories. Searches were seeded with the neighbor-joining tree and phylogenetic reconstruction was performed using JTT+Γ+F with four site rate categories. Accuracy and confidence set sizes reported in Figs. 1 and 2, respectively, are averages over 100 bootstrap replicates of alignments of 1000 sites in length.

Relationship of true topology to maximum likelihood estimated tree and majority rule consensus trees for simulated data. Li refers to the length of the internal branches in expected number of substitutions. LE is a terminal branch length. “True Found” indicates the percentage of times the true tree was contained within the final confidence set. “True is ML*” refers to the fraction of the time that the found true trees were equal to the ML* tree for a given topology. “Consensus Is True” refers to the percentage of the time the majority rule consensus tree equaled the true tree. The topological distance between two trees is the number of branches present in one tree and not the other.

Size of converged confidence sets for topology search using covSEARCH. Li refers to the length of the internal branches in expected number of substitutions. LE is a terminal branch length. Standard deviation is given in parentheses. Statistics for reconstruction both with rates across sites (RAS) variation and assuming homogeneous site rates (–) are shown. Boxes with only lower bounds (“>2K”) indicate that the covSEARCH program was halted before convergence was reached.
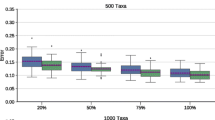
For Fig. 3, 216 single gene alignments were generated using the opisthokont topology shown in the figure as a starting tree. Random selection of the shape parameter, α, was used in the gamma model of rate variation across sites. All branches of the starting tree were scaled proportionately by a factor of 0.05, 0.2, 0.5, 1.0, 1.5, and 2.0. For each scaling factor, 36 alignments were generated: 6 alignments of each of 50, 150, 250, 350, 450, and 550 amino acids. covSEARCH was run with the KH test option on all 216 alignments.

Node support as a function of confidence set size for simulated alignments. a Tree used as the basis for simulated alignments to study node support in confidence sets. The tree shown was inferred from concatenated mitochondrial protein sequences (J. Leigh, unpublished) for the opisthokonts Podospora anserina, Penicillium marneffei, Yarrowia liolytica, Saccharomyces cervisia, Schizosaccharomyces pombe, Schizosaccharomyces japonicus, Cryptococcus neoformans, Crinipellis perniciosa, Spizellomyces punctatus, Harpochytrium sp. 94, Monsiga brevicollis, Metridium senile, and Amoebidium parasiticum. b Number of nodes with >80% support in KH confidence sets for simulations based on the 13-taxon ophisthokont tree pictured in a.
Protein Sequence Data
α-Tubulin sequences were obtained from the National Center for Biotechnology Information Protein database and aligned using ClustalW (Thompson et al. 1994). Five sequences represent yeasts including two Saccharomyces cerevisiae paralogues (accession numbers S50871 and B25076), Candida albicans (AAB53194), and two Schizosaccharomyces pombe paralogues (A25072 and B25072). Five represent microsporidia including Encephalitozoon hellem (P92120), Encephalitozoon cuniculi (NP_586048), Nosema locustae (AAC47419), Glugea plecoglossi (AAN35162), and Trachipleistophora hominis (AAN35139).
Mammal mitochondrial protein sequence alignments were obtained from the Goldman Group web site at http://www.ebi.ac.uk/goldman. This data set consists of 3414 amino acid sites for Homo sapiens (human), Phoca vitulina (harbor seal), Bos taurus (cow), Oryctolagus cuniculus (rabbit), Mus musculus (mouse), and Didelphis virginiana (opossum) taxa.
Results
Efficacy of ML Search
We first examine ML search convergence for real and simulated data using fixed likelihood windows. Table 1. shows the rate of convergence on the ML tree as a function of the likelihood interval used during the search for one real and two simulated data sets. For the 12 taxa ef1α alignment dramatic variation with search breadth is evident. It takes a search pool only 5 units of likelihood in width to practically guarantee recovery of the ML topology. For the longer simulated sequences, containing 1025 sites, single path search proves adequate, finding the ML tree 100% of the time. The results for the 20-taxon simulated data set with 400 sites indicate a less challenging likelihood surface than for the real data case, with single path search finding the global optimum 87% of the time. Nonetheless, some improvement is seen when the search likelihood window is broadened.
In order to test the reliability of the ML* tree for finite data, sequences were simulated from the topologies shown in the first column in Fig. 1 and confidence sets inferred using covSEARCH. Topology B presents the most challenging case for inference with all long terminal branches and five of six short internal branches. The percentage of the time the estimated ML tree was found to equal the true tree is shown in column 6 of Fig. 1. ML* tree agreement with the true tree is perfect, with the exception of topologies with short internal branch lengths corresponding to cases B and F. For these cases, the average accuracy of the estimated ML tree falls to 53.2% and 79.3%, respectively. Thus, for fixed short internal branches, long terminal branches worsen the accuracy of the ML* tree significantly.
Confidence Set Heuristic Search Performance
For topologies where the ML* tree is often not the true tree, the data in column 5 of Fig. 1 show that accuracy in terms of discovering the true tree within the converged covSEARCH confidence sets is generally good. In fact, only in the cases of very short internal branch lengths and when using the ELW test is there significant failure to find the true tree within the final confidence set. Column 8 indicates that improved accuracy is achieved through a broadening of the confidence set topological content with respect to the true tree.
One sees from the data in column 7 of Fig. 1 that the consensus tree is not equal to the true topology under a broader range of circumstances than for the ML* tree. Consensus tree agreement with the true tree not only is poor for topologies with short internal branches, as is the case for the ML estimate, but also exhibits diminished agreement for topologies with only one or two long branches. When only one or two long branches are present and internal branches are not very short, the KH, SH, and ELW confidence sets allow recovery of the true tree using the consensus tree method.
The average sizes of converged confidence sets for various combinations of topology and rejection criteria are listed in Fig. 2. The data show how both relatively long and relatively short branch lengths act to increase the number of accepted candidate trees. For topology E, which has two long terminal branches, most measures find small numbers of trees. RELL-ws and ECDF-site perform exceptionally poorly in this case, generating confidence sets of more than 100 trees. The short internal branches that are ubiquitous in the case of topology F lead to larger confidence sets for all criteria. The worst-case scenario for tree resolution occurs in case B with all long terminal branches and almost all short internal branches. Here we see that the RELL-ws and ECDF-site confidence sets are so large that they preclude running the algorithm to convergence. ELW generates confidence sets with very few trees in all cases. As noted above, however, it achieves poor coverage in challenging inference situations.
Though the consensus tree is often not equal to the true tree for difficult inference problems, a substantial fraction of the nodes in the consensus tree is typically present in most (≥80%) of the topologies for simulated alignments as shown in Fig. 3b. In a 13-taxon tree, 6 of 11 of the nodes are at the 80% support level when there are close to 100 trees in the confidence set. Even for confidence sets containing of the order of 10,000 trees, more than one node exhibits strong support. Thus even for extremely large confidence sets information on specific species bipartitions may still be extracted.
The computational burden involved in evaluating a confidence set of trees at each search step can be significant. The simulation results discussed here were gathered on an AMD Opteron cluster utilizing eight processors for which convergence to confidence sets with up to 2000 trees took only a few minutes. Figure 4 plots covSEARCH running time as a function of the number of trees evaluated during the search for the SH, KH, and ECDF-site tests for the same alignments as in Figs. 1 and 2. ELW times were observed to overlap those of the KH test, while RELL-ws runtime behavior mimics that of ECDF-site. The plots for all tests appear linear (which is to be expected) up to >2000 search trees. The change in slope in the vicinity of 2500 trees for the SH test and 3500 trees for the ECDF-site test is most likely hardware-specific and related to the efficiency of memory handling for large data structures. In general, peak memory usage grows linearly with the maximum candidate pool size, which in turn is a function of both the number of taxa and the difficulty of the search problem.

Time to confidence set convergence as a function of trees evaluated for the SH, KH, and ECDF-site tests on an AMD Opteron cluster utilizing eight processor nodes. Data are the same as those used to generate Figs. 1 and 2 with no RAS for reconstruction. Not shown are data for the ELW test, which overlaps those for the KH test over the observed range of 1 to 500 trees evaluated, and the RELL-ws test, which behaves similarly to the ECDF-site test.
Phylogenetic Examples
Mammalian mitochondrial protein sequences
We first demonstrate the effectiveness of the confidence set heuristics using the mammal mitochondrial protein sequences analyzed by Shimodaira and Hasegawa (1999). As this data set has only six taxa, it is a seemingly trivial search problem with only 105 possible trees. However, because the SPR neighborhood of the initial neighbor joining tree contains only 48 topologies, i.e., the first search step is not exhaustive, this problem is still illustrative of the covSEARCH algorithms. Table 2. lists the top eight topologies in likelihood order and indicates their presence or absence in the confidence sets for the KH, SH, and ELW tests. RELL-ws and ECDF-site results are not listed because the confidence sets contain >90 trees, making them relatively ineffective. The SH, KH, and ELW tests evaluate a total of 89, 79, and 41 candidate trees, respectively. The SH and KH tests result in sets of 8 and 7 trees; these are the same numbers of topologies that would be found by applying the SH and KH tests to the 15 topologies containing the (harbor seal, cow) grouping, as done by Shimodaira and Hasegawa (1999). Search using the ELW test generates the smallest confidence set, including only the ML* tree and the tree with the second highest likelihood. This may indicate a problem of insufficient coverage, as was the case for some simulated data sets. Interestingly, the topology that is present in the SH set but missing in the KH set does not have the lowest likelihood. This demonstrates how the test criterion, through its determination of the search pool, can generate different solutions than consideration of the rank order of likelihood alone.
Microsporidia and ascomycete α-tubulin sequences
Where microsporidia originate within the evolutionary history of eukaryotes is a current point of controversy, therefore finding all plausible trees and quantifying their support is of significant biological interest (Keeling 2003). Resolution for the α-tubulin tree containing the ascomycetes Saccharomyces cerevisiae, Schizosaccaromyces pombe, and Candida albicans is expected to be good, based on prior analyses. Though the number of trees in the converged confidence set varies with the confidence test used, the consensus tree is equal to the ML* tree for all tests. Figure 5 shows the majority consensus tree with labeled nodes. In Table 3 we list the support values for all of the nodes. In addition to the results using heuristic search, ML bootstrap results are compared directly by constructing a confidence set consisting of the four distinct trees found of 100 bootstrap data runs. From this one can see that the microsporidia clade is strongly supported in all cases. Nodes D and G are inadequately supported according to the heuristic confidence set results (<50%) and also show weaker ML bootstrap support. Note that the ML tree is found in >90% of the single-threaded bootstrap data searches so that using the raw percentages as a measure of support without accounting for duplications may be significantly misleading. The advantage in computation time for the heuristic search methods over bootstrapping can be quite large. Here the single KH test breadth search is more than 20 times faster than the accumulated time for 100 bootstrap searches.

The consensus tree (equal to the maximum likelihood tree in this case) for α-tubulin. Internal nodes are labeled in order to associate confidence set and bootstrap support given in Table.
In contrast to the above case, estimated branches for the ascomycete taxa Mycosphaerella graminicola, Rhynchosporium secalis, Sordaria macrospora, Ajellomyces capsulatus, and Emericella nidulans are very short, leading one to expect poor tree resolution. This is confirmed in the confidence set results, where an alignment of these taxa and microsporidia yields a set of nearly 200 trees under the SH criterion (data not shown).
Discussion
Whether local maxima are common features of the likelihood surface for real sequences has been a matter of some debate. While previous simulation studies by Rogers and Swofford (1999) suggested that local maxima are not problematic, Chor et al. (2000) were able to find data sets exhibiting complex likelihood surfaces for branch length optimization. Here we have shown evidence for the existence of multiple topologies that are statistically indistinguishable from the true and/or ML* tree for both real and simulated amino acid alignments. Performing a parallel search on a pool of trees within some likelihood interval of the maximum estimate illuminates the problem of local maxima: when only one path is searched, the estimated peak is unlikely to be optimal. As the search is expanded to parallel threads close in likelihood to the current best tree, performance in terms of finding the global maximum improves steadily.
Disagreement between the ML* tree and the true tree can occur due to the use of finite data for inference or it can also be the result of model misspecification. Note that the simulated data for Fig. 1 were generated under a JTT+Γ+F model but recovered using only JTT+F in an effort to mimic real data model misspecification. Thus in Fig. 1, all simulations are misspecified (due to no RAS), nonetheless there is a clear trend toward ML* inaccuracy in the case of short internal branches. This suggests that the primary cause of inaccuracy is lack of resolving information relative to the true topology in the alignment used for inference.
Low phylogenetic signal creates a difficult search problem in tree space, hence large confidence sets are required to generate a sufficiently broad search to find the global maximum. For fixed short internal branches, the addition of long terminal branches worsens the accuracy of the ML* tree significantly. This is not surprising since short internal branches imply that large amounts of sequence information are required to properly resolve the phylogeny, while long branches indicate that the phylogenetic information per site is low. It is significant because real data sets often have these branch characteristics, as is the case for one of the α-tubulin trees. It is quite encouraging that our simulation results show that the confidence sets nearly always recover the true tree under these circumstances.
In general, there is a tradeoff between coverage and confidence set size, the specifics of which depend on both the tree properties and the confidence set criterion that is used. While ELW yields consistently small confidence sets, its accuracy is relatively poor when the true topology has both short internal branches and long terminal branches. If accuracy is most valued, the KH and SH tests do well with small numbers of retained trees compared to RELL-ws and ECDF-site. In our tests, the KH and SH tests perform very similarly despite the KH test not being intended for the case of multiple tree comparisons to the ML tree. However, SH should be preferred because it is statistically sound in this context. In general, covSEARCH converged confidence sets will be only approximations to the true confidence sets due to the limitations of searching.
Though neither the ML* tree nor the majority rule consensus tree is reliably the true tree, Fig. 3b demonstrates that some biological information is recoverable on a per node basis even for confidence sets of a few thousand trees. Extracting biological answers from these sets is a topic for further research and ongoing work in our group.
Obtaining large output sets of trees radically deviates from the usual ML* tree output that phylogeneticists expect for a ML package. A relatively large number of trees in the output set indicates that only a fraction of the relationship that has to be present in a strictly bifurcated tree is supported by the data. In many cases, the number of trees in the output set is the product of the number of valid topologies within a few subtrees. We suggest focusing the sampling in sequence to more specific questions to avoid the combinatorial increase in the number of trees in the output set.
The time to convergence of the covSEARCH program will generally increase with the size of the required confidence set, which in turn will vary with the degree of phylogenetic resolution allowed by the input alignment. Dependence on the number of taxa and sites in the input alignment will be somewhat complex. Additional taxa and/or sites increase pool update time on a per tree basis, however, the improved phylogenetic resolution that may result from the additional data should decrease the average number of trees in the search pool. Whether adding genes or adding taxa is most beneficial for increasing resolution will vary with the problem under consideration (Zwickl and Hillis 2002; Rokas and Carroll 2005). covSEARCH can optimize multiple genes with constrained topologies as a means to provide more phylogenetic signal. The implications of the latter is the matter of ongoing work.
covSEARCH runtime and memory requirements do not currently allow for application to very large alignments (hundreds of taxa). One possible strategy is to first estimate large phylogenies along with bootstrap support values for each branch using a faster search method (e.g., Phyml or RAXML). Branches with very high bootstrap support may be fixed during the covSEARCH procedure, thus restricting the confidence search to areas of tree space where it can be of most benefit.
The criteria used here are all based on paired site likelihoods and, as such, represent only one category of measures for tree comparison. Some newer likelihood confidence tests have not been considered here (e.g., Shimodaira 2002; Shi et al. 2005) but may be added in the future. Another alternative is parametric bootstrapping (Swofford et al. 1996). Given that models of real data are always somewhat misspecified, however, it seems prudent to choose nonparametric test options. Additionally, parametric bootstrapping tests would likely increase the computational burden dramatically.
The strategy described in this work is a breadth-first approach. This has many downsides from a practical perspective, which impact the runtime of the algorithm as well as the memory requirements. However, it is clear that single-threaded or otherwise greedier approaches may not perform as well in difficult situations. For instance, one might consider starting an NNI-based algorithm from many random initial trees, recording the local maximum found during each run, and applying a confidence test after the fact. This will not work in the case that broad maxima in likelihood are present, i.e., multiple trees near a single maximum cannot be rejected. In order to output all statistically plausible topologies, one must maintain information on trees that are close to optimal in likelihood but are not precisely at a local maximum.
The use of an exhaustive SPR enumeration at each iteration, and for each tree in the current candidate pool, can be seen as a brute force approach. A TBR enumeration would be more exhaustive although practically prohibitive. We are anticipating that larger datasets will further highlight the value of a broad search strategy. However, further work is necessary to refine the algorithm or define new heuristics to make these larger analyses possible on common computer hardware.
Conclusions
ML-based phylogenetic inference can be made more reliable by choosing inference methods that are well matched to the information content of the data. For sequence alignments that provide ample phylogenetic signal relative to the true tree, finding the ML estimated tree may be adequate. In situations where multiple maxima are close in likelihood, as is the case for topologies with short internal branches, a broader search for solutions is necessary. The program covSEARCH provides an efficient set of algorithms in this context and accomplishes robust phylogenetic inference using pool-based topology search guided by confidence sets. Computing phylogenies using this method provides not only the ML estimated tree, but also statistically significant alternative topologies.
One should be cautious in interpreting the majority consensus tree for the confidence sets, as it is not equal to the true tree in many cases. Rather, confidence set output can be used to assess whether there is enough sequence information to resolve specific phylogenetic relationships. Large confidence set solutions indicate that the phylogenetic signal is insufficient to resolve an entire tree. For these situations we recommend focusing on more specific biological questions (i.e., support for particular bipartitions) through judicious taxon and/or gene sampling.
References
Bininda-Emonds OR, Brady SG, Kim J, Sanderson MJ (2001) Scaling of accuracy in extremely large phylogenetic trees. Pac Symp Biocomput 547–558
Blouin C, Butt D, Hickey G, Rau-Chaplin A (2005a) Fast parallel maximum likelihood-based protein phylogeny. ISCA, Las Vegas, USA, September 2005
Blouin C, Butt D, Roger AJ (2005b) The impact of taxon sampling on the estimation of rate of evolution at sites. Mol Biol Evol 22:784–791
Butt D, Roger A, Blouin C (2005) libcov: A C++ bioinformatic library to manipulate protein structures, sequence alignments and phylogeny. BMC Bioinform 6:138
Chang J (1996) Full reconstruction of Markov models on evolutionary trees: Identifiability and consistency. Math Biosci 137:51
Chor B, Hendy M, Holland B, Penny D (2000) Multiple maxima of likelihood in phylogenetic trees. Mol Biol Evol 17:1529–1541
Cummings MP, Otto SP, Wakeley J (1995) Sampling properties of DNA sequence data in phylogenetic analysis. Mol Biol Evol 12:814–822
Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1985) Confidence limits on phylogenies with a molecular clock. Syst Zool 34:152–161
Felsenstein J (2003) Inferring phylogenies. Sinauer Associates, Sunderland, MA
Felsenstein J (2004) PHYLIP (Phylogeny Inference Package), version 3.6. Distributed by the author, Department of Genome Sciences, University of Washington, Seattle
Gascuel O (1997) BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol 14:685–695
Goldman N, Anderson JP, Rodrigo AG (2000) Likelihood-based tests of topologies in phylogenetics. Syst Biol 49:652–670
Hordijk W, Gascuel O (2005) Improving the efficiency of SPR moves in phylogenetic tree search methods based on maximum likelihood. Bioinformatics 21:4338–4347
Keeling PJ (2003) Congruent evidence from α-tubulin and β-tubulin gene phylogenies for a zygomycete origin of microsporidia. Fungal Genet Biol 38:298–309
Kishino H, Hasegawa M (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol 29:170–179
Kishino H, Miyata T, Hasegawa M (1990) Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J Mol Evol 30:151–160
Mossel E, Steel M (2005) How much can evolved characters tell us about the tree that generated them? In: Gascuel O (ed) Mathematics of evolution and phylogeny. Oxford University Press, New York, pp 384–412
Olsen G, Matsuda H, Hagstrom R, Overbeek R (1994) fastDNAML: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. CABIOS 10:41–48
Pollock DD, Zwickl DJ, McGuire JA, Hillis DM (2002) Increased taxon sampling is advantageous for phylogenetic inference. Syst Biol 51:664–671
Rogers JS (2001) Maximum likelihood estimation of phylogenetic trees is consistent when substitution rates vary according to the invariable sites plus gamma distribution. Syst Biol 50:713–722
Rogers J, Swofford D (1999) Multiple local maxima for likelihoods of phylogenetic trees: a simulation study. Mol Biol Evol 16:1079–1085
Rokas A, Carroll SB (2005) More genes or taxa? The relative contribution of gene number and taxon number to phylogenetic accuracy. Mol Biol Evol 22:1337–1344
Rokas A, King N, Finnerty J, Carroll SB (2003) Conflicting phylogenetic signals at the base of the metazoan tree. Evol Dev 5:346–259
Schmidt H, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: Maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Shi X, Gu H, Susko E, Field C (2005) The comparison of the confidence regions in phylogeny. Mol Biol Evol 22:2285–2296
Shimodaira H (2002) An approximately unbiased test of phylogenetic tree selection. Syst Biol 51:492–508
Shimodaira H, Hasegawa M (1999) Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol 16:1114–1116
Stamatakis A, Ludwig T, Meier H (2004) RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21:456–463
Strimmer K, Rambaut A (2002) Inferring confidence sets of possibly misspecified gene trees. Proc R Soc Lond B 269:137–142
Swofford DL (2002) PAUP*: Phylogenetic Analysis Using Parsimony (*and other methods). Sinauer Associates, Sunderland, MA
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Hillis DM, oritz C, Mable BK (eds) Molecular systematics. Sinauer, Sunderland, MA
Thompson JD, Higgins D, Gibson T (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Wolf M, Easteal S, Kahn M, McKay B, Jermin L (2000) TrExML: a maximum-likelihood approach for extensive tree-space exploration. Bioinformatics 16:383–394
Yang Z (2005) Phylogenetic analysis by maximum likelihood (PAML). Available at: http://abacus.gene.ucl.ac.uk/software/paml.html
Zwickl DJ, Hillis DM (2002) Increased taxon sampling greatly reduces phylogenetic error. Syst Biol 51:588–598
Acknowledgments
The authors wish to thank Ed Susko, Associate Editor Nicholas Galtier, and the anonymous reviewers for their suggested improvements to the manuscript and, also, Matthew Spencer for helpful discussion. Thanks go to J. Leigh for providing the tree in Fig. 3a. Shirley Pepke was supported by a Genome Atlantic postdoctoral fellowship. This work was supported by NSERC discovery grant 298397-04 (CB).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Nicolas Galtier]
Rights and permissions
About this article
Cite this article
Pepke, S.L., Butt, D., Nadeau, I. et al. Using Confidence Set Heuristics During Topology Search Improves the Robustness of Phylogenetic Inference. J Mol Evol 64, 80–89 (2007). https://doi.org/10.1007/s00239-006-0072-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-006-0072-4