
Abstract
The evolutionary history of nitrogen fixation has been vigorously debated for almost two decades. Previous phylogenetic analyses of nitrogen fixation genes (nif) have shown support for either evolution by vertical descent or lateral transfer, depending on the specific nif gene examined and the method of analyses used. The debate centers on the placement and monophyly of the cyanobacteria, proteobacteria, and Gram-positive bacteria (actinobacteria and firmicutes). Some analyses place the cyanobacteria and actinobacteria within the proteobacteria, which suggests that the nif genes have been laterally transferred since this topology is incongruent with ribosomal phylogenies, the standard marker for comparison. Other nif analyses resolve and support the monophyly of the cyanobacteria, proteobacteria, and actinobacteria, supporting vertical descent. We have revisited these conflicting scenarios by analyzing nifD from an increased number of cyanobacteria, proteobacteria, and Gram-positive bacteria. Parsimony analyses of amino acid sequences and maximum likelihood analysis of nucleic acid sequences support the monophyly of the cyanobacteria and actinobacteria but not the proteobacteria, lending support for vertical descent. However, distance analysis of nucleic acid sequences placed the actinobacteria within the proteobacteria, supporting lateral transfer. We discuss evidence for both vertical descent and lateral transfer of nitrogen fixation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nitrogen fixation (or diazotrophy) is the process of converting atmospheric nitrogen (N2) to reduced forms such as ammonia (NH3) (Postgate 1982). A small amount of atmospheric dinitrogen is reduced by lightning; however, the majority is reduced by prokaryotes (Postgate 1982; Sprent and Sprent 1990). The enzyme complex nitrogenase is responsible for nitrogen fixation. The subunits of nitrogenase are encoded by the nifHDK operon. Nitrogenase is composed of two components, dinitrogenase reductase (iron protein) and dinitrogenase (molybdenum–iron protein). Dinitrogenase reductase is composed of two identical subunits that are encoded by nifH (Mevarech et al. 1980), while dinitrogenase is a tetramer composed of two subunits encoded by nifD (Lammers and Haselkorn 1984) and two subunits encoded by nifK (Mazur and Chui 1982). Dinitrogenase reductase contains an iron–sulfur (4F–4S) cofactor, which binds the subunits of nitrogenase, and is responsible for mediating the ATP-dependent transfer of electrons to the dinitrogenase tetramer. Dinitrogenase binds atmospheric dinitrogen (N2) and is responsible for the transfer of electrons to it (Postgate 1982).
Other nitrogen fixation operons, in addition to nifHDK, have been identified in the heterocystous cyanobacterium, Nostoc sp. Strain PCC 7120 (Mazur and Chui 1982; Rice et al. 1982; Lammers and Haselkorn 1984). These include nifBSU, fdxN (Mulligan et al. 1988; Mulligan and Haselkorn 1989), and nifENXW (Borthakur et al. 1990; Haselkorn and Buikema 1992). NifB, nifN, and nifE are involved in molybdenum–iron cofactor synthesis. FdxN encodes a bacterial-type ferrodoxin of unknown function, and the functions of nifS, nifU, nifX, and nifW remain unknown. Recent studies have suggested that some nif genes may have arisen via paralogous gene duplication (Fani et al. 2000). In addition to the nif gene family listed above, several alternative nitrogenases have been found and these include Mo-dependent nif2, Va-dependent vnf, and Fe-dependent anf operons (Bishop and Premakumar 1992).
The ability to fix nitrogen is widely distributed in distantly related members of the Eubacteria and the Archaea (Young 1992); however, it is not ubiquitous in occurrence. The evolutionary history of the nitrogen fixation has been debated for some time. The debate has focused on how to explain the random distribution of nitrogen fixation in distantly related lineages of prokaryotes. Several alternative hypotheses have been proposed to explain this haphazard distribution. The first hypothesis is that nitrogen fixation arose once in evolutionary history but has been transferred laterally (i.e., horizontally) to various lineages (Normand and Bousquet 1989). A second hypothesis predicts that it arose early in the evolutionary history of prokaryotes and was at one time ubiquitous among them, but has since been lost by many lineages and retained by a few distantly related ones (i.e., vertical descent, accompanied by multiple losses) (Young 1992; Normand et al. 1992; Hirsch et al. 1995). A third view is that it arose multiple times through convergent evolution (Postgate and Eady 1988).
To evaluate the evolution of nitrogen fixation, we and others (Normand and Bousquet 1989; Normand et al. 1992; Hirsch et al. 1995) have examined the phylogenies of nif genes. We utilized published ribosomal RNA phylogenies (Woese 1987; Olsen and Woese 1993; Olsen et al. 1994) as an independent framework for comparison because rRNA is universally present, highly conserved, believed to be representative of the organismal phylogeny, and not believed to be laterally transferred (Woese 1987; Olsen and Woese 1993; Olsen et al. 1994). Thus, rRNA phylogenies in all likelihood, represent true organismal phylogenies and, therefore, are the most appropriate comparison for exploring alternative hypotheses involving lateral gene transfer. If the nif and the rRNA phylogenies have generally congruent topologies, then vertical descent may be supported. If, on the other hand, the nif and rRNA phylogenies have incongruent topologies, then lateral transfer of nif may have occurred. Convergence, or multiple origins of nif genes, could be indicated by incongruence, although we would also expect significant divergence in nucleotide and amino acid sequences if this were the case. This is in sharp contrast to the high sequence similarity and the conserved organization of nif genes found in nitrogen fixing organisms. Another possibility to explain incongruence is the inadvertent analysis of paralogous gene copies that arose though duplication. Therefore, our study focuses on distinguishing horizontal transfer from vertical descent of nif genes within the Eubacteria. In this paper, we reexamine the evolutionary history of nitrogen fixation by analyzing nifD from an increased number of cyanobacteria, proteobacteria, and Gram-positive bacteria. We analyzed only molybdenum containing nifD genes, and excluded all alternative nitrogenase genes, i.e., anfD and vnfD. Our objective was to compare the topology of our nifD phylogeny to 16S rRNA phylogenies to assess the evolutionary history of nifD and nitrogen fixation within the Eubacteria.
Materials and Methods
Fifty-seven nifD nucleic acid and inferred amino acid sequences were used in this study (Table 1). Inferred amino acid sequences were initially aligned using Clustal W (Thompson et al. 1994) with gaps inserted for optimal alignment. The amino acid alignment was then adjusted manually using MacClade 4.0 (Maddison and Maddison 2000). The nucleotide alignment was made using Codon Align 1.0 (Barry Hall, University of Rochester), which constructs the alignment based on the amino acid alignment, with gaps inserted between codons and not within them. The amino acid alignment was then adjusted manually using MacClade 4.0 (Maddison and Maddison 2000).
The amino acid data matrix was analyzed using parsimony with PAUP* 4.0b10 (Swofford 2002). All trees were rooted with outgroup analysis using six archaean representatives (Table 1). Analyses were performed with the user-defined stepmatrix “PROTPARS,” which is equivalent to Felsenstein’s PROTPARS from PHYLIP (Felsenstein 1981). Parsimony analysis was conducted using the heuristic search option, with gaps treated as missing data, trees obtained using stepwise addition, and the tree-bisection-reconnection (TBR) branch swapping with random sequence addition for 100 replicates, steepest descent not in effect, maxtrees set at 50,000, branches collapsed if the maximum length was zero, multrees option in effect, and no topological constraints enforced. Three parsimony analyses were performed: (1) all characters included, (2) constant and uninformative characters excluded, and (3) constant and uninformative characters plus 51 characters that correspond to an insertion found in nifD genes of Clostridium, Methanosarcina, and Chlorobium tepidum. Bootstrap values were calculated for 500 replicates to evaluate branch support using the parsimony options listed above (Bremer 1994; Felsenstein 1985; Huelsenbeck et al. 1995).
Analysis of the nucleic acid data matrix was performed using maximum likelihood (ML) criteria with PAUP* 4.0b10 (Swofford 2002). We determined the evolutionary model that best described our data using Modeltest 3.06 (Posada and Crandall 1998), which was the F81 model (Felsenstein 1981) with a likelihood score of 22106.18947. Trees were rooted with outgroup analysis using nifD sequences from six archaean representatives (Table 1). ML analysis was conducted using the heuristic search option, with the likelihood settings corresponding to the F81 model (Felsenstein 1981). ML settings were as follows: no molecular clock enforced, starting branch lengths determined via the Rogers–Swofford approximation method, trees with likelihoods that were 5% or further from the target score were rejected, branch-length optimization, equaled one-dimensional Newton Raphson with pass where the limit = 20 and δ = 1e−06, starting trees obtained via stepwise addition, sequence addition as-is, one tree held at each step during stepwise addition, TBR, steepest descent in effect, maxtrees set at 50,000, branches collapsed if the branch lengths equal to or less than 1e−08, multrees option in effect, and topological constraints not enforced. ML analysis was preformed with the third codon excluded, to remove noise resulting from saturation at that position, as well as with and without the ~154-bp insert excluded. Bootstrap values were calculated for 500 replicates to evaluate branch support using parsimony criteria (Bremer 1994; Felsenstein 1985; Huelsenbeck et al. 1995).
Distance analysis of the nucleic acid data matrix was performed using the neighbor-joining method (Saitou and Nei 1987), with all characters included and the DNA distance measure set to LogDet/paralinear to correct for base compositional bias (Lake 1994; Lockhart et al. 1994).
Results
NifD Amino Acid Sequences
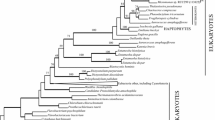
The aligned nifD amino acid data matrix was 599 residues in length and consisted of 349 variable characters. Parsimony analysis with all characters included generated 38 equally most parsimonious trees that were 4389 steps long, with a consistency index (CI) of 0.54, a rescaled consistency index (RC) of 0.39, and a retention index (RI) of 0.71. The 38 most parsimonious trees (MPTs) represented or six islands of similar topology (Fig. 1). The major topological difference between the islands is the placement of Acidithiobacillus ferrooxidans and a clade containing Alcaligenes faecalis, Azotobacter vinelandii, Azoarcus sp. BH72, and Klebsiella pneumoniae. The first island is composed of four trees (Fig. 1A) with a branch containing A. faecalis, A. vinelandii, Azoarcus sp. BH72, K. pneumoniae sister to the cyanobacteria, and Acidithiobacillus ferrooxidans sister to the cyanobacteria and proteobacteria. The second island is composed of 19 trees (Fig. 1B), with the cyanobacteria and proteobacteria as sister, with A. ferrooxidans as sister to the larger clade. Islands 3 through 6, composed of 12, 1, 1, and 1 trees, respectively, place the cyanobacteria and proteobacteria as sister, with A. ferrooxidans embedded within the proteobacteria, and A. faecalis, A. vinelandii, Azoarcus sp. BH72, and K. pneumoniae as sister (Fig. 1C). The islands vary slightly at the terminal branches, but the overall topology of all MPTs is similar. A strict consensus tree of the 38 MPTs from the six islands is presented in Fig. 1D. This tree consists of seven major branches. The cyanobacteria and proteobacteria occur in a single clade, with the cyanobacteria supported as monophyletic and the proteobacteria occurring on several unresolved clades. Neither the α, β, nor γ proteobacterial subgroups are supported as monophyletic.

Trees generated from parsimony analysis of nifD amino acid sequences. The taxnomic groups are indicated; in addition, the proteobacterial subgroups are inbrackets. Branches in boldface are those taxa that occur on different islands (discussed in the text). A Strict consensus tree of the four equally parsimonious trees in island 1. B Strict consensus tree of the 19 equally parsimonious trees in island 2. C Strict consensus tree generated from the 15 equally parsimonious trees generated from islands 3 through 6. D Strict consensus tree of all 38 equally parsimonious trees. Numbers above branches indicate bootstrap values.
In all MPTs of amino acid sequences (Fig. 1), the Gram-positive bacteria (firmicutes and actinobacteria) are not supported as monophyletic. The high G+C Gram-positive bacteria and Frankia strains (actinobacteria) are monophyletic and sister to the proteobacteria and cyanobacteria. The low G+C Gram-positive bacteria, Clostridia species (firmicutes), form a clade sister to the green sulfur bacterium, Chlorobium tepidum. All additional parsimony analyses of the amino acid data matrix (i.e., with and without constant and uninformative characters and the 51 amino acid residue insertion) resulted in trees that are virtually identical in topology to those trees presented in Fig. 1.
NifD Nucleic Acid Analysis
The aligned nifD nucleic acid data matrix was 1794 bp in length, with the 598 third codon positions excluded from the analysis. ML analysis converged on a single tree with a −lnL score of 20239.65577 (Fig. 2) and resolved four major clades. The cyanobacteria are supported as monophyletic and are sister to the proteobacteria. However, ML analysis failed to support the monophyly of the α, β, or γ subgroups of the proteobacteria. Sister to the cyanobacteria and proteobacteria is Acidithiobacillus ferrooxidans. The Gram-positive bacteria (actinobacteria and firmicutes) are not resolved as monophyletic, while the actinobacteria are monophyletic and sister to the cyanobacteria and proteobacteria. Clostridium species occur on a branch with Chlorobium tepidum and Methanosarcina.

Maximum likelihood tree of nifD nucleic acid sequences that had an −InL score of 20239.65577. The different groups of bacterial taxa are bracketed. The proteobacterial subgroups are in brackets. Numbers in boldface above the branches are bootstrap values and numbers below the branches indicate branch lengths.
The neighbor-joining tree (Fig. 3), constructed using LogDet/paralinear distances, resulted in a tree with a topology similar to that of the ML analysis of DNA sequences (Fig. 2) and parsimony analysis of amino acid sequences (Fig. 1); however, Frankia is embedded within the proteobacteria. Distance analysis also differed from the other analyses (Figs. 1 and 2) in that Chlorobium tepidum, Clostridium species, and Methanosarcina species are not placed together.

Distance tree of nifD nucleic acid sequences created with the neighbor-joining method and LogDet/paralinear DNA distances. The different groups of bacterial taxa are bracketed.The proteobacterial subgroups are in brackets. Numbers above the branches indicate branch lengths.
Discussion
The relationships among the cyanobacteria, proteobacteria, and Gram-positive bacteria (actinobacteria and firmicutes) have not been fully resolved from analyses of ribosomal RNAs (Hirsch et al. 1995). Phylogenies based on 16S rRNA differ in the relationships between these eubacterial lineages, with Woese (1987) placing the cyanobacteria and Gram-positive bacteria as sister to each other with the proteobacteria at the base of the tree (Fig. 4A), whereas Olsen et al. (1994) places the Gram-positive bacteria and the proteobacteria as sister to each other with the cyanobacteria at the base of the tree (Fig. 4B). Although these topologies differ, they are consistent in the resolution of these eubacterial lineages as monophyletic and distinct. Therefore, it has been suggested that the relationship among these eubacterial lineages may best be described as an unresolved trichotomy (Hirsch et al. 1995). However, these phylogenies are still useful for comparison because they resolve the three lineages as monophyletic. Thus, incongruence between the nif phylogenies and the 16S rRNA phylogenies may indicate lateral gene transfer. For example, if lateral transfer of nif had occurred, we would not expect to resolve these major lineages as monophyletic. Rather, we would expect that members of one lineage would be placed among members of another lineage (Fig. 4C), which has been reported by others (Normand and Bousquet 1989; Normand et al. 1992; Hirsch et al. 1995). NifH, -D, and -K have all been examined to some extent and have produced conflicting results supporting both lateral transfer and vertical descent.

Published trees redrawn. A 16S rRNA tree modified from Woese (1987). B 16S rRNA tree modified from Olsen et al. (1993). C Phylogeny depicting lateral transfer from the proteobacteria to the cyanobacteria and actinobacteria. Adapted from Hirsch et al. (1995).
Analyses of partial nifH sequences that support lateral transfer have topologies that were incongruent with a 16S rRNA-based phylogeny and placed the cyanobacteria and actinobacteria within the proteobacteria (Normand and Bousquet 1989; Normand et al. 1992; Hirsch et al. 1995; Ueda et al. 1995; Kessler et al. 1997). This suggests that nitrogen fixation was laterally transferred from an ancestral proteobacterium to the most recent common ancestors of cyanobacteria and actinobacteria (Normand and Bousquet 1989; Normand et al. 1992; Hirsch et al. 1995). However, other nifH analyses resolved the cyanobacteria and proteobacteria as sister to each other with the actinobacteria at the base of the tree (Hennecke et al. 1985; Hirsch et al. 1995; Zehr et al. 1998, 2000), consistent with the topology of the 16S rRNA phylogeny supporting vertical descent of nifH.
Previous nifD phylogenies also supported lateral transfer because the cyanobacteria were placed within the proteobacteria (Normand and Bousquet 1989; Hirsch et al. 1995; Kessler et al. 1997). The nifD phylogeny differs from nifH in that the actinobacteria and cyanobacteria do not occur together, and actinobacteria are placed as sister to the proteobacteria and cyanobacteria clade (Normand et al. 1992; Kessler et al. 1997). The failure of nifD to resolve the evolution of nitrogen fixation led some investigators to speculate that it simply does not provide sufficient resolution (Normand et al. 1992; Hirsch et al. 1995).
NifK amino acid sequences have also been analyzed with parsimony and distance methods to resolve the issue (Hirsch et al. 1995). Parsimony analysis resulted in a phylogeny that predicted vertical descent, with the actinobacteria and cyanobacteria as sister to the proteobacteria (Hirsch et al. 1995). In contrast, distance analysis resulted in a phylogeny that supported lateral transfer (Fig. 4C), with the actinobacteria and cyanobacteria sister to each other and embedded within the proteobacteria (Hirsch et al. 1995). Thus, conflicting results may be a combination of three different nif genes being analyzed with different methods of phylogenetic analysis, none of which are model-based methods such as ML.
Our analyses of nifD (Figs. 1,2,3), consistently produced topologies that are congruent with 16S rRNA phylogenies, and with the cyanobacteria, proteobacteria, and actinobacteria fully resolved via reciprocal monophyly and, thus support vertical descent. Our analysis did not result in members of one lineage being place within or among members of another lineage, consistent with 16S rRNA phylogenies supporting vertical descent.
Parsimony analysis of amino acid sequences (Fig. 1) and ML analyses of DNA sequences (Fig. 2), place the firmicutes (Clostridium), Methanosarcina, and Chlorobium tepidum together at the base of the tree; however, distance analysis of DNA sequences (Fig. 3) places them in two clades at the base of the tree. The placement of these taxa together is unexpected; since the firmicutes and Chlorobium tepidum are Eubacteria, and Methanosarcina is a member of the Archaea. Although the placement of these taxa together may be unexpected, analyses of nifH also place Clostridium with Methanosarcina (Zehr et al. 1998, 2000). While this might be attributed to long-branch attraction, Clostridium, Methanosarcina, and Chlorobium tepidum all possess an insertion encoding approximately 50 amino acids not found in any of the other taxa examined. However, this 50-amino acid insert in not conserved in these taxa. Distance analysis using the LogDet/paralinear distances, which compensates for base compositional bias (Lake 1994; Lockhart et al. 1994), did not place these taxa together (Fig. 3). This may suggest that the nifD genes of these taxa share a common evolutionary history due to lateral gene transfer. It also possible that the placement of these two taxa together is due to their nifD sequences being ancient paralogous copies that arose prior to the separation of the Eubacteria and the Archaea (Young 1990, 1992, 2000; Leigh 2001).
Another discrepancy in our results is the placement of Frakia in the parsimony analysis of amino acid sequences (Fig. 1) and ML analysis of DNA sequences (Fig. 2), which place them as sister to the larger clade of cyanobacteria and proteobacteria, in contrast to our distance analysis of DNA sequences (Fig. 3) which places it within the proteobacteria. The placement of Frankia within the proteobacteria (Fig. 3) is consistent with lateral transfer, but its placement as sister to the cyanobacteria and proteobacteria (Fig. 2) is consistent with vertical descent. Although there is conflict regarding the placement of Frankia in the distance tree (Fig. 3), it is more likely that its placement as sister to the cyanobacteria and proteobacteria is correct, supporting vertical descent (Figs. 1 and 2), since this is congruent with 16S rRNA (Woese 1987; Olsen and Woese 1993; Olsen et al. 1994), and the increased probability that parsimony and likelihood methods are superior phylogenetic analyses for more accurately resolving evolutionary relationships.
Our results are consistent with Zehr et al. (1998, 2000) in the placement of the proteobacteria and cyanobacteria as sister, with the actinobacteria at the base. A major difference between Zehr’s parsimony and distance analyses of nifH (Zehr et al. 1998, 2000) and the nifD phylogenies presented here is the placement of the proteobacterial subgroups. (Zehr et al. 1998, 2000) analyses support the monophyly of the proteobacteria as a whole, as well as the monophyly of the α and β subgroups (Zehr et al. 1998, 2000). Our parsimony analysis of amino acid sequences does not support the monophyly of the proteobacteria. However, with the exception of Acidithiobacillus ferrooxidans, the monophyly of the proteobacteria is supported by ML analysis of DNA sequences (Fig. 2). None of the analyses presented here resolve the α, β, and γ proteobacterial subgroups as monophyletic. Recent studies suggest that both nifH (Udea et al. 1995; Hurek et al. 1997, 1998; Laguerre et al. 2001) and nifD (Parker et al. 2002; Qian et al. 2002, 2003) may have been transferred horizontally within the proteobacteria, which would result in failure to differentiate these subgroups. Additionally, it has been suggested that some of the proteobacterial subgroups may not be monophyletic (Garrity and Holt 2001). Although our analyses of nifD vary slightly from those of nifH (Zehr et al. 1998, 2000), the two are similar in overall topology and support vertical descent. Furthermore, the agreement of nifH and nifD can be taken as evidence that nif genes and nitrogen fixation have evolved by vertical descent.
The analyses of nifD presented here are consistent with nifH (Zehr et al. 1998, 2000) and resolve the Gram-positive bacteria (firmicutes and actinobacteria), proteobacteria, and cyanobacteria, which is congruent with 16S rRNA phylogenies (Woese 1987; Olsen and Woese 1993; Olsen et al. 1994), thus supporting the vertical decent of nifD within these lineages. The discrepancies in earlier nif phylogenies may have been due to the number of species that were examined. Previous studies examined a limited number of taxa whereas we examined 57 taxa. These discrepancies could also be due to the methods of analysis used. Previous studies employed only parsimony and/or distance methods. We utilized parsimony, distance, and the model-based maximum likelihood method. It has been suggested that divergent sequences should be analyzed with model-based criteria to more accurately predict phylogeny since noise due to saturation is higher (Swofford et al., 1996; Huelsenbeck and Rannala 1997; Buckely and Cunningham 2002).
References
PE Bishop R Premakumar (1992) Alternative nitrogen fixation systems. G Stacey RH Burris HJ Evans (Eds) Biological Nitrogen Fixation Chapman & Hall New York 736–762
D Borthakur M Basche WJ Buikema P Borthakur R Haselkorn (1990) ArticleTitleExpression, nucleotide sequence, and mutational analysis of two open reading frames in the nif gene region of Anabaena sp. strain PCC 7120. MGG 221 227–234 Occurrence Handle1:CAS:528:DyaK3cXkvFaisbY%3D Occurrence Handle2115111
K Bremer (1994) ArticleTitleBranch support and tree stability. Cladistics 10 295–304 Occurrence Handle10.1006/clad.1994.1019
TK Duckley CW Cunningham (2002) ArticleTitleThe effects of nucleotide substitution model assumptions on estimates of nongrametric bot strap support. Mol Biol Evol 19 394–405 Occurrence Handle11919280
R Fani R Gallo P Lio (2000) ArticleTitleMolecular evolution of nitrogen fixation: The evolutionary history of the nifD, nifK, nifE, and nifN genes. J Mol Evol 51 1–11 Occurrence Handle1:CAS:528:DC%2BD3cXlslyls74%3D Occurrence Handle10903367
J Felsenstein (1981) ArticleTitleEvolutionary trees from DNA sequences: A maximum likelihood approach. J Mol Evol 17 368–376 Occurrence Handle1:CAS:528:DyaL3MXls1Cisr8%3D Occurrence Handle7288891
J Felsenstein (1985) ArticleTitleConfidence limits on phylogenies: An approach using the bootstrap. Evol 39 783–791
GM Garrity JG Holt (2001) The road map to the manual. DR Boone RW Castenholz (Eds) Bergey’s manual of systematic bacteriology, 2nd ed., Vol 1, Springer New York 119–166
K Haukka K Lindstrom JPW Young (1998) ArticleTitleThree phylogenetic groups of nodA and nifH genes in Sinorhizobium and Mesorhizobium isolates from leguminous trees growing in Africa and Latin America. Appl Environ Microbiol 64 419–426 Occurrence Handle1:CAS:528:DyaK1cXpsVSlsA%3D%3D Occurrence Handle9464375
R Haselkorn WJ Buikema (1992) Nitrogen fixation in cyanobacteria. G Stacey R Burris H Evans (Eds) Biological nitrogen fixation. Chapman & Hall New York 43–85
H Hennecke K Kaluzu B Thony M Fuhrmann W Ludwig E Stackebrandt (1985) ArticleTitleConcurrent evolution of nitrogenase genes and 16S RNA in Rhizobium species and other nitrogen fixing bacteria. Arch Microbiol 142 342–348 Occurrence Handle1:CAS:528:DyaL2MXlsleitL4%3D
AM Hirsch HI McKhann A Reddy J Liao Y Fang CR Marshall (1995) ArticleTitleAssessing horizontal transfer of nifHDK genes in Eubacteria: nucleotide sequence of nifK from Frankia strain HFPCcI3. Mol Biol Evol 12 16–27 Occurrence Handle1:CAS:528:DyaK2MXivVahtb0%3D Occurrence Handle7877490
JP Huelsenbeck DM Hillis R Jones (1995) Parametric bootstrapping in molecular phylogenetics: Applications and performance. JS Ferraris SR Palumbi (Eds) Molecular zoology: Strategies and protocols Wiley–Liss New York 19–46
JP Huelsenbeck B Rannala (1997) ArticleTitleMaximum likelihood estimation of phylogeny using stratigraphic data. Paleobiology 23 174–180
L Hurek L Egener B Reinhold Hurek (1997) ArticleTitleDivergence in nitrogenases of Azoarcus spp., Proteobacteria of the beta subclass. J Bacteriol 179 4172–4178 Occurrence Handle1:CAS:528:DyaK2sXkt1Gnu74%3D Occurrence Handle9209030
PS Kessler J McLarnan JA Leigh (1997) ArticleTitleNitrogenase phylogeny and the molybdenum dependence of nitrogen fixation in Methanococcus maripaludis. J Bacteriol 179 541–543 Occurrence Handle1:CAS:528:DyaK2sXltlahtA%3D%3D Occurrence Handle8990309
JA Lake (1994) ArticleTitleReconstructing evolutionary trees from DNA and protein sequences: Paralinear distances. Proc Natl Acad Sci USA 91 1455–1459 Occurrence Handle1:CAS:528:DyaK2cXitFWqt7Y%3D Occurrence Handle8108430
G Laguerre SM Nour V Macheret J Sanjuan P Drouin N Amarger (2001) ArticleTitleClassification of rhizobia based on nodC and nifH gene analysis reveals a close phylogenetic relationship among Phaseolus vulgaris. Microbiology 147 981–993 Occurrence Handle1:CAS:528:DC%2BD3MXjtVeisb8%3D Occurrence Handle11283294
PJ Lammers R Haselkorn (1984) ArticleTitleSequence of the nifD geneogy coding for the alpha subunit of dinitrogenase from the cyanobacterium Anabaena. Cell 44 905–911
JA Leigh (2001) Nitrogen fixation in methanogens—The Archaeal perspective. EW Triplett (Eds) Prokaryotic nitrogen fixation: A model system for analysis of a biological process. Horizon Scientific Press Wymondham, UK 657–669
PJ Lockhart MA Steel MD Hendy D Penny (1994) ArticleTitleRecovering evolutionary trees under a more realistic model of sequence evolution. Mol Biol Evol 11 605–612 Occurrence Handle1:CAS:528:DyaK2cXlsFaks7c%3D
DR Maddison WP Maddison (2000) MacClade version 4.0. Sinauer Associates Sunderland, MA
BJ Mazur CF Chui (1982) ArticleTitleSequence of the gene coding for the beta-subunit of dinitrogenase from the blue-green alga Anabaena. Proc Natl Acad Sci USA 79 6782–6786 Occurrence Handle1:CAS:528:DyaL3sXivVOlsQ%3D%3D
M Mevarech D Rice R Haselkorn (1980) ArticleTitleNucleotide sequence of a cyanobacterial nifH gene coding for nitrogenase reductase. Proc Natl Acad Sci USA 77 6476–6480 Occurrence Handle1:CAS:528:DyaL3MXnvVKntQ%3D%3D
ME Mulligan R Haselkorn (1989) ArticleTitleNitrogen-fixation (nif) genes of the cyanobacterium Anabaena sp. strain PCC 7120. The nifB-fdxN-nifS-nifU operon. J Biol Chem 264 19200–19207 Occurrence Handle1:CAS:528:DyaK3cXhsleitrk%3D Occurrence Handle2553733
ME Mulligan WJ Buikema R Haselkorn (1988) ArticleTitleBacterial-type ferredoxin genes in the nitrogen fixation regions of the cyanobacterium Anabaena sp. strain PCC 7120 and Rhizobium meliloti. J Bacteriol 170 4406–4410 Occurrence Handle1:CAS:528:DyaL1MXhsVyrtr8%3D Occurrence Handle2842320
P Normand J Bousquet (1989) ArticleTitlePhylogeny of mtrogenase sequences in Frankia and other nitrogen-fixing microorganisms. J Mol Evol 29 436–444 Occurrence Handle1:CAS:528:DyaK3cXhtFejt78%3D Occurrence Handle2515293
P Normand M Gouy B Cournoyer P Simonet (1992) ArticleTitleNucleotide sequence of nifD from Frankia alni strain ArI3: phylogenetic inferences. Mol Biol Evol 9 495–506 Occurrence Handle1:CAS:528:DyaK3sXktVWqurk%3D Occurrence Handle1584016
GJ Olsen CR Woese (1993) ArticleTitleRibosomal RNA: A key to phylogeny. FSAEB J 7 113–123 Occurrence Handle1:CAS:528:DyaK3sXhtF2jtro%3D
GJ Olsen CR Woese R Overbeek (1994) ArticleTitleThe winds of (evolutionary) change: Breathing new life into microbiology. J Bacteriol 176 1–6 Occurrence Handle1:STN:280:ByuC3M%2FitFY%3D Occurrence Handle8282683
MA Parker B Lafay JJ Burdon P van Berkum (2002) ArticleTitleConflicting phylogeographic patterns in rRNA and nifD indicate regionally restricted gene transfer in Bradyrhizobium. Microbiol 148 2557–2565 Occurrence Handle1:CAS:528:DC%2BD38XmslSqtr4%3D
D Posada KA Crandall (1998) ArticleTitleModeltest: Testing the model of DNA substitution. Bioinformatics 14 817–818 Occurrence Handle10.1093/bioinformatics/14.9.817 Occurrence Handle1:CAS:528:DyaK1MXktlCltw%3D%3D Occurrence Handle9918953
JR Postgate (1982) The fundamentals of nitrogen fixation. Cambridge University Press New York 1–4
JR Postgate RR Eady (1988) The evolution of biological nitrogen fixation. H Bothe F de Bruijn W Newton (Eds) Nitrogen fixation: Hundred years after. Proceeding of the 7th international congress on nitrogen fixation. Gustav Fischer Stuttgart 31–40
J Qian MA Parker (2002) ArticleTitleContrasting nifD and ribosomal gene relationships among Mesorhizobium from Lotus oroboides in northern Mexico. Syst Appl Mirobiol 25 68–73 Occurrence Handle1:CAS:528:DC%2BD38XmslOksLw%3D
J Qian SW Kwon MA Parker (2003) ArticleTitlerRNA and nifD phylogeny of Bradyrhizobium from sites across the Pacific Basin. FEMS Microbiol Lett 219 13157–13163 Occurrence Handle10.1016/S0378-1097(03)00043-0
D Rice BJ Mazur R Haselkorn (1982) ArticleTitleIsolation and physical mapping of nitrogen fixation genes from the cyanobacterium Anabaena PCC 7120. J Biol Chem 257 13157–13163 Occurrence Handle1:CAS:528:DyaL38XlvFequrc%3D Occurrence Handle6290496
N Saitou M Nei (1987) ArticleTitleThe neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406–425 Occurrence Handle1:STN:280:BieC1cbgtVY%3D Occurrence Handle3447015
J Sprent P Sprent (1990) Nitrogen fixing organisms: pure and applied aspects. Chapman and Hall New York 1–30
D Swofford (2002) PAUP* 4.0: Phylogenetic analysis using parsimony. Sinauer Associates Sunderland, MA
D Swofford GJ Olsen P Waddell DM Hillis (1996) Phylogenetic inference. D Hillis C Mortiz (Eds) Molecular systematics. Sinauer Associates Sunderland, MA 407–513
JD Thompson DG Higgins TJ Gibson (1994) ArticleTitleCLUSTAL W: Improving the sensitivity of progressive multiple alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22 4673–4680 Occurrence Handle7984417
T Ueda Y Suga N Yahiro T Matsuguchi (1995) ArticleTitleRemarkable N2-fixing bacterial diversity detected in rice roots by molecular evolutionary analysis of nifH gene sequences. J Bacteriol 177 1414–1417 Occurrence Handle1:CAS:528:DyaK2MXktVSnu7k%3D Occurrence Handle7868622
CR Woese (1987) ArticleTitleBacterial evolution. Microbiol Rev 51 221–271 Occurrence Handle2439888
JPW Young (1990) The phylogeny of nifH: Gene duplication or lateral transfer? PM Gresshoff LE Roth G Satcey WE Newton (Eds) Nitrogen fixation: Achievements and objectives. Chapman and Hall New York 840
JPW Young (1992) Phylogenetic classification of nitrogen-fixing organisms. G Stacey HJ Evans RH Bums (Eds) Biological nitrogen fixation. Chapman and Hall New York 43–85
JPW Young (1998) Molecular evolution in diazotrophs do the genes agree? FO Pedrosa M Hungria MG Yates (Eds) Nitrogen fixation From molecules to crop productivity. Kluwer Boston 161–164
JP Zehr MT Mellon S Zani (1998) ArticleTitleNew nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of nitrogenase nifH genes. Appl Environ Microbiol 64 3444–3450 Occurrence Handle1:CAS:528:DyaK1cXmtVajtLw%3D Occurrence Handle9726895
JP Zehr EJ Carpenter TA Villareal (2000) ArticleTitleNew perspectives on nitrogen-fixing microorganisms in tropical and subtropical oceans. Trends Microbiol 8 68–73 Occurrence Handle10.1016/S0966-842X(99)01670-4 Occurrence Handle1:STN:280:DC%2BD3c7jtVOktw%3D%3D Occurrence Handle10664599
Acknowledgements
This research was partially funded by the National Science Foundation–POWRE Grant 9973340 to S.R.B., Miami University Committee on Faculty Research Grant to S.R.B, Ohio Sea Grant Development to S.R.B. and L.E.W., and Ohio Board of Regents Academic Challenge Grant to B.J.H. We also acknowledge the help of Chris Wood and The Center for Functional Genomics and Bioinformatics at Miami University, Eric Stahlberg with the Ohio Super Computer, and Dr. Guillermo Orti for providing advice on the analyses. This paper represents, in part, the senior author’s master’s thesis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Henson, B.J., Watson, L.E. & Barnum, S.R. The Evolutionary History of Nitrogen Fixation, as Assessed by NifD . J Mol Evol 58, 390–399 (2004). https://doi.org/10.1007/s00239-003-2560-0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s00239-003-2560-0