
Abstract
SINEs are short interspersed retrotransposable elements that invade new genomic sites. Their retrotransposition depends on reverse transcriptase and endonuclease activities encoded by partner LINEs (long interspersed elements). Recent genomic research has demonstrated that retroposons account for at least 40% of the human genome. Hitherto, more than 30 families of SINEs have been characterized in mammalian genomes, comprising ∼4600 extant species; the distribution and extent of SINEs in reptilian genomes, however, are poorly documented. With more than 7400 species of lizards and snakes, Squamata constitutes the largest and most diverse group of living reptiles. We have discovered and characterized a novel SINE family, Sauria SINEs, whose members are widely distributed among genomes of lizards, snakes, and tuataras. Sauria SINEs comprise a 5′ tRNA-related region, a tRNA-unrelated region, and a 3′ tail region (containing short tandem repeats) derived from LINEs. We distinguished eight Sauria SINE subfamilies in genomes of four major squamate lineages and investigated their evolutionary relationships. Our data illustrate the overall efficacy of Sauria SINEs as novel retrotransposable markers for elucidation of squamate evolutionary history. We show that all Sauria SINEs share an identical 3′ sequence with Bov-B LINEs and propose that they utilize the enzymatic machinery of Bov-B LINEs for their own retrotransposition. This finding, along with the ubiquity of Bov-B LINEs previously demonstrated in squamate genomes, suggests that these LINEs have been an active partner of Sauria SINEs since this SINE family was generated more than 200 million years ago.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
SINEs (short interspersed elements) are nonviral retrotransposable repetitive sequences with a length of 70–500 bp that are widespread among eukaryotic genomes (Weiner et al. 1986; Okada 1991; Schmid and Maraia 1992; Deininger and Batzer 1993). While some SINEs are derived from 7SL RNA (Ullu and Tschudi 1984) or 5S rRNA (Kapitonov and Jurka 2003), most SINEs are derived from tRNA (for a list of references see Ohshima and Okada 2005). Hence, the tRNA-like secondary structure as well as the conserved RNA polymerase III–specific internal promoter sequences (designated A and B boxes) allows new SINE elements to be distinguished from other repetitive elements in the genome. SINEs can amplify nonautonomously by a copy-and-paste mechanism, in which the initial amplification of a SINE at the parent locus is followed by integration of a SINE copy at the genomic target site. This retrotransposition of SINEs is dependent on autonomous partner LINEs (long interspersed elements) that encode reverse transcriptase (RT) and endonuclease (EN) for their own amplification (Eickbush 1992; Luan et al. 1993). Since most LINE/SINE pairs share an identical 3′ tail sequence in eukaryotic genomes (Ohshima et al. 1996; Okada et al. 1997), RTs encoded by LINE partners recognize the matching 3′ tails of SINEs and thus initiate SINE replication via retrotransposition in trans. This mechanism of SINE amplification was demonstrated experimentally by our group for active LINE/SINE pairs in the eel genome (Kajikawa and Okada 2002; Kajikawa et al. 2005). Okada et al. (1997) proposed that LINEs can be divided into two groups, the stringent type and the relaxed type, depending on the relative specificity of recognition of their own RT 3′ end. Most LINE family RTs strictly recognize a specific sequence at their 3′ tail, whereas in the mammalian LINE family, L1, no such 3′ end-specific region (except the poly[A] tail) is needed for RT recognition (Moran et al. 1996). The L1-encoded RT-dependent retrotranspositional mechanism in trans is also believed to be the driving force of Alu-SINE amplification in primates (Jurka 1997; Esnault et al. 2000; Ohshima et al. 2003) and CYN t-SINE amplification in flying lemurs (Piskurek et. al. 2003; Piskurek and Okada 2005). Retrotransposed CYN t-SINEs in flying lemurs are composed exclusively of tRNA-related regions, along with a poly(A) tail that is proposed to serve as a recognition site for L1 RTs. Recently, Churakov et al. (2005) characterized tRNA-related mobile elements with poly(A) tails in the genome of armadillos and proposed that DAS elements recruited the enzymatic machinery of L1 LINEs as well.
The retroposition site of offspring copies is almost random, although a slight sequence preference for the target site has been reported (Jurka 1997). Also, the copy-and-paste mechanism of retrotransposition is intrinsically unidirectional. In addition, no mechanism is known for the precise removal of SINEs from any genome (Shedlock and Okada 2000; Batzer and Deininger 2002). Thus, in phylogenetic studies the insertion of a SINE at a specific genomic location represents the derived character state for all species that share a SINE at an orthologous genome site. In contrast, the ancestral state is the absence of a SINE at a particular genomic location. Because of these characteristics, SINEs are thought to be homoplasy-free molecular markers for evolutionary studies (Okada et al. 1997; Shedlock and Okada 2000). In the last decade, a growing body of literature has demonstrated that SINEs are extremely effective markers for elucidating evolutionary history (Murata et al. 1993; Takahashi et al. 1998; Nikaido et al. 1999; Schmitz et al. 2001; Terai et al. 2004; Ray et al. 2005). Their limitations for divergence times up to 150–200 Myr were addressed by several other investigators as well (Hamada et al. 1998; Hillis 1999; Miyamoto 1999; Okada et al. 2004; Shedlock et al. 2004). Mammalian orders have received special attention with respect to the detection and characterization of novel SINE families. To date, tRNA-derived SINE families are one of the most abundant genomic components in species of all four major placental mammalian clades proposed by Murphy et al. (2001). They are present in genomes of laurasiatherians, which include carnivores, cetartiodactyls, chiropterans, eulipothyphlans, perissodactyls, and pholydotans (e.g., see Shimamura et al. 1997, 1999; Nikaido et al. 1999; Kawai et al. 2002), in genomes of the Euarchontoglires, namely, primates, dermopterans, scandentians, rodents, and lagomorphs (Cheng et al. 1984; Britten et al. 1988; Schmid 1996; Nishihara et al. 2002; Schmitz and Zischler 2003; Piskurek et al. 2003; Ray et al. 2005), as well as in genomes of the afrotherian clade (Nikaido et al. 2003; Nishihara et al. 2005) and in genomes of xenarthrans (Churakov et al. 2005).
In contrast, very little progress in SINE research has been made for reptile genomes, although the Sauropsida represent the sister group of mammals. Whereas there are approximately 4600 species of mammals, more than 16,000 extant species of birds, crocodiles, lizards, snakes, and turtles are known. Hitherto, the only example of reptile SINEs that were characterized and applied as molecular markers to infer reptilian phylogeny of one turtle family (Bataguridae) is the tortoise polIII/SINE in hidden-necked turtles, the discovery of which dates back about 20 years (Endoh and Okada 1986; Endoh et al. 1990; Ohshima et al. 1996; Sasaki et al. 2004). While the Bataguridae represent the major group of turtles (about 60 extant species), squamate reptiles, with more than 7400 extant species of lizards and snakes, are the largest and most diverse group of living reptiles (Zug et al. 2001). The order Sphenodontia represents the sister group of the Squamata and consists of only two surviving species of tuatara (genus Sphenodon) from New Zealand (Zardoya and Meyer 1998; Rieppel and Reiz 1999; Rest et al. 2003). Sphenodontia and Squamata together form the Lepidosauria.
In this study we describe a novel tRNA-derived SINE family that is widely distributed among lepidosaurian genomes. We first discovered mobile elements in the genome of the common wall lizards (Podarcis muralis, suborder Sauria) and subsequently characterized tRNA-derived SINEs of the same family in two additional major lineages of lizards and in the genome of snakes. We designate this new SINE family as Sauria SINE. We examine and discuss Sauria SINE evolution, such as the genealogy of certain SINE subfamilies in genomes of lizards and snakes, a possible secondary structure for the tRNA-like 5′ end, and we discuss the short 3′ sequence that originated from the Bov-B LINE. Furthermore, we give an example of how to use the Sauria SINE family as a marker for the evolution of monitor lizards.
Materials and Methods
DNA Isolation of Tissue Samples
Genomic DNA from all major Squamata groups, additional reptiles, and outgroups was isolated (Table 1) by phenol-chloroform extraction as described by Blin and Stafford (1976).
Construction and Screening of Genomic Libraries and Sequencing of Cloned DNA
Our group previously detected many SINE families by in vitro transcription of total genomic DNA (Endoh and Okada 1986). However, this method has been progressively replaced by high-throughput technologies designed for sequencing large amounts of DNA data in a short time (Okada et al. 2004). Considering that typical SINEs are often present in numbers that exceed 104 copies per genome, a sufficient amount of SINE sequences can usually be gained with 0.6 Mb genomic sequence data. Genomic libraries from three lizards (Podarcis muralis, Anolis carolinensis, Varanus indicus) and one snake (Azemiops feae) were constructed by complete digestion of genomic DNA with HindIII, followed by sedimentation through a sucrose gradient and selection of DNA fragments of up to 2 kbp. The size-fractionated genomic DNA was ligated into HindIII-digested pUC18 plasmids at 37°C overnight. Aliquots of the ligation reactions were transformed into Escherichia coli DH5-α cells. Colonies were transferred to membranes for screening. The first five SINE loci were identified by random selection and sequencing of approximately 0.8 Mb genomic sequence data of Podarcis muralis.
Additional Podarcis loci were screened using internal SINE primers (POM1F, CTAGGGCTTGCTGATCAGAAG; POM1R, GGCCAATAAAGCGAGATGAG; POM2F, TGTGGGTTAA AGCCRCAGCG; POM2R, ACGGGCAGGGGTACCTTTAC) labeled by primer extension in the presence of [α-32P]dCTP. [γ-32P]dATP-labeled internal primer sequences were also used to further investigate the evolution of this novel SINE family. Hybridization was performed at 42°C overnight in a solution of 6× SSC containing 1% SDS, 2× Denhardt’s solution, and 100 μg/ml herring sperm DNA and washed at 50°C for 10 min in a solution of 2× SSC containing 1% SDS. Positive plasmid clones that appeared to contain SINE loci were isolated, and the inserts were sequenced using universal primers M4 and RV (TaKaRa). Sequencing was performed with an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). Sauria SINE sequences reported in this paper have been deposited in GenBank under accession numbers DQ023333–DQ023415.
PCR with Internal Sauria SINE Primers and Sequencing of Internal SINE Regions
Podarcis SINE primers POM1F, POM1R, POM2F, and POM2R were used for PCR to amplify internal SINE regions from representatives of all major Squamata groups (Table 1). After initial denaturation for 3 min at 94°C, 33 cycles were performed, consisting of 30 s denaturation at 94°C, 60 s annealing at 50°C, and 40 s elongation at 72°C. Amplified PCR products were cloned and sequenced with an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems), and partial SINE regions of additional Squamata species were aligned with SINEs already identified in the common wall lizard. Subsequently, universal SINE primers were designed for Sauria SINEs in lizards and snakes (SQ1F, CCCWG CTCCTGCCAACCTAGC; SQ1R, TAGTCATGCTGGCCA CATGACC) and used to screen SINE loci in genomes of Anolis carolinensis, Varanus indicus, and Azemiops feae, as described above.
Genomic DNA of all major Squamata groups, a tuatara (Sphenodon punctatus), additional reptiles, and mammalian outgroups (Table 1) was amplified by PCR using internal Sauria SINE primers (SQ1F and SQ1R). PCR conditions were as follows: after initial denaturation for 3 min at 94°C, 33 cycles were performed consisting of 30 s denaturation at 94°C, 50 s annealing at 50°C, and 30 s elongation at 72°C.
PCR Using Primers for Sauria SINE Flanking Regions
Genomic DNA of Varanus salvator, Varanus indicus, and Varanus jobiensis was amplified by PCR using primers for Sauria SINE flanking regions (VIN1for, CTAACACTGGACCCATGCTAG; VIN1rev, AGGATTCAAGCTGATTCTGC; VIN2for, AGAGG GCGATGGATTACTGG; VIN2rev, AGAAGGTAGCCAGACG GTGG; VIN6for, TTGGTCTCAGCCTCATCTTC; VIN6rev, GGATCCTGACCTGAAAGATG). PCR conditions were as follows: after initial denaturation for 3 min at 94°C, 33 cycles were performed, consisting of 30 s denaturation at 94°C, 60 s annealing at 51°C, and 60 s elongation at 72°C.
Sequence Analyses
Multiple sequence alignments were constructed using CLUSTAL W (Thompson et al. 1994), and sequence analyses were performed with BioEdit (Hall 1999). Database searches were performed with BLASTN (Altschul et al. 1997). The Sauria SINE 5′ end was compared with tRNA sequences obtained from the tRNA compilation of Sprinzl and Vassilenko (2005). A tRNA cloverleaf structure was constructed with the tRNAscan-SE program (Lowe and Eddy 1997). Using TREE-PUZZLE 5.0 (Strimmer and von Haeseler 1996), a maximum likelihood analysis based on the HKY85 model was performed (Hasegawa et al. 1985) using the discrete gamma distribution (eight categories) for site heterogeneity (Yang 1996). Puzzling supports were based on 25,000 replicates. Pairwise distance calculations based on the Kimura two-parameter model were conducted using MEGA version 3.0 (Kumar et al. 2004). Frequently encountered CpG sites were not included in the analyses of diagnostic nucleotides and the maximum likelihood analysis.
Results
Identification of Novel SINEs in Lizards and Snakes
In order to identify novel SINEs in reptiles, we used the strategy suggested by Okada et al. (2004) and randomly sequenced 0.8 Mb of genomic sequence data of the common wall lizard (Podarcis muralis). This procedure identified the first five copies of tRNA-derived SINEs having a length of approximately 350 nucleotides and belonging to the same family. Following the initial characterization of these newly discovered repetitive sequences in lizards, 49 additional loci were detected by screening the entire Podarcis genome (see Materials and Methods). These mobile elements have the typical characteristics of SINEs. Like most SINEs described to date, they are composed of a 5′ tRNA-related region (including A and B boxes for internal RNA polymerase III promoters), a tRNA-unrelated region, and a 3′ AT-rich region (Okada 1991; Okada et al. 1997). The sharing of an identical 3′ sequence of a partner LINE family (Ohshima et al. 1996) is a typical feature for nonmammalian tRNA-related SINEs (see below). Also, they are dispersed in the genome of Podarcis and flanked by characteristic direct repeats, suggesting that they were amplified through retrotransposition (see Supplemental Fig. 1, available at the publisher’s web site). Following the design of universal SINE primers for Squamata species, we screened for additional SINE loci in other lizards and snakes (see Materials and Methods). The common wall lizard (Podarcis muralis) is a member of the Scincomorpha, the green anole (Anolis carolinensis) was chosen as representative of the Iguania, the mangrove monitor (Varanus indicus) is an Anguimorph, and Fea’s viper (Azemiops feae) is a member of the Serpentes (snakes). Very large and diverse Squamata taxa belong to these four reptile groups. We detected 19, 4, and 6 additional SINE loci by screening the genomes of Anolis, Varanus, and Azemiops, respectively. Subsequently, we designated these novel SINE sequences as Sauria SINEs. An alignment of eight subfamily consensus sequences (see below) is shown in Fig. 1.

Alignment of consensus sequences of eight Sauria SINE subfamilies isolated from the genomes of Podarcis muralis (POM), Varanus indicus (VIN), Azemiops feae (AFE), and Anolis carolinensis (ACA). The tRNA-derived consensus sequence for the Sauria SINE 5′ end as well as the short Bov-B LINE-derived Sauria SINE 3′ end is shown (see also Fig. 2). Proposed double-stranded regions are boxed. Ac, acceptor stem; D, D loop stem; An, anticodon stem; Ps, TψC stem; Serp, Serpentes (Vipera ammodytes; accession number AF332697); Bos, Bos taurus (accession number AC089992).

A General cloverleaf structure for tRNAs (Gauss et al. 1979; Lowe and Eddy 1997). Gray circles represent conserved and semiconserved nucleotides, whereas dots represent variable sequence regions. B Predicted cloverleaf structure of tRNA-related Sauria SINE consensus sequence (tRNAscan-SE program [Lowe and Eddy 1997]). All conserved and semiconserved nucleotides found in tRNAs are present. C Predicted secondary structure of the Bov-B LINE-derived Sauria SINE region. The gray box represents the common 3′ sequence of published Bov-B LINEs and all Sauria SINEs subfamilies characterized in this study. This sequence is proposed to be important for the process of retrotransposition.
The Sauria SINE 5′ End Is Derived from tRNA
We were able to construct a tRNA-like cloverleaf structure with conserved and semiconserved nucleotides, as described by Gauss et al. (1979), for all characterized 5′ ends of Sauria SINE subfamilies. Thus, the 5′ sequence of the Sauria SINE is clearly derived from a tRNA (Figs. 1 and 2). Moreover, the long variable arm in all lizard and snake subfamilies resembles the characteristic long variable arm of eukaryotic class II tRNAs. While the secondary structure is mostly conserved in tRNA regions of lizards, it is partially disrupted by insertions and deletions in tRNA regions of snakes. However, when the proposed nucleotide insertions just 3′ of the D loop stem (T) and in the acceptor arm (AA) are deleted, a homology search with the tRNAscan-SE program (Lowe and Eddy 1997), based on the most recent Sprinzl tRNA database (Sprinzl and Vassilenko 2005), predicts a thermodynamically stable tRNASer-like secondary structure (Fig. 2B). The obvious conservation of tRNA-related regions with promoter sequences A and B boxes in all Sauria SINE subfamilies suggests the requirement for transcription by polymerase III (Okada and Ohshima 1995).
The Sauria SINE 3′ End Is Derived from a LINE
In each subfamily, the tRNA-related region was followed by a central domain that is nearly as conserved as the Sauria SINE 3′ tail (Table 2A). Interestingly, the 3′ tail of the Sauria SINE is highly similar in all eight subfamilies (except for a 9-bp deletion in snakes) and is composed of an approximately 40-bp conserved stem-loop region and a well-conserved region of short tandem repeats (ACCTTT), as illustrated in Figure 2C. We recently suggested such stem-loop structures for the 3′ end of LINE/SINE pairs of the eel genome (Kajikawa and Okada 2002; Kajikawa et al. 2005). Our previous studies demonstrated that the conserved stem-loop structures as well as the conserved short tandem terminal repeat regions are required for retrotransposition. In this study we showed that a short sequence of the 3′ Sauria SINE tail is identical with the 3′ sequence of Bov-B LINEs (Figs. 1 and 2C). We postulate that the common 3′ sequence of Bov-B LINEs and Sauria SINEs as well as the tandem repeat region are important for retrotransposition (see Discussion).
Evolution of Sauria SINE Subfamilies
Analysis of the Sauria SINE family led us to divide repetitive sequences of lizards and snakes into different subfamilies. The genome of Podarcis includes three Sauria SINE subfamilies. Seven loci belong to the POMα-type, 5 loci form the POMβ-type, and the remaining 42 Sauria SINE sequences were characterized as part of the POMγ-type subfamily (see Supplemental Fig. 1, available at the publisher’s web site). Likewise, three distinct subfamilies are present in the genome of Anolis. An assemblage of 13 loci illustrates the structure of the ACAα-type, 2 loci belong to subfamily ACAβ-type, and 4 loci cluster together and form the ACAγ-type (see Supplemental Fig. 2, available at the publisher’s web site). The four Sauria SINE loci in the genome of Varanus and the six loci detected in Azemiops represent, in each case, one subfamily of Sauria SINEs (Fig. 1).
An analysis of the distribution of diagnostic nucleotides, together with the distribution of Sauria SINE subfamilies among major squamate lineages, allows us to infer the genealogy of novel Sauria SINE subfamilies in genomes of scincomorph lizards, iguanian lizards, anguimorph lizards, and snakes. The POMβ-type and POMγ-type are more closely related to each other than either subfamily is to the POMα-type. Numerous diagnostic nucleotides (Supplemental Fig. 1; sites 28, 32, 34, 37, 60, 94, 150, 153–155, 175, 182, 183, 216, 234, 242, 271, 293, 297, 298, 323, 331, 338, 343, 350, 353, 354, 359) and a maximum likelihood analysis (see below) using Sauria subfamilies of other squamate groups to root the tree support this sister group relationship with convincing puzzle support values (Figs. 3 and 4A). This result suggests that the POMβ-type and POMγ-type source genes originated from POMα-type sequences. The POMγ-type can be distinguished most obviously from the other two subfamilies by a 10-bp deletion in the tRNA-unrelated region (Fig. 3; Supplemental Fig. 1, sites 275–284). The presence of these 10 nucleotides and a number of other diagnostic sites distinguishes the POMβ-type from the POMγ-type subfamily (Fig. 3; Supplemental Fig. 1, sites 27, 45, 47, 226, 268, 269, 304, 339, 352, 357, 369). Furthermore, many nucleotides indicate that POMα-type members 7 and 74 are part of a subsubfamily that is more closely related to POMβ-type members than the rest of the POMα-type group (Fig. 4A; Supplemental Fig. 1, sites 32, 94, 125, 153-155, 175, 338, 343). It is also possible to further divide the POMβ-type subfamily into two subsubfamilies (Fig. 4A; Supplemental Fig. 1, sites 27, 41, 42). POMβ-type members 11 and 87 share a small deletion of two nucleotides in the tRNA-related region that is also present in all 42 POMγ-type members (Fig. 3). Therefore, it can be assumed that the POMγ-type source gene originated from a common ancestor of Sauria SINE members with a structure most likely similar to POMα-type members 7 and 74 and POMβ-type members 11 and 87. Based on the random and frequent detection of POMγ-type sequences, it is obvious that the POMγ-type represents the most abundant subfamily in the genome of Podarcis. Only a few members of the POMγ-type group cluster together to form subsubfamilies. They can be distinguished from all other members of this group by a 9-bp (Supplemental Fig. 1, sites 233–241), a 5-bp (Supplemental Fig. 1, sites 212–216), and an 8-bp (Supplemental Fig. 1, sites 5–12) deletion in loci POM 8 and POM 121, POM 80 and POM 111, and loci POM 95 and POM 127, respectively (Fig. 4A). The low level of variation among POMγ-type members seems to indicate that the POMγ-type source gene is young. Pairwise distance calculations of SINEs in the genome of the common wall lizard support this finding (data not shown). Moreover, the highest rate of amplification of POMγ-type SINE copies shows that this source gene is predominantly active in its retrotransposition of offspring copies in the genome of lacertid lizards.

Genealogy of Sauria SINE subfamilies in the genomes of three lizards and one snake. Diagnostic nucleotides are shown either as white bars (deletions of two or more nucleotides), as black bars (insertions of two or more nucleotides), or as numbers below the four major Sauria SINE regions of schematic subfamily structures. Numbers on the left side of the schematic subfamily structures indicate the quantity of Sauria SINE loci sequenced from a certain subfamily. Numbers in parentheses on the right side of each schematic subfamily structure indicate the number of sub-subfamilies identified in a given genome (see also Evolution of Sauria SINE Subfamilies, in text, and Fig. 4A).

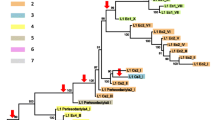
A Evolutionary relationships among Sauria SINE subfamilies of four major squamate lineages analyzed using the maximum likelihood method. Numbers corresponding to internal nodes represent puzzle support values. The puzzle support value for the sister group cluster of ACAβ-type and ACAγ-type increases to 68 if subfamilies AFE and VIN are used as outgroups. Branch length represents nucleotide substitutions per site. Large arrows illustrate Sauria SINE subfamilies, whereas small arrows represent sub-subfamilies (see Evolution of Sauria SINE Subfamilies, in text, for explanations). B Evolutionary relationships of major squamate lineages (Townsend et al. 2004). The tree topology of major snake lineages is boxed (Vidal and Hedges 2004). We analyzed Sauria SINEs of the indicated Squamata species obtained in this study and from database searches (Table 3).
We also divided Sauria SINEs into three distinct subfamilies in the genome of Anolis. Twelve diagnostic nucleotides support the sister group cluster of ACAβ-type and ACAγ-type members to ACAα-type sequences (Fig. 3; Supplemental Fig. 2, sites 25, 32, 64, 75–79, 91, 101, 107, 128, 324). This implies that the ACAβ-type and ACAγ-type source genes were derived from ACAα-type sequences, as revealed subsequently by a maximum likelihood analysis (Fig. 4A). The ACAβ-type subfamily can easily be distinguished from ACAγ-type members by a 3-bp duplication in the tRNA-unrelated region (Fig. 3; Supplemental Fig. 2, sites 231–233). Three additional diagnostic nucleotides discriminate members of the ACAβ-type from ACAγ-type sequences (Fig. 3; Supplemental Fig. 2, sites 93, 108, 338). The ACAα-type source gene appears to generate most Sauria SINE sequences in the genome of Anolis and thus was likely the most successful in its retrotransposition of offspring copies. It is possible to distinguish two sub-subfamilies in the ACAα-type subfamily based on four diagnostic nucleotides (Fig. 4A; Supplemental Fig. 2, sites 11, 20, 100, 121). We identified several examples in the genome of Anolis in which Sauria SINEs inserted at the 3′ end into the tandem repeat region of preexisting SINE loci. Previous studies based on mammalian genomes have mentioned that various retroposons show a common tendency to insert near or within sequence regions where other mobile elements have previously inserted (Slagel et al. 1987; Krane et al. 1991; Piskurek et al. 2003).
The number of Sauria SINE members in anguimorph lizards and snakes is too limited to distinguish different subfamilies in their genomes. However, consensus sequences of both species represent distinct subfamilies of the Sauria SINE family. Moreover, subfamilies in monitor lizards (VIN) and snakes (AFE) are more closely related to each other than to Sauria SINE subfamily members of scincomorph and iguanian lizards (Fig. 4A). The most obvious way to distinguish both VIN and AFE subfamilies from all other subfamilies is the consensus region at nucleotide positions 220–247 (Fig. 1). A 15-bp deletion discriminates both subfamilies from Sauria SINEs in scincomorph lizards (Fig. 1, sites 220–235), whereas Sauria SINEs in iguanian lizards have an even larger 27-bp deletion at this location (Fig. 1, sites 220–247). Three other diagnostic nucleotides verify a close relation between Sauria SINE subfamilies VIN and AFE (Fig. 1, sites 50, 187, 269). Besides, 16 unambiguous diagnostic nucleotides support ACA subfamilies as a sister group to a clade including the subfamily cluster VIN/AFE (Fig. 1, sites 19, 40, 43, 93, 152, 153, 155, 202, 210, 269, 309, 312, 317, 331, 375, 376), whereas eight diagnostic nucleotides are specific for ACA subfamilies in iguanian lizards (Fig. 1, sites 5, 6, 10, 113, 163, 218, 302, 303). In other words, it can be proposed that the 15-bp deletion (Fig. 1, sites 220–235) happened in a common ancestor of source genes, AFE, VIN, and ACA, while a second 12-bp deletion occurred separately in the ancestral source gene of ACAα-type, ACAβ-type, and ACAγ-type in the genome of iguanian lizards (Fig. 3).
Furthermore, relationships of Sauria SINE subfamilies were examined using all subfamily consensus sequences in a maximum likelihood analysis (Fig. 4A). Sauria SINE subfamilies of lacertid lizards were used to root the tree (Townsend et al. 2004; Vidal and Hedges 2004). The tree topology of Sauria SINE subfamilies from all four major squamate infraorders we investigated is clearly identical to the tree topology of major Squamata groups based on large nuclear data sets (Townsend et al. 2004). Although SINE subfamilies do not necessarily represent actual evolutionary relationships of species, it seems quite evident, considering the detailed examination of Sauria SINE subfamilies in different squamate genomes, that in this case there is a strong correlation (Fig. 4B). Townsend et al. (2004) proposed a close relation of snakes and anguimorph lizards, which together represent the sister group of iguanian lizards. Sauria SINE subfamilies are related to each other in such a phylogenetic pattern (Fig. 4A; see Discussion).
Distribution, Sequence Divergence, and Copy Number of Sauria SINEs
To examine the distribution of Sauria SINEs among reptilian genomes, we isolated genomic DNA from 48 species and analyzed it by PCR using this DNA as template and two oligonucleotide primers that were specific to the tRNA-unrelated region (see Materials and Methods). Sauria SINEs are widely distributed among genomes of all major groups of lizards and snakes as well as in the genome of the tuatara (Fig. 5). This result was supported through database searches with BLASTN (Altschul et al. 1997), since we detected several partial and complete Sauria SINEs in additional squamate species (Fig. 4B, Table 3). This result suggests that Sauria SINEs might have been generated in a common ancestor of lepidosaurian genomes approximately 230 million years ago (Benton 1993).

PCR analysis with primers directed toward internal Sauria SINE sequences. Genomic DNA from reptilian and outgroup sources was amplified by PCR using primers SQ1F and SQ1R, as described under Materials and Methods. M, marker (φX174-HincII digest). See Table 1 for the abbreviations of lizards, snakes, and other amniotes.
Sauria SINE subfamily consensus sequences in squamate lineages, some of which diverged more than 100 million years ago, are surprisingly identical (4–34%; see Table 2B). While the mean sequence divergence of Sauria SINE members within lizard genomes is relatively low (POM, 13.9%; VIN, 9.6%; ACA, 17.7%), it is a little higher in the snake genome (AFE, 27.6%). We strengthened this result with pairwise distance calculations of all subfamily members against subfamily consensus sequences (Fig. 6).

Pairwise distances of Sauria SINE members within four different Squamata genomes. The sequence divergence in the genome of lizards (POM, ACA, VIN) is slightly lower than the sequence divergence in the snake genome (AFE).
We estimated the copy number of Sauria SINEs on the basis of the random isolation frequency of retrotransposable elements obtained from the Podarcis muralis genome. The mean genome size of Squamata species is postulated to be 2.1 × 109 bp (http://www.genomesize.com). Therefore, the predicted copy number of Sauria SINEs is 130,000 per haploid genome. However, the copy number varied greatly depending on the genome analyzed (data not shown), probably because of differences among retrotranspositional activity of Sauria SINEs in the genomes of various lizards and snakes.
Using Sauria SINEs as Markers for Evolution
In order to test if Sauria SINEs provide an evolutionary marker system in reptile genomes, we performed PCR experiments with primers specific for sequences flanking SINEs for a group of anguimorph lizards. Monitor lizards, genus Varanus, represent a monophyletic group within Anguimorpha, which is believed to be closely related to snakes (Lee 2000). Within Varanus, three major lineages, African, Indo-Asian, and Indo-Australian, are delineated based on their biogeographical distribution (Fuller et al. 1998; Ast 2001). The Indo-Asian lineage comprises two distinct clades with a proposed divergence time of more than 112 Myr (Schulte et al. 2003; Hugall and Lee 2004). Terrestrial Asian forms and the water monitors of the Varanus salvator complex belong to Indo-Asian clade A, whereas the mangrove monitors of the Varanus indicus complex belong to Indo-Asian clade B (Ast 2001). We investigated Sauria SINE loci in three species of the Indo-Asian lineage, namely, in the common water monitor (Varanus salvator) as representative of clade A as well as in the mangrove monitor (Varanus indicus) and the peach-throated monitor (Varanus jobiensis) as representatives of clade B. The presence of the SINE sequences, VIN1, VIN2, and VIN6, in the latter two species and the absence of these three Sauria SINEs at orthologous genome sites in Varanus salvator clearly indicate that Varanus indicus and Varanus jobiensis belong to a monophyletic group (Figs. 7A and B). This example verifies that Sauria SINEs in squamate genomes can be used to track evolutionary events in reptile lineages.

A Alignment of three Sauria SINE loci and their flanking sequences in monitor lizards. The absence of these three SINE loci in the genome of Varanus salvator (VSA) and the presence of SINE sequences at orthologous genome locations in Varanus indicus (VIN) and Varanus jobiensis (VJO) support a monophyletic origin of the latter two species. A and B boxes of the tRNA-related sequence regions are shown and the clearly recognizable flanking direct repeats are boxed. B PCR analysis with flanking Sauria SINE primers. Genomic DNA from monitor lizards was amplified by PCR using primers VIN1for+VIN1rev, VIN2for+VIN2rev, and VIN6for+VIN6rev, as described under Materials and Methods. PCR products that illustrate the presence and absence of SINE loci are boxed. M, size marker.
Discussion
Structural Aspects of Sauria SINEs and Their Connection to Bov-B LINEs
tRNAs can be divided into two distinct classes. In comparison to class I tRNAs, which contain a variable region with only four or five nucleotides, eukaryotic class II tRNAs, including tRNALeu and tRNASer, contain more than twice as many nucleotides, thus forming an additional stem-loop, which is known as the long extra arm. Apart from the Sauria SINE, tRNASer has been proposed to be the most likely candidate for the origin of equine SINEs (Sakagami et al. 1994).
We found that Sauria SINEs and Bov-B LINEs have a short common 3′ sequence. Luan et al. (1993) proposed the “target-primed reverse transcription” (TPRT) as the mechanism of LINE retrotransposition. In the TPRT, the RT synthesizes cDNA in situ using a 3′ OH of the DNA generated by a nick introduced through the EN as a primer. Later, Ohshima et al. (1996) discovered that 3′ ends of the tortoise PolIII/SINE and the CR1 LINE, also present in the tortoise genome, are almost identical. This finding prompted us to generalize the observation of Luan et al. (1993) and conclude that 3′ ends of SINE families are actually derived from 3′ ends of corresponding LINE families. Thus, we proposed that SINEs are amplified through the TPRT using RTs encoded by LINEs in trans (Ohshima et al. 1996; Okada et al. 1997; Kajikawa and Okada 2002; Kajikawa et al. 2005; Ohshima and Okada 2005). Therefore, the function of the illustrated Sauria SINE stem-loop structure probably correlates with the recognition of the RT encoded by the Bov-B LINE. This was experimentally demonstrated for the eel LINE UnaL2 (Baba et al. 2004). While the stem-loop structure suggested for LINE/SINE pairs of the eel genome is straightforward (Kajikawa and Okada 2002; Kajikawa et al. 2005), it is of a more sophisticated nature in LINE/SINE pairs in the genome of sharks (Ogiwara et al. 1999).
Recently there has been a controversial discussion about the Bov-B family, and many questions concerning its genomic origin and evolution remain unanswered. Originally, the Bov-B family was thought to be a SINE family (Lenstra et al. 1993) until Szemraj et al. (1995) identified a full-length (3.1-kbp) element of Bov-B (BDDF for bovine dimer-driven family). Subsequently, the Bov-B family was designated the Bov-B LINE family (Okada and Hamada 1997). The Kordis group discovered Bov-B elements in squamate genomes (Kordis and Gubensek 1995) and examined their distribution (Kordis and Gubensek 1998; Zupunski et al. 2001; see below). The stem-loop structure and the conserved terminal repeat region in Sauria SINEs confirm the stringent type character of Bov-B LINEs. It was previously suggested that Bov-B-LINE RTs strictly recognize the specific 3′ tail of their partner SINE family Bov-tA in the genome of ruminants (Okada and Hamada 1997; Okada et al. 1997). Gilbert and Labuda (1999) strengthened this finding when they reported MIR-like SINEs (Mar-1) in marsupial genomes that share approximately 95 bp of their 3′ end with Bov-B LINEs. The shared common 3′ end of Sauria SINEs and Bov-B LINEs is approximately 20 nucleotides in length (Fig. 2C). It is not possible to align the following 20 bp right before the terminal tandem repeat sequence of Sauria SINEs and Bov-B LINEs, which might be a sign of distinct, yet unknown, Bov-B LINE subfamilies in squamate genomes (Fig. 1). After all, Sauria SINEs in genomes of lizards and snakes represent another interesting example in which an active LINE has donated its 3′ end for retrotransposition of its partner SINE during genomic evolution.
Relationship of Sauria SINEs and Partner Bov-B LINEs to Other Widely Distributed SINE Families
We identified Sauria SINE subfamilies in all major lineages of lizards and snakes. The Sauria SINE copy number in the genome of the common wall lizard indicates that these novel retrotransposable elements account for up to 1% of the total genomic information. However, whereas the genome size of Podarcis represents the mean genome size of Squamata species, some skinks have just half the genome size, whereas the giant girdled lizard has a genome size that is about twice that of lacertid lizards (http://www.genomesize.com).
Interestingly, Sauria SINEs in different squamate lineages are nearly identical, although substitution rates in squamate genomes are higher than in other sauropsids (Hughes and Mouchiroud 2001). For example, nuclear-encoded Squamata genes evolve approximately 30–40% faster than those of the chicken genome. Hughes and Mouchiroud (2001) also found a slightly higher substitution rate for snakes compared with lizards, which we mentioned earlier as well. Nonetheless, the relatively low sequence divergence, about 19%, between subfamily consensus sequences of snakes and anguimorph lizards (Table 2B) in comparison with obviously higher sequence divergences between subfamily consensus sequences of different lizard genomes suggests that substitution rates are very similar in Squamata lineages. Thus, we might expect that, in the period since the generation of the Sauria SINE family approximately 230 million years ago, these SINEs have been highly active in squamate genomes and have been retrotransposed through RT and EN encoded by partner Bov-B LINEs. The Bov-B LINE distribution described by the Kordis group (Kordis and Gubensek 1995, 1998; Zupunski et al. 2001) proves that the proposed partner LINE family of Sauria SINEs is equally present in lizards and snakes, which is a requirement for the successful retrotransposition of Sauria SINEs, as explained earlier. Kordis and Gubensek (1995) suggested a horizontal transfer of Bov-B LINEs from the ancestral snake lineage to the ancestor of ruminants. However, subsequently Bov-B LINEs were detected (Gilbert and Labuda 1999) and sequenced (Zupunski et al. 2001) in marsupials, calling into question the hypothesis of horizontal transfer. Thus, Bov-B LINEs may have originally been present in a common ancestor of all mammals or even in an ancestor of all amniotes. Although the research about the distribution of Bov-B LINEs is still ongoing, it seems clear that they represent a perfect partner LINE family for widely distributed Sauria SINEs in lepidosaurian reptiles. Another ancient and widely distributed SINE family was designated MIR, for mammalian-wide interspersed repeats (Jurka et al. 1995; Smit and Riggs 1995). MIRs proliferated not only before the mammalian radiation but possibly even before the amniote radiation. About 10,000 faint matches to MIRs were reported in the chicken genome (International Chicken Genome Sequencing Consortium 2004), and they are also present in crocodile genomes (A. Shedlock, pers. comm.). An approximately 70-bp central segment of MIRs was named the core region since it appears to be highly conserved in all sequences. It was shown that the core region survived in different lineages such as mammals, reptiles, birds, fish, and even invertebrates like mollusks (Gilbert and Labuda 1999). Therefore, Gilbert and Labuda (1999) proposed to call this widely spread class of SINE families CORE-SINEs. CORE-SINEs and Sauria SINEs are not related, although the CORE-SINE family Mar-1 also shares an identical 3′ tail with Bov-B LINEs (Gilbert and Labuda 1999). Another superfamily of SINEs, V-SINEs, is widespread in vertebrate genomes (Ogiwara et al. 2002). V-SINEs also contain a central region that is fairly well conserved. Although there is no relation between V-SINEs and Sauria SINEs, a central domain that is more conserved than other SINE regions (except the 3′ tail region) precedes the 5′ end tRNA-related sequence of Sauria SINEs as well (Table 2A). Finally, another superfamily of SINEs in vertebrates, recently characterized by our group, is not identical to Sauria SINE sequences in genomes of lizards and snakes (unpublished data of Nishihara and Okada).
Sauria SINE Subfamilies and Their Evolutionary Implications
Despite the fact that iguanian lizards are traditionally not nested within Scleroglossa (which represent all lizard species besides those belonging to Iguania), previous morphological studies have proposed a sister group relationship between Anguimorpha and Serpentes (Estes et al. 1988). Our tree topology, which is based on diagnostic nucleotides of eight Sauria SINE subfamilies, is identical to this species topology. Lee (2000) placed snakes as nested within Anguimorpha and close to monitor lizards. In contrast, Vidal and Hedges (2004) predicted a terrestrial origin for snakes when discussing a close relation between iguanian lizards and snakes. Kumazawa (2004), using complete mitochondrial genomes, placed snakes at the base of the squamate tree as a sister group to all lizard taxa. Also, Townsend et al. (2004) obtained different results for the squamate tree when combining their nuclear RAG-1 and c-mos data with their mitochondrial ND2 data set. Schmitz et al. (2005) proposed that the longer the divergence time and the higher the evolutionary rate differences between genes, the less convincing the phylogenetic tree on the basis of a mixed set of both mitochondrial and nuclear sequences. Furthermore, they discussed problematic aspects of mitochondrial data sets versus nuclear sequences for phylogenetic analyses.
Phylogenetic analyses based on SINEs as genetic markers have been performed extensively in recent years in mammalian genomes (see Introduction). However, in reptiles, only one example from turtle genomes is known using SINEs as markers to map evolutionary history (Sasaki et al. 2004). Since we might expect to find Sauria SINE loci at orthologous genome sites in anguimorph lizards and snakes (see above), we performed flanking PCRs for SINE loci in monitor lizards and demonstrated that Sauria SINEs can be used as evolutionary markers for future studies to infer the phylogenetic relationships of squamate reptiles.
A New Approach to Solve the Origin of Snakes
Sauria SINEs as genetic markers for evolution might eventually bring a resolution to the problem of the phylogenetic placement of snakes. However, the high mutational rate of SINE loci in nonfunctional regions of the genome can make the detection of insertion patterns difficult after ∼150–200 Myr of divergence, which is approximately the time frame of the split between snakes and their closest relatives. On the other hand, Sauria SINE subfamilies in the genomes of monitor lizards and snakes are closely related (Fig. 4A), and the flanking sequences in monitor lizards are fairly well conserved (Fig. 7A), which might be associated with their conserved overall morphology. These aspects of Sauria SINEs make these SINEs ideal for the identification of shared SINE loci in lizards and snakes, which may resolve the vagaries of squamate genome evolution and finally give molecular proof that “snakes are lizards too” (Pianka and Vitt 2003).
We examined the generation and evolution of eight Sauria SINE subfamilies in genomes of four major squamate lineages and demonstrated that they are noticeably conserved over more than 200 Myr of evolution. Besides the fact that we have now established Sauria SINEs as effective evolutionary markers for reptile evolution, the slower mutation rate of certain sequences in Sauria SINEs (Table 2A), as we previously mentioned for other widely distributed SINE families, might be associated with an as yet unidentified function.
References
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipmann DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Ast JC (2001) Mitochondrial DNA evidence and evolution in Varanoidea (Squamata). Cladistics 17:211–226
Baba S, Kajikawa M, Okada N, Kawai G (2004) Solution structure of an RNA stem-loop derived from the 3’ conserved region of eel LINE UnaL2. RNA 10:1380–1387
Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379
Benton MJ (1993) The fossil record 2. Chapman and Hall, New York
Blin N, Stafford DW (1976) A general method for isolation of high molecular weight DNA from eukaryotes. Nucleic Acid Res 3:2303–2308
Britten RJ, Baron WF, Stout DB, Davidson EH (1988) Sources and evolution of human Alu repeated sequences. Proc Natl Acad Sci USA 85:4770–4774
Cheng JF, Printz R, Callaghan T, Shuey D, Hardison RC (1984) The rabbit C family of short, interspersed repeats. Nucleotide sequence determination and transcriptional analysis. J Mol Biol 176:1–20
Churakov G, Smit AF, Brosius J, Schmitz J (2005) A novel abundant family of retroposed elements (DAS-SINEs) in the nine-banded armadillo (Dasypus novemcinctus). Mol Biol Evol 22:886–893
Deininger PL, Batzer MA (1993) Evolution of retroposons. Evol Biol 27:157–196
Eickbush TH (1992) Transposing without ends: the non-LTR retrotransposable elements. New Biol 4:430–440
Endoh H, Okada N (1986) Total DNA transcription in vitro: a procedure to detect highly repetitive and transcribable sequences with tRNA-like structures. Proc Natl Acad Sci USA 83:251–255
Endoh H, Nagahashi S, Okada N (1990) Highly repetitive and transcribable sequence in the tortoise genome is probably a retroposon. Eur J Biochem 189:25–31
Esnault C, Maestre J, Heidmann T (2000) Human LINE retrotransposons generate processed pseudogenes. Nat Genet 24:363–367
Estes R, de Queiroz K, Gauthier J (1988) Phylogenetic relationships within Squamata. In: Estes R, Pregill G (eds). Phylogenetic relationships of the lizard families. Stanford University Press, Stanford, CA, pp 119–281
Fuller S, Baverstock P, King D (1998) Biogeographic origins of goannas (Varanidae): a molecular perspective. Mol Phylogenet Evol 9:294–307
Gauss DH, Grüter F, Sprinzl M (1979) Compilation of tRNA sequences. In: Abelson JN (eds). Transfer RNA: structure, properties, and recognition. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, pp 518–537
Gilbert N, Labuda D (1999) CORE-SINEs: eukaryotic short interspersed retroposing elements with common sequence motifs. Proc Natl Acad Sci USA 96:2869–2874
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hamada M, Takasaki N, Reist JD, DeCicco AL, Goto A, Okada N (1998) Detection of the ongoing sorting of ancestrally polymorphic SINEs toward fixation or loss in populations of two species of charr during speciation. Genetics 150:301–311
Hasegawa M, Kishino H, Yano T (1985) Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Hillis DM (1999) SINEs of the perfect character. Proc Natl Acad Sci USA 96:9979–9981
Hugall AF, Lee MS (2004) Molecular claims of Gondwanan age for Australian agamid lizards are untenable. Mol Biol Evol 21:2102–2110
Hughes S, Mouchiroud D (2001) High evolutionary rates in nuclear genes of squamates. J Mol Evol 53:70–76
International Chicken Genome Sequencing Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432:695–716
Jurka J (1997) Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci USA 94:1872–1877
Jurka J, Zietkiewicz E, Labuda D (1995) Ubiquitous mammalian–wide interspersed repeats (MIRs) are molecular fossils from the mesozoic era. Nucleic Acids Res 23:170–175
Kajikawa M, Okada N (2002) LINEs mobilize SINEs in the eel through a shared 3′ sequence. Cell 111:433–444
Kajikawa M, Okada N (2005) Isolation and characterization of active LINE and SINEs from the eel. Mol Biol Evol 22:673–682
Kapitonov VV, Jurka J (2003) A novel class of SINE elements derived from 5S rRNA. Mol Biol Evol 20:694–702
Kawai K, Nikaido M, Harada M, Matsumura S, Lin L-K, Wu Y, Hasegawa M, Okada N (2002) Intra- and interfamily relationships of Vespertilionidae inferred by various molecular markers including SINE insertion data. J Mol Evol 55:284–301
Kordis D, Gubensek F (1995) Horizontal SINE transfer between vertebrate classes. Nat Genet 10:131–132
Kordis D, Gubensek F (1998) Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc Natl Acad Sci USA 95:10704–10709
Krane DE, Clark AG, Cheng J-F, Hardison RC (1991) Subfamily relationships and clustering of rabbit C repeats. Mol Biol Evol 8:1–30
Kumar S, Tamura K, Nei M (2004) MEGA3: integrated software for molecular evolutionary genetics analyses and sequence alignments. Briefings in Bioinformatics 5:150–163
Kumazawa Y (2004) Mitochondrial DNA sequences of five squamates: phylogenetic affiliation of snakes. DNA Res 11:137–144
Lee MSY (2000) Soft anatomy, diffuse homoplasy, and the relationships of lizards and snakes. Zool Scr 29:101–130
Lenstra JA, van Boxtel JA, Zwaagstra KA, Schwerin M (1993) Short interspersed nuclear element (SINE) sequences of the Bovidae. Anim Genet 24:33–39
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Luan DD, Korman MH, Jakubczak JL, Eickbush TH (1993) Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell 72:595–605
Miyamoto MM (1999). Molecular systematics: Perfect SINEs of evolutionary history? Curr Biol 9:R816–R819
Moran JV, Holmes SE, Naas TP, DeBeradinis RJ, Boeke JD, Kazazian HH Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927
Murata S, Takasaki N, Saitoh M, Okada N (1993) Determination of the phylogenetic relationships among Pacific salmonids using short interspersed elements (SINEs) as temporal landmarks of evolution. Proc Natl Acad Sci USA 90:6995–6999
Murphy WJ, Eizirik E, Johnson WE, Zhang YP, Ryder OA, O’Brien SJ (2001) Molecular phylogenetics and the origins of placental mammals. Nature 409:614–618
Nikaido M, Rooney AP, Okada N (1999) Phylogenetic relationships among cetartiodactyls based on insertions of short and long interspersed elements: Hippopotamuses are the closest extant relatives of whales. Proc Natl Acad Sci USA 96:10261–10266
Nikaido M, Nishihara H, Yukio H, Okada N (2003) Ancient SINEs from African endemic mammals. Mol Biol Evol 20:522–527
Nishihara H, Terai Y, Okada N (2002) Characterization of novel Alu- and tRNA-related SINEs from the tree shrew and evolutionary implications of their origins. Mol Biol Evol 19:1964–1972
Nishihara H, Satta Y, Nikaido M, Thewissen JG, Stanhope MJ, Okada N (2005) A retroposon analysis of afrotherian phylogeny. Mol Biol Evol 22:1823–1833
Ogiwara I, Miya M, Ohshima K, Okada N (1999) Retropositional parasitism of SINEs on LINEs: identification of SINEs and LINEs in elasmobranchs. Mol Biol Evol 16:1238–1250
Ogiwara I, Miya M, Ohshima K, Okada N (2002) V-SINEs: a new superfamily of vertebrate SINEs that are widespread in vertebrate genomes and retain a strongly conserved segment within each repetitive unit. Genome Res 12:316–324
Ohshima K, Okada N (2005) SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res 110:475–490
Ohshima K, Hamada M, Terai Y, Okada N (1996) The 3′ends of tRNA-derived short interspersed repetitive elements are derived from the 3′ends of long interspersed repetitive elements. Mol Cell Biol 16:3756–3764
Ohshima K, Hattori M, Yada T, Gojobori T, Sakaki Y, Okada N (2003) Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol 4:R74
Okada N (1991) SINEs: short interspersed repeated elements of the eukaryotic genome. Trends Ecol Evol 6:358–361
Okada N, Hamada M (1997) The 3’ ends of tRNA-derived SINEs originated from the 3′ ends of LINEs: a new example from the bovine genome. J Mol Evol 44:52–56
Okada N, Ohshima K (1995) Evolution of tRNA-derived SINEs. In: Maraia RJ (eds). The impact of short interspersed elements (SINEs) on the host genome. Springer, R.G. Landes, Austin, TX, pp 61–79
Okada N, Hamada M, Ogiwara I, Ohshima K (1997) SINEs and LINEs share common 3′ sequence: a review. Gene 205:229–243
Okada N, Shedlock AM, Nikaido M (2004) Retroposon mapping in molecular systematics. In: Miller WJ, Capy P (eds). Mobile genetic elements. Protocols and genomic applications. Humana Press, Totowa, NJ, pp 189–226
Pianka ER, Vitt LJ (2003) Lizards: windows to the evolution of diversity. University of California Press, Berkeley/Los Angeles
Piskurek O, Okada N (2005) Simple and complex SINEs: a brief critical comment. Gene (in press)
Piskurek O, Nikaido M, Boeadi, Baba M, Okada N (2003) Unique mammalian tRNA-derived repetitive elements in dermopterans: the t-SINE family and its retrotransposition through multiple sources. Mol Biol Evol 20:1659–1668
Ray DA, Xing J, Hedges DJ, Hall MA, Laborde ME, Anders BA, White BR, Stoilova N, Fowlkes JD, Landry KE, Chemnick LG, Ryder OA, Batzer MA (2005) Alu insertion loci and platyrrhine primate phylogeny. Mol Phylogenet Evol 35:117–126
Rieppel O, Reiz R (1999) The origin and early evolution of turtles. Annu Rev Ecol Syst 30:1–22
Rest JS, Ast JC, Austin CC, Waddell PJ, Tibbetts EA, Hay JM, Mindell DP (2003) Molecular systematics of primary reptilian lineages and the tuatara mitochondrial genome. Mol Phylogenet Evol 29:289–297
Sakagami M, Ohshima K, Mukoyama H, Yasue H, Okada N (1994) A novel tRNA species as an origin of short interspersed repetitive elements (SINEs): equine SINEs may have originated from tRNA(Ser). J Mol Biol 239:731–735
Sasaki T, Kazuhiko T, Nikaido M, Miura S, Yasukawa Y, Okada N (2004) First application of the SINE (short interspersed repetitive element) method to infer phylogenetic relationships in reptiles: an example from the turtle superfamily Testudinoidea. Mol Biol Evol 21:705–715
Schmid CW (1996) Alu: structure, origin, evolution, significance and function of one-tenth of human DNA. Prog Nucleic Acids Res Mol Biol 53:283–319
Schmid CW, Maraia R (1992) Transcriptional regulation and transpositional selection of active SINE sequences. Curr Opin Genet Dev 2:874–882
Schmitz J, Zischler H (2003) A novel family of tRNA-derived SINEs in the colugo and two new retrotransposable markers separating dermopterans from primates. Mol Phylogenet Evol 28:341–349
Schmitz J, Ohme M, Zischler H (2001) SINE insertions in cladistic analyses and the phylogenetic affiliations of Tarsius bancanus to other primates. Genetics 157:777–784
Schmitz J, Piskurek O, Zischler H (2005) 40 million years of independent evolution: a mitochondrial gene and its corresponding nuclear pseudogene in primates. J Mol Evol 61:1–11
Schulte II JA, Melville J, Larson A (2003) Molecular phylogenetic evidence for ancient divergence of lizard taxa on either side of Wallace’s Line. Proc Biol Sci 270:597–603
Shedlock AM, Okada N (2000) SINE insertions: powerful tools for molecular systematics. Bioessays 22:148–160
Shedlock A, Takahashi K, Okada N (2004) SINEs of speciation: tracking lineages with retroposons. Trends Ecol Evol 19:545–553
Shimamura M, Yasue H, Ohshima K, Abe H, Kato H, Kishiro T, Goto M, Munechika I, Okada N (1997) Molecular evidence from retroposons that whales form a clade within even-toed ungulates. Nature 388:666–670
Shimamura M, Abe H, Nikaido M, Ohshima K, Okada N (1999) Genealogy of families of SINEs in cetaceans and artiodactyls: the presence of a huge superfamily of tRNAGlu-derived families of SINEs. Mol Biol Evol 16:1046–1060
Slagel V, Flemington E, Traina–Dorge V, Bradshaw H, Deininger P (1987) Clustering and subfamily relationships of the Alu family in the human genome. Mol Biol Evol 4:19–29
Smit AF, Riggs AD (1995) MIRs are classic, tRNA-derived SINEs that amplified before the mammalian radiation. Nucleic Acids Res 23:98–102
Sprinzl M, Vassilenko S (2005) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 33:139–140
Strimmer K, von Haeseler A (1996) Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol 13:964–969
Szemraj J, Plucienniczak G, Jaworski J, Plucienniczak A (1995) Bovine Alu-like sequences mediate transposition of a new site-specific retroelement. Gene 152:261–264
Takahashi K, Terai Y, Nishida M, Okada N (1998) A novel family of short interspersed repetitive elements (SINEs) from cichlids: the patterns of insertion of SINEs at orthologous loci support the proposed monophyly of four major groups of cichlid fishes in Lake Tanganyika. Mol Biol Evol 15:391–407
Terai Y, Takezaki N, Mayer WE, Tichy H, Takahata N, Klein J, Okada N (2004) Phylogenetic relationships among East African haplochromine fish as revealed by short interspersed elements (SINEs). J Mol Evol 58:64–78
Thompson JD, Higgens DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Townsend T, Larson A, Louis E, Macey JR (2004) Molecular phylogenetics of squamata: the position of snakes, amphisbaenians, and dibamids, and the root of the squamate tree. Syst Biol 53:735–757
Ullu E, Tschudi C (1984) Alu sequences are processed 7SL RNA genes. Nature 312:171–172
Vidal N, Hedges SB (2004) Molecular evidence for a terrestrial origin of snakes. Proc Biol Sci 271 (Suppl 4):S226–S229
Weiner AM, Deininger PL, Efstratiadis A (1986) Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu Rev Biochem 55:631–661
Yang Z (1996) Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol 11:367–372
Zardoya R, Meyer A (1998) Complete mitochondrial genome suggests diapsid affinities of turtles. Proc Natl Acad Sci USA 95:14226–14231
Zug GR, Vitt LJ, Caldwell JP (2001) Herpetology, 2nd ed. Academic Press, San Diego, CA
Zupunski V, Gubensek F, Kordis D (2001) Evolutionary dynamics and evolutionary history in the RTE clade of non-LTR retrotransposons. Mol Biol Evol 18:1849–1863
Acknowledgments
This work was funded by research grants from the Monbukagakusho (Ministry of Education, Culture, Sports, Science and Technology, Government of Japan; MEXT) to O.P. and N.O. and from the National Science Foundation to C.C.A. We thank Mathias Sprinzl for his help in identifying the tRNA-like 5′ end of the Sauria SINEs. We thank the California Academy of Sciences (Jens Vindum), the Museum of Vertebrate Zoology (Jim McGuire), and the Museum of Natural Science at Louisiana State University for tissues.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Martin Kreitman]
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Piskurek, O., Austin, C.C. & Okada, N. Sauria SINEs: Novel Short Interspersed Retroposable Elements That Are Widespread in Reptile Genomes. J Mol Evol 62, 630–644 (2006). https://doi.org/10.1007/s00239-005-0201-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-005-0201-5