
Abstract
The plastids of red algae, green plants, and glaucophytes may have originated directly from a cyanobacterium-like prokaryote via primary endosymbiosis. In contrast, the plastids of other lineages of eukaryotic phototrophs appear to be the result of secondary or tertiary endosymbiotic events involving a phototrophic eukaryote and a eukaryotic host cell. Although phylogenetic analyses of multiple plastid genes from a wide range of eukaryotic lineages have been carried out, the phylogenetic positions of the secondary plastids of the Chromista (Heterokontophyta, Haptophyta and Cryptophyta) are ambiguous in a range of different analyses. This ambiguity may be the result of unusual substitutions or bias in the plastid genes established by the secondary endosymbiosis. In this study, we carried out phylogenetic analyses of five nuclear genes of cyanobacterial origin (6-phosphogluconate dehydrogenase [gnd], oxygen-evolving-enhancer [psbO], phosphoglycerate kinase [pgk], delta-aminolevulinic acid dehydratase [aladh], and ATP synthase gamma [atpC] genes), using the genome sequence data from the primitive red alga Cyanidioschyzon merolae 10D. The sequence data robustly resolved the origin of the cyanobacterial genes in the nuclei of the Chromista (Heterokontophyta and Haptophyta) and Dinophyta, before the divergence of the extant red algae (including Porphyra [Rhodophyceae] and Cyanidioschyzon [Cyadidiophyceae]). Although it is likely that gnd genes in the Chromista were transmitted from the cyanobacterium-like ancestor of plastids in the primary endosymbiosis, other genes might have been transferred from nuclei of a red algal ancestor in the secondary endosymbiosis. Therefore, the results indicate that the Chromista might have originated from the ancient secondary endosymbiosis before the divergence of extant red algae.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The origin and diversity of plastids (chloroplasts) in eukaryotic cells can be attributed to two types of endosymbiotic events: primary endosymbiosis, by which the primary plastids (with two bounding membranes) originated directly from a cyanobacterium-like prokaryote, and secondary or tertiary endosymbiosis, involving a phototrophic eukaryote and a eukaryotic host cell giving rise to secondary or tertiary plastids (surrounded by three or four bounding membranes) (e.g., Bhattacharya and Medlin 1995; Delwiche 1999; McFadden 2001; Cavalier-Smith 2002; Yoon et al. 2002a). Based on phylogenetic analyses of plastid-coding genes, and the similarity of pigment compositions and plastid genome organization, the secondary plastids have been assigned to either the red lineage with the red algal plastids or the green lineage with green plant plastids (e.g., Bhattacharya and Medlin 1995; Delwiche 1999; McFadden 2001; Cavalier-Smith 2002; Nozaki et al. 2003b). The Chromista, which are comprised of the Heterokontophyta, Haptophyta, and Cryptophyta, have secondary plastids of the red lineage (e.g., Delwiche 1999; McFadden 2001; Cavalier-Smith 2002, 2003). Although phylogenetic analyses using multiple plastid genes from a wide range of eukaryotic lineages suggest that the secondary plastids in the Chromista originated after the divergence of extant red algae (Rhodophyceae and Cyanidiophyceae), the phylogenetic positions of secondary plastids have been found to be ambiguous in a range of different phylogenetic analyses (Martin et al. 2002; Maul et al. 2002; Yoon et al. 2002a, b; Ohta et al. 2003). This ambiguity might be derived from unusual substitutions or bias, related to differences between the primary and the secondary plastids during evolution.
Cyanobacterial genes are encoded in the nuclear genomes in a wide range of primary photosynthetic organisms, and these genes are thought to have been transmitted from a cyanobacterium-like ancestor of plastids by primary endosymbiosis. These genes are usually located in the nuclear genomes of the secondary photosynthetic organisms and are, therefore, useful as phylogenetic markers for primary and secondary endosymbiosis that has occurred during the evolution of eukaryotes. Andersson and Roger (2002) demonstrated that 6-phosphogluconate dehydrogenase (gnd) genes with cyanobacterial affinity are present in the nuclear genomes of primary and secondary photosynthetic organisms, including Heterokontophyta, green plants, and the red alga Porphyra. Ishida and Green (2002) investigated the phylogenetic relationships between the plastids from Chromista and Dinophyta based on sequences of the nuclear-encoded plastid gene encoding oxygen-evolving-enhancer (psbO). More recently, Archibald et al. (2003) analyzed several cyanobacterial nuclear genes from various eukaryotes, including the chlorarachniophyte Bigelowiella. However, since most of these phylogenetic analyses included only a single red algal operational taxonomic unit (OTU) (Porphyra from the Rhodophyceae), the phylogenetic position of secondary plastids in the Chromista could not be resolved with respect to divergence of the extant red algae (Andersson and Roger 2002; Ishida and Green 2002; Archibald et al. 2003). Therefore, the study of cyanobacterial nuclear genes from other red algae, especially those of the Cyanidiophyceae, is needed to resolve this problem.
We recently launched a genome project to investigate the primitive red alga Cyanidioschyzon merolae (Cyanidiophyceae). This project has resulted in the complete nuclear genome sequence for this taxon (Matsuzaki et al. unpublished). In the present study, we aimed to determine the plastid primary and secondary endosymbiosis based on sequences from nuclear genes of cyanobacterial origin. The analysis of these genes has given robust support to the origin of the cyanobacterial genes in the Chromista nuclei, before the divergence of the extant red algae.
Materials and Methods
Phylogenetic Analyses of Protein Sequences from Each Gene
The data matrices of the five nuclear genes of cyanobacterial origin were found to be essentially the same as those reported in Andersson and Roger (2002) and Archibald et al. (2003), except that they included the sequences listed in Table 1 and excluded amitochondrial eukaryotic organisms, which are known to have unusual gene substitutions due to parasitism or the absence of mitochondria (see Stiller et al. 1998, 2001). The four nuclear genes (gnd, psbO, phosphoglycerate kinase [pgk], delta-aminolevulinic acid dehydratase [aladh], and ATP synthase gamma [atpC]) used by Archibald et al. (2003) were selected for the present study, as OTUs from the Chromista and red algae were included in the data matrices in that study, and two representatives of the red algae (Cyanidioschyzon of the Cyanidiophyceae and Porphyra/Gracilaria of the Rhodophyceae) were analyzed using the sequences provided in Table 1. Since the pgk and aladh genes of the chlorarachniophyte Bigelowiella can be considered to be results of the lateral gene transfers from the red algae, these genes were excluded from the present phylogenetic analyses. The sequences were aligned using CLUSTAL X (Thompson et al. 1997) with default options. Ambiguous portions with large gaps in the alignment were excluded from the data matrices (available from HN on request).
Maximum parsimony (MP) trees were constructed using a heuristic search (with the tree bisection-reconnection [TBR] branch-swapping algorithm), using PAUP 4.0 b10 (Swofford 2002). Neighbor-joining (NJ) trees (Saitou and Nei 1987) were constructed using CLUSTAL X, and genetic distances were calculated according to the method of Kimura (1983). Maximum likelihood (ML) analyses were carried out using Proml of PHYLIP 3.6 (Felsenstein 2002) based on the JTT model (Jones et al. 1992) and the discrete gamma model for site heterogeneity (using five discrete rate categories) with alpha parameters that were calculated by TREE-PUZZLE 5.0 (Strimmer and von Haeseler 1996). The calculations of bootstrap values (Felsenstein 1985) for the MP and NJ analyses were based on 1000 replications. For the ML method, bootstrap values were calculated based on 100 replications using Seqboot and Proml of PHYLIP 3.6 (Felsenstein 2002).
Phylogenetic Analyses of Limited Eukaryotic OTUs Using Concatenated Protein Sequences from Four Nuclear Genes
Sequence data for 10 representative OTUs (Spinacea, monocotyledons [Oryza, Triticum, and Hordeum], Arabidopsis, Chlamydomonas, Rhodophyceae [Porphyra and Gracilaria], Cyanidioschyzon [Cyanidiophyceae], diatoms [Phaeodactylum and Odontella], Anabaena sp. PCC 7120, Synechocystis sp. PCC 6803, and Prochlorococcus marinus CCMP 1986) were extracted from the data matrices of the four genes (psbO, pgk, aladh, and atpC). In these four nuclear genes, plastid secondary endosymbiosis possibly resulted in the transference of genes to the nuclei of the Chromista (see below). These four amino acid sequences were concatenated to form a single data matrix consisting of 1217 amino acids (available from H.N. on request), which was analyzed phylogenetically as described above except that bootstrap values for ML analysis were calculated based on 1000 replications. In addition, all 15 possible trees for green plants (Spinacea, monocotyledons, Arabidopsis, and Chlamydomonas,), Rhodophyceae, Cyanidioschyzon, diatoms, and cyanobacteria (Anabaena sp. PCC 7120, Synechocystis sp. PCC 6803, and Prochlorococcus marinus CCMP 1986; designated as the outgroup) were examined by PROTML of MOLPHY 2.3 (Adachi and Hasegawa 1996) and AAML of PAML (Yang 1997) without and with the discrete gamma model for site heterogeneity (the relationships within green plants and cyanobacteria were fixed as shown in Fig. 6). Bootstrap values were calculated by the RELL method (Kishino et al. 1990) with 10,000 replications. The Kishino and Hasegawa (KH; 1989), weighted Shimodaira and Hasegawa (WSH; 1999), and approximately unbiased (AU) tests were performed using the program CONSEL (Shimodaira and Hasegawa 2001).
Results
Phylogeny of gnd Genes
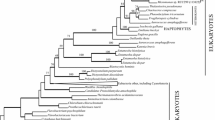
The gnd genes of the eukaryotic organisms analyzed in this study formed two distinct lineages, one closely related to the cyanobacterial lineage and the other with higher affinity to the proteobacteria (Fig. 1). The former lineage was composed of primary and secondary plastid-containing organisms (green plants, red algae, heterokontophytes, and apicomplexans), as well as plastid-lacking organisms (heterokont Phythophora and Heterolobosea) belonging to the Bikonta (Cavalier-Smith 2003) or Plantae (Nozaki et al. 2003a). Although monophyly of the red algae, green plants, and Heterokonta was resolved, the phylogenetic positions of these three groups were ambiguous within the former eukaryotic lineage (Fig. 1). The latter eukaryotic lineage was composed of the Opisthokonta (Metazoa and Fungi), red algae, and cellular slime mold Dictyostelium, whereas red algae were resolved as a robust monophyletic sister group to the Opisthokonta, with Dictyostelium occupying the most basal position (Fig. 1).

One of the seven most parsimonious trees (with tree length of 4930 and consistency index of 0.4069) based on 443 amino acid sequences from gnd genes of 52 OTUs representing a wide range of prokaryotic and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by AUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and/or maximum likelihood calculation (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without or with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary plastids, respectively.
Phylogenetic Analyses of Other Nuclear Genes
The psbO protein sequences analyzed in this study demonstrated that the red and green lineages are resolved as two monophyletic groups with high bootstrap support (95–100%) using all three phylogenetic methods (Fig. 2). In the red lineage, the red algae (Porphyra and Cyanidioschyzon) formed a robust monophyletic group (with 73–91% bootstrap values) that was sister to the lineage composed of Chromista (Haptophyta and Heterokontophyta) and Dinophyta (Fig. 2). Other phylogenetic results of psbO gene phylogeny were essentially the same as those reported by Ishida and Green (2002) and Archibald et al. (2003).

One of the two most parsimonious trees (with tree length of 1406 and consistency index of 0.6031) based on 228 amino acid sequences from psbO genes of 30 OTUs representing cyanobacterial and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by PAUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and/or maximum likelihood calculation (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without or with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary/tertiary plastids, respectively.
In the phylogenetic analyses of pgk and atpC, the red lineage was resolved as a robust monophyletic group in all of the three phylogenetic methods (with more than 80% bootstrap values) (Figs. 3 and 4). However, only the NJ and ML analysis resolved the red lineage with more than 50% bootstrap values in the phylogeny of aladh gene sequences (Fig. 5). Although the analysis of atpC gene sequences suggested the robust monophyly of the red algae (Porphyra and Cyanidioschyzon), with 67–93% bootstrap values (Fig. 4), phylogenetic results for pgk suggested the moderate nonmonophyly of the red algae (Fig. 3) and monophyly of the red algae was supported by only the ML method with 50% or more bootstrap values in aladh gene phylogeny (Fig. 5).

Single most parsimonious tree (with tree length of 3232 and consistency index of 0.4709) based on 372 amino acid sequences from pgk genes of 46 OTUs representing various prokaryotic and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by PAUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and/or maximum likelihood calculation (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without or with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary plastids, respectively.

Single most parsimonious tree (with tree length of 1247 and consistency index of 0.6576) based on 307 amino acid sequences from atpC genes of 21 OTUs representing various prokaryotic and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by PAUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and/or maximum likelihood calculation (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without or with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary plastids, respectively.

One of the three most parsimonious trees (with tree length of 1605 and consistency index of 0.5707) based on 310 amino acid sequences from aladh genes of 32 OTUs representing various prokaryotic and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by PAUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and/or maximum likelihood calculation (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without or with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary plastids, respectively.
Phylogenetic analyses of limited eukaryotic OTUs using concatenated protein sequences from four nuclear genes (psbO, pgk, aladh, and atpC) demonstrated the robust monophyly of the red (Rhodophyceae, Cyanidioschyzon, and Heterokontophyta) and green lineages, with 100% bootstrap values by all three phylogenetic methods. In the red lineage, the red algae were resolved as a robust monophyletic group (with 84–99% bootstrap values) to which the Heterokontophyta was sister.
From both the PROTML and the AAML analyses, the highest likelihood tree turned out to be Tree 1: (green plants, ((Rhodophyceae, Cyanidioschyzon), diatoms), Cyanobacteria) (Table 2). A clade consisting of Rhodophyceae and Cyanidioschyzon was supported with 97.8 and 92.7% bootstrap values by the PROTML and AAML analyses, respectively. By contrast, the groupings of diatoms and Rhodophyceae/Cyanidioschyzon (Trees 2 and 3; Table 2) were rejected with only 0.2–7.2% bootstrap support by the PROTML and AAML analyses. These groupings were also rejected at the 5% level by the KH and AU tests for the PROTML analysis, although they could not be rejected by the conservative WSH test for the PROTML analysis (highest P value was 0.089) and by the three tests for the AAML analysis (highest P values were 0.078–0.239) (Table 2).
Discussion
Phylogeny of gnd Genes
Andersson and Roger (2002) showed that gnd genes with a high cyanobacterial affinity are present in plastid-lacking organisms belonging to the Heterokonta and Heterolobosea (Discicristata), and these genes form a monophyletic group with those from the plastid-containing Apicomplexa, Heterokonta, green plants, and the red alga Porphyra. Therefore, two alternative hypotheses have been suggested: either (1) primary plastids might have been introduced into the eukaryotic cells of a common ancestor of the three primary photosynthetic eukaryotic groups as well as Discicristata, Heterokonta, and Alveolata, or (2) secondary plastids might have mediated the gnd gene transfer (Andersson and Roger 2002; Cavalier-Smith 2002). The results of our phylogenetic analyses suggest that monophyly of the extant red algae, as represented by Porphyra (Rhodophyceae) and Cyanidioschyzon (Cyanidiophyceae), might have resulted from gnd gene transfer before the divergence of the extant red algae (Fig. 1). This is consistent with the hypothesis recently proposed by Nozaki et al. (2003a), that primary endosymbiosis might have occurred in a common ancestor of the three primary photosynthetic eukaryotes as well as the organisms lacking primary plastids (Discicristata, Heterokonta, and Alveolata). More recently, Nozaki et al. (2003b) proposed that the gnd genes in Heterolobosea (Discicristata) could have resulted from gene transfer in an ancient primary endosymbiosis because their cladistic analysis of plastid gene loss suggested the relatively recent acquisition of the secondary plastid of Euglena (after the divergence of Chlorophyceae and Trebouxiophyceae).
In this study, we demonstrated that the gnd genes of Cyanidioschyzon and Porphyra, with a high proteobacterial affinity, form a monophyletic group that is sister to the Opisthokonta in the gnd gene phylogeny (Fig. 1). However, such genes have not been reported in other plastid-containing or plastid-lacking organisms (green plants, Heterokonta, Discicristata, and Apicomplexa) belonging to Bikonta (Cavalier-Smith 2003) or Plantae (Nozaki et al. 2003a) (Fig. 1). Therefore, the gnd genes with a high proteobacterial affinity might have been lost in these organisms, and this loss might have taken place only once, as suggested by the results of Nozaki et al. (2003a), resolving the red algae as a basal lineage to the monophyletic group composed of other organisms (green plants, Heterokonta, Discicristata, etc.) in Bikonta (Cavalier-Smith 2003) or Plantae (Nozaki et al. 2003a). Alternatively, such genes might have been transmitted to the red algae by the horizontal gene transfer from the common ancestor of the Opisthokonta (Metazoa and Fungi) before the divergence of the extant red algae.
Although the question of which organism is the most basal eukaryote has been the subject of a long-standing debate, recent phylogenetic studies have suggested two candidates: excavates (including amitochondrial eukaryotes, diplomonads, and parabasalids) and Amoebozoa. Previous phylogenetic studies of eukaryotes based on nuclear protein-coding genes usually included the amitochondrial eukaryotic organisms such as Diplomonadida (e.g., Giardia lambia) and Parabasala (e.g., Trichomonas), with the archaebacterial homologous genes designated as the outgroup (e.g., Hasegawa et al. 1993; Baldauf and Doolittle 1997; Stiller et al. 1998; Moreira et al. 2000; Bapteste et al. 2002). In these studies, amitochondrial organisms are consistently positioned as the most basal group within the eukaryotes (Hasegawa et al. 1993; Baldauf and Doolittle 1997; Stiller et al. 1998; Moreira et al. 2000; Bapteste et al. 2002). However, the basal phylogenetic position of amitochondrial eukaryotes can possibly be considered an artifact, due to the unusual gene substitutions of amitochondrial eukaryotes and the large genetic distance between the Archaebacteria and Eukaryotes (see Nozaki et al. 2003a). Recently, Cavalier-Smith (2002) and Stechmann and Cavalier-Smith (2002) suggested that the Amoebozoa represent the most basal eukaryotic lineage, based on the diversification of the microtubular cytoskeleton and/or presence/absence of dihydrofolate reductase-thymidylate synthase gene fusion. In addition, the present phylogenetic results and the analysis of the gnd genes with a high proteobacterial affinity demonstrate the basal position of the cellular slime mold (Dictyostelium) to the red algae and Opisthokonta (Fig. 1). The basal position of Dictyostelium has also been suggested in some phylogenetic trees based on the presence of recently (after eukaryotic origin) duplicated genes in the nuclear genome. Rivero et al. (2001) showed that 15 Rho (Ras-like small GTPases)-related proteins from Dictyostelium hold basal positions to the Metazoa and Fungi, as well as to the green plant Arabidopsis. Nozaki et al. (2003a) resolved the cellular slime molds as the most basal mitochondria-containing eukaryotes, based on a paralogous comparison of the concatenated alpha- and beta-tubulin genes of mitochondria-containing organisms.
Phylogenetic Analyses of Other Nuclear Genes
Recent phylogenetic analyses of the plastid multigene sequences have demonstrated that secondary plastids are positioned within the red algal lineages, suggesting secondary endosymbiosis after the divergence of extant red algae (Rhodophyceae and Cyanidiophyceae)(Marin et al. 2002; Maul et al. 2002; Yoon et al. 2002a, b; Ohta et al. 2003). In contrast, the present phylogenetic analyses using nuclear cyanobacterial genes suggest that secondary endosymbiosis in red algae could have taken place before the divergence of the extant red algae, based on the robust monophyly of the extant red algae (Figs. 2 and 4-6, Table 2) and the rejection of Trees 2 and 3 at the 5% level by KH and AU tests for PROTML analysis (Table 2). Itoh et al. (2002) compared the complete genome sequences of two different endosymbionts and demonstrated that the rate of amino acid substitution is twice as high in symbionts as in their relatives, suggesting that the elevated evolutionary rate is mainly due to an enhanced mutation rate in endosymbionts. The available evidence suggests that secondary plastid genes may carry accelerated and/or unusual gene substitutions compared with those of primary plastids, because the secondary plastids can be considered to be the plastids of reduced eukaryotic endosymbionts (e.g., Delwiche 1999; McFadden 2001). Thus, phylogenetic results of nuclear genes of a high cyanobacterial affinity should be useful for representing natural phylogenetic relationships because such genes in secondary photosynthetic eukaryotes have experienced the same evolutionary environment as those in the primary photosynthetic eukaryotes, even following secondary endosymbiosis, when the genes should have been transferred from the engulfed photosynthetic eukaryote to the host nucleus. However, the multigene phylogeny presented in this study (Fig. 6) analyzed a limited number of taxa, especially in the red lineages. Analysis of additional OTUs/sequences (especially other members of the Chromista) might better resolve the phylogenetic relationships between plastids within the red lineage.

Single most parsimonious tree (with tree length of 2578 and consistency index of 0.8181) based on concatenated 1217 amino acid sequences from four genes (psbO, pgk, aladh, and atpC) of 10 OTUs representing cyanobacterial and eukaryotic taxa. The tree was constructed based on the heuristic search using stepwise addition of 50 random replications (with the TBR branch-swapping algorithm) by PAUP 4.0b10 (Swofford 2002). Branch lengths are proportional to numbers of amino acid substitutions, which are indicated by the scale bar below the tree. Numbers above branches are the bootstrap values (50% or more) based on 1000 replications of the full heuristic analysis (with simple addition sequence). Branches resolved with 50% or more bootstrap values by the neighbor-joining method (based on the Kimura distances) and maximum likelihood analysis (based on JTT model with rate heterogeneity) are also shown by numbers under the branches without and with parentheses, respectively. Single and double asterisks indicate taxa with primary and secondary plastids, respectively.
In contrast to the eukaryotic gnd genes, which may have originated directly from cyanobacterium-like ancestors as a result of primary endosymbiosis (see above), transmission of four other genes to the nuclei of the Chromista possibly occurred during secondary endosymbiosis, because the red lineage was found to be monophyletic (Archibald et al. 2003) (Figs. 2–4, 5). Such genes are not recognized in plastid-lacking organisms (Ishida and Green 2002; Archibald et al. 2003). In addition, analysis of these genes demonstrated that secondary endosymbiosis in the red lineage preceded the divergence of the extant red algae (Figs. 2 and 4 – 6, Table 2). Such ancient secondary endosymbiosis has also been suggested by Fast et al. (2001) and Harper and Keeling (2003). These authors provided evidence that the plastids of Alveolata (Apicomplexa and Dinophyta) and Chromista (Heterokontophyta, Cryptophyta, and Haptophyta) appear to be the result of a single secondary endosymbiotic event on the basis of the distribution and phylogeny of the plastid-targeted glyceraldehyde-3-phosphate dehydrogenase gene. Yoon et al. (2002b), on the basis of the broadly sampled data set and molecular dating of the chloroplast multiple genes, also suggested the single, ancient origin of secondary plastids in the red lineage. In contrast, secondary endosymbiosis in the green lineage (Chlorarachniophyta and Euglenophyta) seems to be a relatively recent event (after the divergence of Chlorophyta and Streptophyta [Nozaki et al. 2003b]) based on phylogenetic analyses of the nuclear-encoded cyanobacterial genes (Archibald et al. 2003) and cladistic analysis of plastid gene losses (Nozaki et al. 2003b). In the psbO gene phylogeny, the chlorarachniophyte Bigelowiella is robustly resolved as a sister lineage to the Volvocales (Volvox, Chlamydomonas, and Dunaleiella) within the Chlorophyta (Archibald et al. 2003) (Fig. 2). Although MP and NJ analyses of the chloroplast multiple genes robustly resolved the most basal position of the Euglena plastid within the green lineage (e.g., Martin et al. 2002; Ohta et al. 2003), the ML method of the same data matrix suggested the close relationship between Euglena and Chlorella plastids (Martin et al. 2002) and the recent cladistic analysis of plastid gene losses robustly demonstrated the secondary endosymbiosis for the Euglena plastid after the divergence of Chlamydomonas (Chlorophyceae) and Chlorella (Trebouxiophyceae) within the Chlorophyta (Nozaki et al. 2003b).
References
J Adachi M Hasegawa (1996) Computer Science Monographs, No. 28, MOLPHY Version 2.3: Programs for Molecular Phylogenetics Based on Maximum Likelihood Institute of Statistical Mathematics Tokyo
JM Archibald MB Rogers M Toop K Ishida PJ Keeling (2003) ArticleTitleLateral gene transfer and the evolution of plastid-targeted proteins in the secondary plastid-containing alga Bigelowiella natans Proc Natl Acad Sci USA 100 7678–7683 Occurrence Handle10.1073/pnas.1230951100 Occurrence Handle1:CAS:528:DC%2BD3sXlt1Wqsrs%3D Occurrence Handle12777624
JO Andersson AJ Roger (2002) ArticleTitleA cyanobacterial gene in nonphotosynthetic protests—An early chloroplast acquisition in Eukaryotes? Curr Biol 12 115–119 Occurrence Handle10.1016/S0960-9822(01)00649-2 Occurrence Handle1:CAS:528:DC%2BD38XhtVKru7o%3D Occurrence Handle11818061
SL Baldauf WF Doolittle (1997) ArticleTitleOrigin and evolution of the slime molds (Mycetozoa) Proc Natl Acad Sci USA 94 12007–12012 Occurrence Handle10.1073/pnas.94.22.12007 Occurrence Handle1:CAS:528:DyaK2sXntFSmsb8%3D Occurrence Handle9342353
E Bapteste H Brinkmann JA Lee DV Moore CW Sensen P Gordon L Duruflé T Gaasterland P Lopez M Müller H Philippe (2002) ArticleTitleThe analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba Proc Natl Acad Sci USA 99 1414–1419 Occurrence Handle10.1073/pnas.032662799 Occurrence Handle1:CAS:528:DC%2BD38Xht1Cls7c%3D Occurrence Handle11830664
D Bhattacharya L Medlin (1995) ArticleTitleThe phylogeny of plastids: A review based on comparisons of small subunit ribosomal RNA coding regions J Phycol 31 489–498 Occurrence Handle1:CAS:528:DyaK2MXos1Gls7Y%3D
T Cavalier-Smith (2002) ArticleTitleChloroplast evolution: Secondary symbiogenesis and multiple losses Curr Biol 12 R62–R64 Occurrence Handle10.1016/S0960-9822(01)00675-3 Occurrence Handle1:CAS:528:DC%2BD38XhtVKrurg%3D Occurrence Handle11818081
T Cavalier-Smith (2003) ArticleTitleGenomic reduction and evolution of novel genetic membranes and protein-targeting machinery in eukaryote-eukaryote chimaeras (meta-algae) Phil Trans R Soc Lond B Biol Sci 358 109–133 Occurrence Handle10.1098/rstb.2002.1194 Occurrence Handle1:CAS:528:DC%2BD3sXktVWhsro%3D
CF Delwiche (1999) ArticleTitleTracing the thread of plastid diversity through the tapestry of life Am Nat 154 164–177 Occurrence Handle10.1086/303291
NM Fast JC Kissinger DS Roos PJ Keeling (2001) ArticleTitleNuclear-encoded, plastid-targeted genes suggest a single common origin for apicomplexan and dinoflagellate plastids Mol Biol Evol 18 418–426 Occurrence Handle1:CAS:528:DC%2BD3MXhvVKrt78%3D Occurrence Handle11230543
J Felsenstein (1985) ArticleTitleConfidence limits on phylogenies: an approach using bootstrap Evolution 38 16–24
J Felsenstein (2002) PHYLIP (Phylogeny Inference Package) version 3.6a3. Distributed by the author, Department of Genome Sciences University of Washington Seattle
JT Harper PJ Keeling (2003) ArticleTitleNucleus-encoded, plastid-targeted glyceraldehyde-3-phosphate dehydrogenase (GAPDH) indicates a single origin for chromalveolate plastids Mol Biol Evol 20 1730–1735 Occurrence Handle10.1093/molbev/msg195 Occurrence Handle1:CAS:528:DC%2BD3sXotlWjsL0%3D Occurrence Handle12885964
M Hasegawa T Hashimoto J Adachi N Iwabe T Miyata (1993) ArticleTitleEarly branchings in the evolution of eukaryotes: Ancient divergence of Entamoeba that lacks mitochondria revealed by protein sequence data J Mol Evol 36 380–388 Occurrence Handle1:CAS:528:DyaK3sXitV2hur8%3D Occurrence Handle8315658
K Ishida BR Green (2002) ArticleTitleSecond- and third-hand chloroplasts in dinoflagellates: phylogeny of oxygen-evolving enhancer 1 (PsbO) protein reveals replacement of a nuclear-encoded plastid gene by that of a haptophyte tertiary endosymbiont Proc Natl Acad Sci USA 99 9294–9299 Occurrence Handle10.1073/pnas.142091799 Occurrence Handle1:CAS:528:DC%2BD38XlsVGgsLk%3D Occurrence Handle12089328
T Itoh W Martin M Nei (2002) ArticleTitleAcceleration of genomic evolution caused by enhanced mutation rate in endocellular symbionts Proc Natl Acad Sci USA 99 12944–12948 Occurrence Handle10.1073/pnas.192449699 Occurrence Handle1:CAS:528:DC%2BD38XnvFGht7s%3D Occurrence Handle12235368
DT Jones WR Taylor JM Thornton (1992) ArticleTitleThe rapid generation of mutation data matrices from protein sequences CABIOS 8 275–282 Occurrence Handle1:CAS:528:DyaK38Xlt1Okt7w%3D Occurrence Handle1633570
M Kimura (1983) The neutral theory of molecular evolution Cambridge University Press Cambridge
H Kishino M Hasegawa (1989) ArticleTitleEvaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea J Mol Evol 29 170–179 Occurrence Handle1:CAS:528:DyaL1MXkvFCnsbc%3D Occurrence Handle2509717
H Kishino T Miyata M Hasegawa (1990) ArticleTitleMaximum likelihood inference of protein phylogeny, and the origin of chloroplasts J Mol Evol 31 151–160 Occurrence Handle1:CAS:528:DyaK3cXlt1yqtLw%3D
W Martin T Rujan E Richly A Hansen S Cornelsen T Lins D Leister B Stoebe M Hasegawa D Penny (2002) ArticleTitleEvolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus Proc Natl Acad Sci USA 99 12246–12251 Occurrence Handle10.1073/pnas.182432999 Occurrence Handle1:CAS:528:DC%2BD38XntlCks70%3D Occurrence Handle12218172
JE Maul JW Lilly L Cui CW dePamphilis W Miller EH Harris DB Stern (2002) ArticleTitleThe Chlamydomonas reinhardtii plastid chromosome: islands of genes in a sea of repeats Plant Cell 14 2659–2679 Occurrence Handle10.1105/tpc.006155 Occurrence Handle1:CAS:528:DC%2BD38XovF2qtbg%3D Occurrence Handle12417694
GI McFadden (2001) ArticleTitlePrimary and secondary endosymbiosis and the origin of plastids J Phycol 37 951–959 Occurrence Handle10.1046/j.1529-8817.2001.01126.x
D Moreira H Le Guyader H Philippe (2000) ArticleTitleThe origin of red algae and the evolution of chloroplasts Nature 405 69–72 Occurrence Handle10.1038/35011054 Occurrence Handle1:STN:280:DC%2BD3c3mvFemtA%3D%3D Occurrence Handle10811219
I Nikaido E Asamizu M Nakajima Y Nakamura N Saga S Tabata (2000) ArticleTitleGeneration of 10,154 expressed sequence tags from a leafy gametophyte of a marine red alga, Porphyra yezoensis DNA Res 7 223–227 Occurrence Handle10907854
H Nozaki M Matsuzakil M Takahara O Misumil H Kuroiwa M Hasegawa T Shin-i Y Kohara N Ogasawara T Kuroiwa (2003a) ArticleTitleThe phylogenetic position of red algae revealed by multiple nuclear genes from mitochondria-containing eukaryotes and an alternative hypothesis on the origin of plastids J Mol Evol 56 485–497 Occurrence Handle10.1007/s00239-002-2419-9 Occurrence Handle1:CAS:528:DC%2BD3sXisV2qtr8%3D
H Nozaki N Ohta M Matsuzaki O Misumi T Kuroiwa (2003b) ArticleTitlePhylogeny of plastids based on cladistic analysis of gene loss inferred from complete plastid genome sequences J Mol Evol 57 377–382 Occurrence Handle10.1007/s00239-003-2486-6 Occurrence Handle1:CAS:528:DC%2BD3sXovFCgt7o%3D
N Ohta M Matsuzaki O Misumi S Miyagishima H Nozaki K Tanaka T Shin-i Y Kohara T Kuroiwa (2003) ArticleTitleAnalyses of the complete nucleotide sequence of the plastid genome of the unicellular red alga Cyanidioschyzon merolae: Genome structure, gene organization, and gene content DNA Res 10 67–77 Occurrence Handle1:CAS:528:DC%2BD3sXjsFCqsLs%3D Occurrence Handle12755171
F Rivero H Dislich G Glockner AA Noegel (2001) ArticleTitleThe Dictyostelium discoideum family of Rho-related proteins Nucleic Acids Res 29 1068–1079 Occurrence Handle10.1093/nar/29.5.1068 Occurrence Handle1:CAS:528:DC%2BD3MXhvFaqs7c%3D Occurrence Handle11222756
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic trees Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
H Shimodaira M Hasegawa (1999) ArticleTitleMultiple comparisons of log-likelihoods with applications to phylogenetic inference Mol Biol Evol 16 1114–1116 Occurrence Handle1:CAS:528:DyaK1MXltVyksrg%3D
H Shimodaira M Hasegawa (2001) ArticleTitleCONSEL: For assessing the confidence of phylogenetic tree selection Bioinformatics 17 1246–1247 Occurrence Handle10.1093/bioinformatics/17.12.1246 Occurrence Handle1:STN:280:DC%2BD38%2FgtFOlsw%3D%3D Occurrence Handle11751242
A Stechmann T Cavalier-Smith (2002) ArticleTitleRooting the eukaryote tree by using a derived gene fusion Science 297 89–91 Occurrence Handle10.1126/science.1071196 Occurrence Handle1:CAS:528:DC%2BD38XltFGntrw%3D Occurrence Handle12098695
JW Stiller ECS Duffield BD Hall (1998) ArticleTitleAmitochondriate amoebae and the evolution of DNA-dependent RNA polymerase II Proc Natl Acad Sci USA 95 11769–11774 Occurrence Handle10.1073/pnas.95.20.11769 Occurrence Handle1:CAS:528:DyaK1cXmsV2hs70%3D Occurrence Handle9751740
JW Stiller J Riley BD Hall (2001) ArticleTitleAre red algae plants? A critical evaluation of three key molecular data sets J Mol Evol 52 527–539 Occurrence Handle1:CAS:528:DC%2BD3MXksFOiuro%3D Occurrence Handle11443356
K Strimmer A Haeseler ParticleVon (1996) ArticleTitleQuartet puzzling: A quartet maximum-likelihood method for reconstructing tree topologies Mol Biol Evol 13 964–969 Occurrence Handle1:CAS:528:DyaK28XltlSmsLk%3D
DL Swofford (2002) PAUP* 4.0: Phylogenetic analysis using parsimony, version 4.0b10 Computer program distributed by Sinauer Associates Fitchburg
JD Thompson TJ Gibson F Plewniak F Jeanmougin DG Higgins (1997) ArticleTitleThe CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools Nucleic Acids Res 25 4876–4882 Occurrence Handle10.1093/nar/25.24.4876 Occurrence Handle1:CAS:528:DyaK1cXntFyntQ%3D%3D Occurrence Handle9396791
Z Yang (1997) ArticleTitlePAML: A program package for phylogenetic analysis by maximum likelihood CABIOS 13 555–556 Occurrence Handle1:CAS:528:DyaK2sXntlGnu7s%3D Occurrence Handle9367129
HS Yoon JD Hackett D Bhattacharya (2002a) ArticleTitleA single origin of the peridinin- and fucoxanthin-containing plastids in dinoflagellates through tertiary endosymbiosis Proc Natl Acad Sci USA 99 11724–11729 Occurrence Handle10.1073/pnas.172234799 Occurrence Handle1:CAS:528:DC%2BD38XntFWqtrw%3D
HS Yoon JD Hackett G Pinto D Bhattacharya (2002b) ArticleTitleThe single, ancient origin of chromist plastids Proc Natl Acad Sci USA 99 15507–15512 Occurrence Handle10.1073/pnas.242379899 Occurrence Handle1:CAS:528:DC%2BD3sXjvVOg
Acknowledgments
This study was supported by Grant-in-Aid for Scientific Research on Priority Areas (c) “Genome Biology” from the Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 1320611 to T.K.), and by the Program for the Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN; to T.K., T.H., and H.N.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nozaki, H., Matsuzaki, M., Misumi, O. et al. Cyanobacterial Genes Transmitted to the Nucleus Before Divergence of Red Algae in the Chromista. J Mol Evol 59, 103–113 (2004). https://doi.org/10.1007/s00239-003-2611-1
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2611-1