
Abstract
It is well known that stop codons play a critical role in the process of protein synthesis. However, little effort has been made to investigate whether stop codon usage exhibits biases, such as widely seen for synonymous codon usage. Here we systematically investigate stop codon usage bias in various eukaryotes as well as its relationships with its context, GC3 content, gene expression level, and secondary structure. The results show that there is a strong bias for stop codon usage in different eukaryotes, i.e., UAA is overrepresented in the lower eukaryotes, UGA is overrepresented in the higher eukaryotes, and UAG is least used in all eukaryotes. Different conserved patterns for each stop codon in different eukaryotic classes are found based on information content and logo analysis. GC3 contents increase with increasing complexity of organisms. Secondary structure prediction revealed that UAA is generally associated with loop structures, whereas UGA is more uniformly present in loop and stem structures, i.e., UGA is less biased toward having a particular structure. The stop codon usage bias, however, shows no significant relationship with GC3 content and gene expression level in individual eukaryotes. The results indicate that genomic complexity and GC3 content might contribute to stop codon usage bias in different eukaryotes. Our results indicate that stop codons, like synonymous codons, exhibit biases in usage. Additional work will be needed to understand the causes of these biases and their relationship to the mechanism of protein termination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Termination of protein synthesis, one of the most important steps in gene expression, mainly involves stop codons, release factors, and the ribosome (Kisselev et al. 2003). In general, when one of the there stop codons is translocated into the ribosomal A-site and decoded by the release factor, the termination process is triggered. But the mechanism for the coexistence of three alternative stop codons in the same organism still remains unclear. In 1988, Sharp and Bulmer (1988) investigated the frequency of stop codons in several bacteria and yeast using the limited sequence data available at the time. With many eukaryotic sequences now available, it is possible to examine stop codon usage bias across a much broader phylogenetic scale.
Though stop codons play a key role in the process of protein synthesis, it has been shown that trinucleotides (stop codons) alone are insufficient for efficient translation termination (Tate et al. 1996). Nucleotides in both the upstream region (i.e., the coding region) and the downstream region (i.e., 3′ uncoding region) around stop codons are significantly biased and affect gene translation efficiency in many eukaryotes (Cassan and Rousset 2001; McCaughan et al. 1995; Mottagui-Tabar and Isaksson 1997; Namy et al. 2001). Therefore correlation of characteristics of sequence patterns around stop codons with stop codon usage would provide clues for understanding the stop codon usage bias.
In fact, three stop codons have the same function in the termination process so they can be regarded as synonymous codons too. Previous research on synonymous codon usage often analyzed GC3 content (the GC level at their third codon positions) and gene expression level to reveal the correlation between codon bias and gene expression level or GC content and, further, to investigate the effectiveness of natural selection (Duan and Antezana 2003; Duret and Mouchiroud 1999; Gillespie 2000; Herbeck et al. 2003; Kliman et al. 2003; Vinogradov 2003). In addition, when a stop codon enters the ribosome and triggers the termination process, it must interact with proteins or/and rRNA, thus it might be informative to know the structural features of the three nucleotides within a stop codon.
In the present work, we systematically studied stop codon usage bias in various eukaryotes as well as its relationships with its context, secondary structure features, GC3 isochores, and gene expression level. The results show that UAA is overrepresented in eukaryotes with lower complexity, while UGA is overrepresented in eukaryotes with higher complexity. Different conserved patterns around each stop codon in different eukaryotic classes were found. GC3 contents increase with increasing complexity of organisms. Secondary structure prediction revealed that UAA is generally associated with loop structures, whereas UGA is more uniformly present in loop and stem structures, i.e., UGA is less biased toward having a particular structure. Stop codon usage bias, however, shows no significant relationship with GC3 contents and gene expression level in individual eukaryotes.
The results indicate that the genomic complexity and GC3 content might contribute to the stop codon usage bias in different eukaryotes. They also indicate that stop codons, like synonymous codons, exhibit biases in usage. Additional work will be needed to understand the causes of these biases and their relationship to the mechanism of protein termination.
Materials and Methods
Sequence Retrieval
In order to accurately determine the position of the stop codon in a sequence, we first downloaded 3′ untranslated sequences from the 3UTRdb-15 database (Pesole et al. 2002). The database is specialized for 3′ untranslated sequences of genes in eukaryotic organisms and eukaryotic viruses. In order to determine the upstream context of stop codons, we abstracted the accession numbers from the above data and downloaded the corresponding CDSs (complete coding sequences) from EMBL (Stoesser et al. 2003) through the sequence retrieval system (SRS) (entries without counterparts in EMBL were deleted). All sequences were sorted the same as the UTRdb databank classes (viral sequences discarded). Sequences of each class were sorted into UAA group genes, UAG group genes, and UGA group genes, respectively, according to the stop codon types. The numbers and classes of all sequences analyzed are listed in Table 1.
Because each organism has its own codon usage table, it is necessary to select a representative for each class when measuring the gene expression level. Therefore we selected five model organisms, i.e., fission yeast, fruit fly, thale cress, zebrafish, and mouse, and human for each class except for the “other mammal” class. The gene sequences of these six organisms were downloaded from the corresponding databases (Table 2).
Calculation of Synonymous Codon Usage Value
The relative synonymous codon usage (RSCU) value is often used to compare synonymous codon usage bias among different genes or organisms (Sharp et al. 1986). In fact, three stop codons share the same function during the process of protein synthesis so they can be regarded as synonymous codons. So we used the modified RSCU formula to study the stop codon bias in seven eukaryotes classes, which is represented as:
where i = UAA, UAG or UGA and X i is the number of occurrences of stop codon i.
Calculation of Information Content and Logo Analysis
In order to analyze nucleotide patterns around stop codons, we extracted neighboring sequences (50 nucleotides upstream and 50 nucleotides downstream) around stop codons. To locate the position of the highly conserved pattern around each translation termination site, we calculated the information content (Schneider et al. 1986) for each position with respect to the stop codons.
Information content is calculated as follows:
where L is the position with respect to the stop codon, P(i,L) is the average probability of nucleotide i in the genes being analyzed, and P i is the random average probability of each of the four nucleotides appearing.
Based on the conserved regions derived from information content analyses, logo analyses were performed on the 20 nucleotides around stop codons using the Web site tool, WebLogo (Crooks et al. 2004), which can generate graphical representations of the patterns within a multiple sequence alignment.
Calculation of Gene Expression Level and GC3 Content
When measuring gene expression level, we used the codon adaptation index (CAI) as the indicator for less complex organisms such as fission yeast, fruit fly, thale cress, and zebrafish (Sharp and Li 1987). We applied the program Cai in the EMBOSS package (version 2.8.0) (Rice et al. 2000) to create an individual codon usage table for each organism and calculated the corresponding CAI value of each gene.
However, it is not suitable to use the CAI to estimate the gene expression level for more complex organisms such as mouse and human since it has been shown that highly expressed vertebrates genes do not display high codon usage bias (Duret 2002). Thus for mouse and human we used the tissue number relative probability (Arhondakis et al. 2004; Konu and D.Li 2002) calculated from microarray data to estimate the gene expression level. Affimatrix GPL91 of human and Affimetrix GPL81 of mouse were downloaded from Gene Expression Omnibus in GenBank. A gene was regarded as expressed if its signal level exceeded a conservative threshold of 200 arbitrary units (Su et al. 2002). The tissue number relative probability was calculated as the number of tissues that a gene expressed / the total number of tissues. The data on characterized genes expressed in at least one normal tissue were used (Table 2). The samples and replicates representing the same tissue were averaged.
Prediction of Secondary Structure
We used 50 nucleotides, including the termination site (23 upstream nucleotides and 24 downstream nucleotides) of all CDSs, to predict the secondary structures using the RNA secondary structure prediction program, Vienna RNA Package 2.0, with the default parameters (including the temperature parameter 37°C for all species) (Hofacker 2003). Then we statistically analyzed the states of the three nucleotides of the stop codon on the predicted secondary structures. Each nucleotide is either within the loop (abbreviated L) or within the stem (abbreviated S), so there may be eight states for three nucleotides, abbreviated LLL, LLS, LSL, LSS, SLL, SSL, SLS, and SSS, respectively.
The statistical analyses of all data were analyzed using the software Matlab 6.5.
Results
Stop Codon Usage Bias in Eukaryotes
The usage of three stop codons in seven eukaryotic organism classes varies as revealed by the RSCU values in Fig. 1. Variance analysis showed that the three stop codon usages are significantly different among seven eukaryotic classes (p<0.01) and coefficient correlation analysis showed that there is a highly negative correlation between UAA usage and UGA usage (R = –0.9925). Statistically, codon UAA is overrepresented in less complex eukaryotes such as yeast and invertebrate; stop codon UGA is overrepresented in more complex eukaryotes, especially in vertebrates; and stop codon UAG is less used in all organisms compared with UAA and UGA.

RSCU values of three stop codons in various eukaryotes. The vertical axis represents the RSCU value of the stop codon. The horizontal axis represents the seven species.
Relative to the preferred stop codon, the two other stop codons, UGA and UAG in yeast and invertebrates and UAA and UAG in other mammals, basically have no difference in their usages. Interestingly, in plants and other vertebrates, although UAG is least used, UGA and UAA are essentially equally used. All the observations indicate that different organisms select their own preferred stop codon. Moreover, this kind of selection gradually changes from fungi to human, namely, the usage of stop codon UAA gradually decreases from less complex organisms to more complex organisms, while the usage of stop codon UGA gradually increases from less complex to more complex organisms.
Conserved Patterns Around Stop Codons
Since each class of eukaryotes selects its individual preferred stop codons, it is natural to ask if there exist different conserved patterns around different stop codons and, if so, where they are and what they are. To answer these questions, we analyzed the information content of each position around stop codons in seven eukaryotic classes (Figure 2). Variance analysis of the information content values of 100 positions showed that not only are the information content values of the three stop codon groups in each class significantly different (p<0.01), but also the information content values of the same stop codon groups in seven eukaryotic classes are significantly different (p<0.01). Further analyses revealed that these differences are mainly caused by the first position in the downstream region (+1 position) and the last two positions in the upstream region (−1 position and −2 position), as their information content values are much higher than those of other positions. Therefore, these positions have more conserved sequence patterns than other positions do.

Information content values of 100 nucleotides around the stop codons in seven classes of eukaryotes. Information content values of the three stop codons were 2, but for mapping convenience, the information contents of the stop codon is designated 0. The vertical axis represents the information content values. The horizontal axis represents the nucleotide position, that is, −1 means immediately before the stop codon, while +1 means immediately after the stop codon; 0 indicates the position of the second nucleotide of the stop codon. The blue curve represents the UAA group genes, the green curve represents the UAG group genes, and the red curve represents the UGA group genes.
In order to determine which nucleotide is the main contributor to the high information content value, we conducted logo analysis on only 20 nucleotides around the stop codon (including the stop codon) (Fig. 3). Figure 3 shows that the bits which indicate the sequence conservation for the four nucleotides A, T, G, and C at the last two positions in the upstream region (referred to as the −1 and −2 positions hereafter, but corresponding to the −2 and −3 positions in Fig. 3) and the first position in the downstream region (+1 position, but corresponding to the +2 position in Fig. 3) around preferred stop codons in various classes are significantly different. In fungus, invertebrate, and plant classes, the bits of nucleotide A at both the −1 position and the +1 position around the preferred stop codon UAA are highest. In other mammal, rodent, and human classes, the bit of nucleotide C at the −1 position and the bit of nucleotide G at the +1 position around the preferred stop codon UGA are highest. However, in other vertebrates, the bits of nucleotide A at the −2 position and the bits of nucleotide C at the −1 position are highest, and the bit of nucleotide G at the +1 position is highest. Thus different conserved patterns for each stop codon in different eukaryotic classes can be deduced from the above observations.

Logo analyses of 20 nucleotides around the stop codon (including the stop codon) in seven classes of eukaryotes. The vertical axis represents the conservation at a certain position (measured in bits) (Crooks et al. 2004). The highest bit is 2 for the stop codon position. For mapping convenience, the stop codons were removed from the resultant map. The horizontal axis represents the nucleotide position around the stop codon. Here the positions of the three nucleotides of the stop codon are indicated as –1, 0, and 1; the last two nucleotides upstream are –2 and –3; the first nucleotide downstream is 2; and so on. In the text, however, for consistency with the description of the information content, the last two nucleotides upstream are indicated as –2 and –1, the first nucleotide downstream is +1, and so on.
Figure 3 also shows that the bits of the conserved positions around stop codon UAA become smaller and smaller from fungi to human, while, on the contrary, the bits of the conserved positions around stop codon UGA become larger. In addition to the difference in conserved patterns among different classes, the conserved patterns of the three stop codon groups in the same class also differ. Except for the plant and fungus classes, the bits of conserved patterns around the preferred stop codons are higher than for the other two codon groups. These results indicate that the conserved patterns around the stop codon have a certain relationship with stop codons bias, namely, for each organism, the preferred stop codon has its own special preferred conserved pattern around itself.
Relationships Among Stop Codon Usage, GC3 Content, and Gene Expression Level
Table 2 presents the average GC3 contents and average gene expression level indicator values for different stop codon groups of the five model organisms and human. In the same organism the GC3 content does not differ significantly among the three stop codon groups (p > 0.05), whereas the GC3 content increases more and more from fungi to human. Table 2 also shows that the average gene expression level among the three stop codon groups in the five model organisms and human does not differ significantly (p>0.05).
Predicted Secondary Structures of Stop Codons
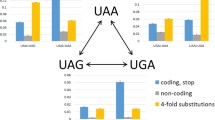
Figure 4 presents the statistic analysis of the predicted secondary structure states of the three nucleotides of stop codons in seven eukaryotic classes. Variance analysis showed that the secondary structure states of the three stop codons are significantly different in each class (p < 0.01), while there are no significant differences among the seven eukaryotic classes(p > 0.05). The three nucleotides of stop codon UAA are mainly located at the loop; the three nucleotides of stop codon UAG appeared mainly as three states, LLL, LLS, and SSS, respectively; and the three nucleotides of stop codon UGA appeared mainly as five states, LLL, LSS, SLL, SSL, and SSS, respectively. This indicates that different propensities of the three stop codon bases exist, i.e., UAA is biased toward being located within loop structures, whereas UGA is less biased toward having a particular structure.

The secondary structure types of the three nucleotides of the stop codon in various species. The horizontal axis represents the types of secondary structures. The vertical axis represents the frequencies of the secondary structure types.
Discussion
It is well known that translation termination codons (stop codons) trigger the termination of protein synthesis and most organisms have three stop codons. It is natural to ask whether the three alternative termination codons are used equally in all organisms. To answer this question, Sharp and Blurmer (1988) examined the usage frequency of three stop codons in 165 Escherichia coli genes, 52 Bacillus subtilis genes, and 106 Saccharomyces cerevisiae genes. They found that stop codon UAA was the most preferred stop codon in these genes and proposed that such selection could result from the greater availability of a UAA-cognate release factor(s) or from a less complex frequency of translation readthrough at UAA.
Previous studies have revealed that both the among-gene variance in expression-associated selection and compositional bias contribute to variance in codon usage in Drosophila melanogaster and human (Duret 2002; Kliman and Hey 1994; Knight et al. 2001; Maside et al. 2004; Sueoka and Kawanishi 2000). Here we symmetrically investigated the stop codon usage in various eukaryotes and tried to determine the reason for the bias in eukaryotes. The frequencies of the three stop codon usages in various eukaryotes are indeed different as described under Results. The change tendency of the preferred stop codons and conserved patterns around the stop codons from yeast to human suggested that the selection of preferred stop codons in different organisms might be related to the complexity of the organisms. It is difficult to draw a conclusion about the relationship between stop codon usage biases and molecular evolution, as the various eukaryotes analyzed here have no strict evolutionary relationship. However, reclassifying the sequence data based on the evolutionary relationships of the organisms to which the individual sequences belong might reveal whether stop codon usage biases are driven by evolution. Our results that GC3 contents increase from yeast to human also indicate that compositional bias might contribute to the stop codon usage bias in various eukaryotes as that did to synonymous codons usage bias (Knight et al. 2001; Sueoka and Kawanishi 2000).
It has been observed that the nucleotides surrounding stop codons affect the efficiency of translation termination in Escherichia coli, Saccharomyces cerevisiae, and mammals (Bonetti et al. 1995; McCaughan et al. 1995; Mottagui-Tabar and Isaksson 1997; Mottagui-Tabar et al. 1998). And Brown et al. 1990 suggested that tetranucleotides signal the termination of protein synthesis in eukaryotes. Our results show that the context of stop codons has conserved sequence patterns and the termination signals may contain more than four nucleotides.
Additionally, our results provide a glimpse of the changes in context from less complex to more complex eukaryotes. The difference between upstream and downstream conserved nucleotides indicates that these conserved nucleotides play different roles during the process of protein synthesis in different eukaryotes.
It is well known that highly expressed genes exhibit a strong bias for particular synonymous codons in many bacteria (Karlin et al. 1998, 2001) as well as in less complex eukaryotes (Akashi 1994; Coghlan and Wolfe 2000). But few studies have been done on the relationship between stop codon usage and gene expression level in eukaryotes. There is no universal indicator for gene expression level measurement in various organisms. Here we used the codon adaptation index (CAI) as the indicator for less complex organisms, such as fission yeast, fruit fly, thale cress, and zebrafish, and the tissue number relative probability calculated from microarray-based data to estimate the gene expression level in human and mouse. Our data revealed an obvious bias in stop codon usage in different eukaryotes; the gene expression level, however, showed no obvious relationships with the stop codon bias in eukaryotes. These results might be explained if the stop codon just triggers the termination of the protein synthesis process and is not the major determinant of the accuracy and efficiency of protein synthesis, which might be determined by the context around the stop codon and interaction between the termination signal and release factors and the ribosome.
Despite considerable progress in the study of translation termination, many details are still unclear, especially the subtle difference in the three stop codons. The use of a reconstituted translation system and high-resolution crystal of ribosome in complex with releasing factor will be necessary to understand the translation termination mechanism more fully.
References
H Akashi (1994) ArticleTitleSynonymous codon usage in Drosophila melanogaster: Natural selection and translational accuracy Genetics 136 927–935 Occurrence Handle8005445
S Arhondakis F Auletta G Torelli G D’Onofrio (2004) ArticleTitleBase composition and expression level of human genes Gene 325 165–169 Occurrence Handle10.1016/j.gene.2003.10.009 Occurrence Handle14697521
B Bonetti L Fu J Moon DM Bedwell (1995) ArticleTitleThe efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences insaccharomyces cerevisiae J Mol Biol 251 334–345 Occurrence Handle10.1006/jmbi.1995.0438 Occurrence Handle7650736
C Brown P Stockwell C Trotman W Tate (1990) ArticleTitleSequence analysis suggests that tetra-nucleotides signal the termination of protein synthesis in eukaryotes Nucleic Acids Res 18 6339–6345 Occurrence Handle2123028
M Cassan JP Rousset (2001) ArticleTitleUAG readthrough in mammalian cells: Effect of upstream and downstream stop codon contexts reveal different signals BMC Mol Biol 2 3 Occurrence Handle10.1186/1471-2199-2-3 Occurrence Handle11242562
A Coghlan KH Wolfe (2000) ArticleTitleRelationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae Yeast 16 1131–1145 Occurrence Handle10.1002/1097-0061(20000915)16:12<1131::AID-YEA609>3.0.CO;2-F Occurrence Handle10953085
GE Crooks G Hon J-M Chandonia SE Brenner (2004) ArticleTitleWebLogo: A sequence logo Generator Genome Res 14 1188–1190 Occurrence Handle10.1101/gr.849004 Occurrence Handle15173120
J Duan MA Antezana (2003) ArticleTitleMammalian mutation pressure, synonymous codon choice, and mRNA degradation J Mol Evol 57 694–701 Occurrence Handle10.1007/s00239-003-2519-1 Occurrence Handle14745538
L Duret (2002) ArticleTitleEvolution of synonymous codon usage in metazoans Curr Opin Genet Dev 12 640–649 Occurrence Handle10.1016/S0959-437X(02)00353-2 Occurrence Handle12433576
L Duret D Mouchiroud (2002) ArticleTitleExpression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis Proc Natl Acad Sci USA 96 4482–4487 Occurrence Handle10.1073/pnas.96.8.4482
JH Gillespie (2000) ArticleTitleGenetic drift in an infinite population. The pseudohitchhiking Model Genetics 155 909–919 Occurrence Handle10835409
JT Herbeck DP Wall JJ Wernegreen (2003) ArticleTitleGene expression level influences amino acid usage, but not codon usage, in the tsetse fly endosymbiont Wigglesworthia Microbiology 149 2585–2596 Occurrence Handle10.1099/mic.0.26381-0 Occurrence Handle12949182
IL Hofacker (2003) ArticleTitleVienna RNA secondary structure server Nucleic Acids Res 31 3429–3431 Occurrence Handle10.1093/nar/gkg599 Occurrence Handle12824340
S Karlin J Mrazek AM Campbell (1998) ArticleTitleCodon usages in different gene classes of the Escherichia coli genome Mol Microbiol 29 1341–1355 Occurrence Handle10.1046/j.1365-2958.1998.01008.x Occurrence Handle9781873
S Karlin J Mrazek A Campbell D Kaiser (2001) ArticleTitleCharacterizations of highly expressed genes of four fast-growing bacteria J Bacteriol 183 5025–5040 Occurrence Handle10.1128/JB.183.17.5025-5040.2001 Occurrence Handle11489855
L Kisselev M Ehrenberg L Frolova (2003) ArticleTitleNew EMBO member’s review: Termination of translation: Interplay of mRNA, rRNAs and release factors? EMBO J 22 175–182 Occurrence Handle10.1093/emboj/cdg017 Occurrence Handle12514123
RM Kliman J Hey (1994) ArticleTitleThe effects of mutation and natural selection on codon bias in the genes of Drosophila Genetics 137 1049–1056 Occurrence Handle7982559
RM Kliman N Irving M Santiago (2003) ArticleTitleSelection conflicts, gene expression, and codon usage trends in yeast J Mol Evol 57 98–109 Occurrence Handle10.1007/s00239-003-2459-9 Occurrence Handle12962310
Knight RD, Freeland SJ, Landweber LF (2001) A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol 2: research 0010.1-10.13.
D Konuo M Li (2002) ArticleTitleCorrelations between mRNA expression levels and GC contents of coding and untranslated regions of genes in rodents J Mol Evol 54 35–41 Occurrence Handle11734896
X Maside AW Lee B Charlesworth (2004) ArticleTitleSelection on codon usage in Drosophila Americana Curr Biol 20 150–154 Occurrence Handle10.1016/j.cub.2003.12.055
K McCaughan C Brown M Dalphin M Berry W Tate (1995) ArticleTitleTranslational termination efficiency in mammals is influenced by the base following the stop codon Proc Natl Acad Sci USA 6 5431–5435
S Mottagui-Tabar LA Isaksson (1997) ArticleTitleOnly the last amino acids in the nascent peptide influence translation termination in Escherichia coli genes FEBS Lett 414 165–170 Occurrence Handle10.1016/S0014-5793(97)00978-2 Occurrence Handle9305752
S Mottagui-Tabar MF Tuite LA Isaksson (1998) ArticleTitleThe influence of 5′ codon context on translation termination in Saccharomyces cerevisiae Eur J Biochem 257 249–254 Occurrence Handle10.1046/j.1432-1327.1998.2570249.x Occurrence Handle9799126
O Namy I Hatin J-P Rousset (2001) ArticleTitleImpact of the six nucleotides downstream of the stop codon on translation termination EMBO Rep 2 787–793 Occurrence Handle10.1093/embo-reports/kve176 Occurrence Handle11520858
G Pesole S Liuni G Grillo F Licciulli F Mignone C Gissi C Saccone (2002) ArticleTitleUTRdb and UTRsite: specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Update 2002 Nucleic Acids Res 30 335–340 Occurrence Handle10.1093/nar/30.1.335 Occurrence Handle11752330
P Rice I Longden A Bleasby (2000) ArticleTitleEMBOSS: The European Molecular Biology Open Software Suite Trends Genet 16 276–277 Occurrence Handle10.1016/S0168-9525(00)02024-2 Occurrence Handle10827456
TD Schneider GD Stormo L Gold A Ehrenfeuch (1986) ArticleTitleThe information content of binding sites on nucleotide sequences J Mol Biol 188 415–431 Occurrence Handle10.1016/0022-2836(86)90165-8 Occurrence Handle3525846
P Sharp W Li (1987) ArticleTitleThe codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications Nucleic Acids Res 15 1281–1295 Occurrence Handle3547335
P Sharp T Tuohy K Mosurski (1986) ArticleTitleCodon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes Nucleic Acids Res 14 5125–5143 Occurrence Handle3526280
PM Sharp M Bulmer (1988) ArticleTitleSelective differences among translation termination codons Gene 63 141–145 Occurrence Handle10.1016/0378-1119(88)90553-7 Occurrence Handle3133285
G Stoesser W Baker A Broek Particlevan den M Garcia-Pastor C Kanz T Kulikova R Leinonen Q Lin V Lombard R Lopez R Mancuso F Nardone P Stoehr MA Tuli K Tzouvara R Vaughan (2003) ArticleTitleThe EMBL Nucleotide Sequence Database: major new developments Nucleic Acids Res 31 17–22 Occurrence Handle10.1093/nar/gkg021 Occurrence Handle12519939
AI Su MP Cooke KA Ching Y Hakak JR Walker T Wiltshire AP Orth RG Vega LM Sapinoso A Moqrich A Patapoutian GM Hampton PG Schultz JB Hogenesch (2002) ArticleTitleLarge-scale analysis of the human and mouse transcriptomes Proc Natl Acad Sci USA 99 4465–4470 Occurrence Handle10.1073/pnas.012025199 Occurrence Handle11904358
N Sueoka Y Kawanishi (2000) ArticleTitleDNA G+C content of the third codon position and codon usage biases of human genes Gene 261 53–62 Occurrence Handle10.1016/S0378-1119(00)00480-7 Occurrence Handle11164037
WP Tate ES Poole ME Dalphin LL Major DJG Crawford SA Mannering (1996) ArticleTitleThe translational stop signal: Codon with a context, or extended factor recognition element? Biochimie 78 945–952 Occurrence Handle10.1016/S0300-9084(97)86716-8 Occurrence Handle9150871
AE Vinogradov (2003) ArticleTitleIsochores and tissue-specificity Nucleic Acids Res 31 5212–5220 Occurrence Handle10.1093/nar/gkg699 Occurrence Handle12930973
Acknowledgments
This work was supported by the 863 Hi-Tech Program of China (Grant 2002AA234021), the State Key Program of Basic Research of China (Grant 2002CB512800), and the Sci & Tech Committee of Shanghai (Grant K02D 105501).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Reviewing Editor: Dr. Manyuan Long]
Rights and permissions
About this article
Cite this article
Sun, J., Chen, M., Xu, J. et al. Relationships Among Stop Codon Usage Bias, Its Context, Isochores, and Gene Expression Level in Various Eukaryotes. J Mol Evol 61, 437–444 (2005). https://doi.org/10.1007/s00239-004-0277-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-004-0277-3