
Abstract
Purpose
Morning glory disc anomaly (MGDA) is a rare congenital ophthalmologic disorder. Historically it has been diagnosed fundoscopically, with little in the literature regarding its imaging findings. The purpose of this study is to further characterize the orbital and associated intracranial magnetic resonance imaging (MRI) findings of MGDA in our tertiary pediatric center.
Methods
A retrospective review was performed of fundoscopically-diagnosed cases of MGDA, that had been referred for MRI. All MRI studies were scrutinized for orbital and other intracranial abnormalities known to occur in association with MGDA.
Results
18 of 19 cases of MGDA showed three characteristic MRI findings: funnel-shaped morphology of the posterior optic disc, abnormal soft tissue associated with the retrobulbar optic nerve, and effacement of adjacent subarachnoid spaces. The ipsilateral (intraorbital) optic nerve was larger in one patient and smaller in six. The ipsilateral optic chiasm was larger in two patients and smaller in one.
Conclusion
This study represents a comprehensive radiological-led investigation into MGDA. It describes the most frequently-encountered MRI findings in MGDA and emphasizes the importance of MRI in this cohort, i.e., in distinguishing MGDA from other posterior globe abnormalities, in assessing the visual pathway, and in screening for associated intracranial abnormalities – skull base/cerebral, vascular, and facial. It hypothesizes neurocristopathy as an underlying cause of MGDA and its associations. Caliber abnormalities of the ipsilateral optic nerve and chiasm are a frequent finding in MGDA. Optic pathway enlargement should not be labeled “glioma”. (239/250).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Morning glory disc anomaly (MGDA) is a rare congenital abnormality of the optic disc, the etiology of which is incompletely understood. It is usually sporadic, unilateral, and an isolated ocular abnormality [1, 2]. However, various pathologies, ranging from ocular and skull base/cerebral to vascular and facial, have been reported in association with this anomaly.
MGDA is typically an ophthalmologic diagnosis. It is characterized, fundoscopically, by a funnel-shaped excavation of the posterior globe, which incorporates the optic disc. Much of the excavated disc is filled with glial tissue. There is an annulus of chorioretinal pigmentary change. The surrounding retinal vessels have an unusual, straightened orientation and are narrowed. Its fundoscopic appearance resembles the morning glory flower [3] (Fig. 1).

11 month old girl with right MGDA diagnosed fundoscopically. a Normal fundoscopic appearance of the left eye. b Abnormal fundoscopic appearance of the right eye, with an enlarged, excavated optic disc, central glial tuft (arrowhead), halo of abnormal chorioretinal pigmentation (dashed circle) and radial orientation of retinal blood vessels (arrows). c Morning glory flower, after which this condition is named
It is not widely known that radiologists can identify MGDA on imaging. However, magnetic resonance imaging (MRI) of the brain and orbits can be useful in its diagnosis, especially where fundoscopic examination is difficult and/or in rare instances where there is ophthalmological uncertainty regarding the diagnosis. MRI not only aids in distinguishing MGDA from other posterior globe abnormalities, including coloboma and staphyloma but also facilitates a comprehensive assessment of the visual pathway. Crucially, it allows for screening of associated intracranial abnormalities.
Despite its clinical significance, the radiological literature regarding MGDA is sparse, consisting mostly of case reports and case series [1, 4,5,6].
Ellika et al. described three characteristic MRI findings in six cases of MGDA [4]. These include:
-
1.
Funnel-shaped morphology of the posterior optic disc with elevation of the adjacent retinal surface
-
2.
Abnormal soft tissue associated with the retrobulbar optic nerve and effacement of the adjacent subarachnoid spaces
-
3.
Posterior discontinuity of the uveoscleral coat.
The purpose of this study is to further characterize the MRI findings of MGDA and its associations—skull base/cerebral, vascular, and facial—in our center.
Materials and methods
Ethical approval was obtained from Western Australia Health. A retrospective review of all cases of MGDA from our institution—Perth Children’s Hospital, formerly Princess Margaret Hospital, Western Australia's specialist pediatric hospital and trauma center—was carried out. A radiological report keyword search was performed on our state-wide radiology information system using the terms “morning” and “glory” to identify ophthalmologically-diagnosed cases of MGDA, that had been referred for MRI between January 2004 and September 2023. The only inclusion criterion was age < 18 years.
Patient characteristics—age, gender, side of MGDA—were documented. All MRIs, frequently multiple studies per patient, were systematically reviewed by two of the authors (CNL, pediatric radiology fellow and RW, specialist pediatric neuroradiologist with 14 years of radiological experience) and any disagreements resolved by consensus. For all studies, we documented the magnetic field strength of the MRI machine (1.5 or 3 Tesla [T]), study quality (degree of motion degradation) and protocol used (high-resolution, T2-weighted, fat-saturated imaging of the orbits, contrast-enhanced T1-weighted imaging of the brain/orbits, angiographic sequence of the brain).
All MRIs were scrutinized for the following findings, which have previously been reported in cases of MGDA [4, 6, 7]: funnel-shaped morphology of the optic disc at the optic nerve head insertion, thickening/corrugation of the adjacent retinal surface, retinal T1 hyperintensity, retinal detachment, abnormal soft tissue associated with the ipsilateral retrobulbar optic nerve, effacement of perioptic nerve subarachnoid spaces, discontinuity in the posterior globe wall, fatty infiltration in the retrobulbar optic nerve sheath, contrast enhancement in the retrobulbar optic nerve, globe caliber, optic nerve caliber (ipsi- and contralateral), optic chiasm caliber, intracranial abnormalities, in particular midline (including skull) defects and vascular abnormalities, known to occur in association with MGDA. Where no dedicated angiographic sequence was available for review, the signal flow voids of the proximal intracranial arteries were assessed for vasculopathy using T2-weighted imaging.
Results
The radiological keyword search yielded 25 results. Four adult/elderly patients were excluded; here, “morning glory” referred to the morning glory sign of progressive nuclear palsy [8], as opposed to MGDA. Three children were also excluded—one underwent computed tomography (CT) only and in the other two, MRI was non-diagnostic due to motion.
In total, 18 children with the condition and 19 cases of MGDA (one child had bilateral MGDA) were included in the study. 13 of 18 children affected were girls. Mean age at the time of first MRI was 4 years and 10 months (2 days—12 years 10 months). In nine of 18 children, the right eye was affected, in eight the left and in one, both.
Imaging was performed at 1.5 T in eight patients and at 3 T in ten. High-resolution, T2-weighted, fat-saturated imaging of the orbits was performed in 17 patients, contrast-enhanced T1-weighted imaging of the brain/orbits in 6 and an angiographic sequence in 14, on at least one occasion.
The following MRI findings were seen in 18 of 19 cases (94.7%): funnel-shaped morphology of the posterior optic disc, abnormal soft tissue associated with the retrobulbar optic nerve and effacement of adjacent subarachnoid spaces. Other common findings were discontinuity in the posterior globe wall (evident in 16 of 19 cases) and thickening of the retinal surface (seen in 15 of 19 cases). Of the seven cases where contrast was given, there was discontinuous enhancement in the posterior globe wall in five patients and enhancement in the retrobulbar optic nerve in four (Fig. 2).

a Axial high-resolution, T2-weighted, fat-saturated MRI of the left orbit in a 31 month old boy with left MGDA diagnosed fundoscopically demonstrates funnel-shaped morphology of the optic disc at the optic nerve head insertion, with discontinuity in the posterior globe wall (bracket), thickening/corrugation of the adjacent retinal surface (arrows), abnormal soft tissue associated with the ipsilateral retrobulbar optic nerve (arrowhead) and effacement of perioptic nerve subarachnoid spaces. b Axial T1-weighted imaging of the right orbit in a 5 year 7 month old boy with right MGDA diagnosed fundoscopically demonstrates T1 high signal in the retrobulbar optic nerve sheath (arrowhead), consistent with fat. c Axial T1-weighted, fat-saturated, post-gadolinium imaging of the left orbit in a 4 year old girl with left MGDA diagnosed fundoscopically demonstrates discontinuous posterior globe wall enhancement and abnormal enhancement in the retrobulbar optic nerve (dashed line)
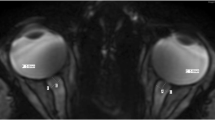
The affected globe was smaller in five of 19 cases. The ipsilateral (intraorbital) optic nerve was larger in one patient, smaller in six (including the child with bilateral MGDA) and normal in ten. The ipsilateral optic chiasm was larger in two patients and smaller in one (Fig. 3).

a Coronal high-resolution, T2-weighted, fat-saturated imaging of the orbits in an 11 month old girl with right MGDA diagnosed fundoscopically demonstrates a normal left and abnormal, thickened right intraorbital optic nerve (dashed circle). Note homogenous T2 signal within the enlarged right intraorbital optic nerve; there was no associated enhancement nor progression on 6 month follow-up MRI. b Coronal high-resolution, T2-weighted, fat-saturated imaging of the orbits in a 7 year 10 month old girl with left MGDA diagnosed fundoscopically demonstrates a normal right and abnormal, thinned left intraorbital optic nerve (dashed circle). c Coronal high-resolution, T2-weighted, fat-saturated imaging of the orbits in the same 11 month old girl as in Fig. 3a demonstrates abnormal, asymmetric enlargement of the right optic chiasm (arrowhead). Note normal T2 signal within the enlarged right optic chiasm. d Coronal high-resolution, T2-weighted, fat-saturated imaging of the orbits in a 9 year 11 month old girl with left MGDA diagnosed fundoscopically demonstrates mild, asymmetric thinning of the left optic chiasm (arrowhead)
Three children had intracranial abnormalities—one had Chiari 1 deformity and one had Chiari 2 malformation (Fig. 4).

a Midline sagittal T1-weighted imaging of the brain in a 4 year old girl with left MGDA diagnosed fundoscopically demonstrates caudal descent of the cerebellar tonsils (arrow), > 5 mm below the foramen magnum; the tonsils have an abnormal “peg-like” morphology. Findings are consistent with Chiari 1 deformity. b Midline sagittal T1-weighted imaging of the brain in a 3 year 5 month old boy with bilateral MGDA diagnosed fundoscopically demonstrates caudal descent of the cerebellar tonsils, which lie at the level of C3 (arrow), a small posterior fossa (bracket), abnormally low attachment of the tentorium cerebelli and low-lying torcula (dashed arrow), towering cerebellum (thick arrow), slit-like fourth ventricle (arrowhead), tectal beaking (open arrowhead), a large massa intermedia (dashed circle) and thin corpus callosum (dashed line). The child also had a lower lumbar myelomeningocele (not shown). Findings are consistent with Chiari 2 malformation
There was one complex congenital case, whereby unilateral MGDA was seen, in conjunction with ipsilateral dysplastic hemimegalencephaly, an enhancing internal auditory meatus (IAM) mass, multiple intradural, extramedullary lipomatous spinal lesions, a low-lying conus medullaris, pathological nerve root enlargement and enhancement and several non-neuroaxis anomalies, including thoracic vasculopathy, aortic coarctation and bilateral renal dysplasia (Fig. 5).

2 day old girl with congenital verrucous epidermal naevus syndrome, seizures and cranial ultrasound suggesting right hemimegalencephaly and right temporal lobe polymicrogyria. a Axial T2-weighted imaging of the brain demonstrates asymmetric enlargement of the right cerebral hemisphere and right lateral ventricle and suspected polymicrogyria involving the right posterior perisylvian cortex (arrowhead). b Axial T1-weighted, fat-saturated, post-contrast imaging of the brain demonstrates an enhancing mass in the right IAM (arrowhead). c Midline sagittal T1-weighted imaging of the brain demonstrates a lipomatous lesion at the dorsal cervicomedullary junction (arrow). d Sagittal T1-weighted imaging of the spine demonstrates further intradural, extramedullary lipomatous lesions in the dorsal spinal canal from T1-T5 and from T7-T9 (arrows). e Sagittal T2-weighted imaging of the spine demonstrates a low-lying conus, at the inferior endplate of L4 (arrow). f Axial T1-weighted, fat-saturated, post-contrast imaging of the cervical spine demonstrates enlargement of and abnormal enhancement within dorsal root ganglia/nerve roots (arrowheads). g Axial T1-weighted, fat-saturated, post-contrast imaging of the spine demonstrates enlargement of and abnormal enhancement within right cauda equina nerve roots (arrowhead). Note also low signal/non-enhancing, cystic foci in the right kidney (arrows), consistent with renal dysplasia. Findings raised suspicion for a systemic overgrowth syndrome, i.e. CLOVES
One child had stenosis of the right terminal internal carotid artery (ICA). There was no other definite vascular abnormality (Table 1).
Discussion
This study represents a comprehensive radiological-led investigation into patients with MGDA, illustrating the most frequently-encountered MRI findings.
The etiology of MGDA is disputed. Traboulsi et al. hypothesized that a mesenchymal defect leads to faulty closure of the posterior sclera and lamina cribrosa, permitting posterior herniation of retinal and neural tissue [9]. However, a concomitant defect in the posterior embryonic fissure is possible [9].
MGDA is typically an isolated ocular abnormality that is diagnosed fundoscopically. However, MRI can be helpful in confirming the diagnosis, especially when fundoscopic exam is difficult and/or non-diagnostic. For example, persistent fetal vasculature, which may be associated with MGDA, can make fundoscopic diagnosis challenging [1, 6, 10].
MRI is also helpful in differentiating MGDA from other optic disc abnormalities, i.e., coloboma and staphyloma. Coloboma, unlike MGDA, is typically genetic, and associated with multisystem abnormalities, e.g., CHARGE syndrome (coloboma, heart disease, choanal atresia, retardation, genital hypoplasia and ear anomalies), while MGDA is almost never familial [11]. Thus, the distinction between MGDA and coloboma has important prognostic/genetic counselling implications for the child and their family. In MGDA, the optic disc lies symmetrically and centrally within the funnel-shaped excavation of the posterior globe. By contrast, optic nerve coloboma, which occurs due to incomplete closure of the embryonic optic fissure, manifests at the inferonasal aspect of the optic disc or below it [12]. The funnel-shaped excavation of the optic disc seen with coloboma is typically vertically oblong [11]. Crucially, coloboma lacks the abnormal retrobulbar soft tissue that characterizes MGDA [4]. MGDA and optic nerve colobomas are congenital anomalies, whereas staphyloma refers to acquired globe ectasia [7] and on MRI is characterized by focal bulging and thinning of the globe wall, which most commonly occurs posteriorly [13]. The retrobulbar optic nerve is normal (Fig. 6).

a Note typical MRI findings of MGDA, as described in Fig. 2a above. b Coloboma. Note a defect in the posteroinferior left globe, with a normal left retrobulbar optic nerve. c Staphyloma. Note broad-based thinning of the posterior globe, remote from the optic nerve head insertion
MRI also identifies ocular abnormalities that occur in association with MGDA, for example, retinal detachment [1, 6, 14,15,16], which we encountered in four of our cases and decreased globe caliber [6, 17], which we observed in five of 19 cases.
Several case reports have described enlarged optic nerves/chiasms in the setting of MGDA as optic pathway “gliomas” (OPGs) [18, 19]. However, more recent research has shown that optic pathway caliber abnormalities – both thickening and thinning, affecting anywhere from orbit to chiasm – are common in patients with MGDA [1, 5, 6]. Optic pathway caliber abnormalities were frequent in our cohort, with either thickening or thinning, affecting nerve and/or chiasm in nine of our 18 patients.
Optic pathway enlargement (OPE) in MGDA does not display the characteristic features of OPGs. In our cohort, all cases of OPE demonstrated homogeneously isointense T2 signal, without contrast enhancement (where given) or progression on follow-up imaging. These findings align with the largest MGDA radiological study by Poillon et al. (n = 40), wherein all cases of optic nerve and/or chiasm enlargement displayed homogeneously isointense T1 and homogeneously iso-/hyperintense T2 signal, without abnormal enhancement, restricted diffusion or progression [6]. Sporadic gliomas of the optic nerves are rare and more commonly occur in the setting of neurofibromatosis type 1. OPGs, unlike OPE in MGDA, tend to be heterogenous in signal, with enhancement and progression on follow-up studies [20].
In addition to optic nerve/chiasm enlargement, small caliber optic nerves were common in our cohort and have been previously described by other groups. Nguyen et al., in a series of nine patients with MGDA reported optic nerve/chiasm thickness abnormalities in all cases, with a thinner optic nerve in one patient and irregular optic pathway thickness in all other cases [1]. Ceynowa et al. also described optic nerve thinning in the context of MGDA [21].
Notably, all cases of optic pathway caliber abnormality, both in our cohort and in prior studies [1, 5, 6], occurred ipsilateral to the MGDA. Thus, research to date suggests that optic pathway caliber abnormalities, both thickening and thinning, in the setting of MGDA likely reflect malformative/developmental or dysplastic abnormalities. However, as surgical resection is considered a “last resort” in the treatment of optic nerve gliomas [22], there is a lack of histopathological data at present to support the theory that OPE in MGDA reflects a developmental or dysplastic, as opposed to neoplastic, process. As radiologists, we must be aware of this association, as it may justify an increased interval between follow-up MRIs, particularly in younger children, requiring general anaesthetic for same. It may also obviate the need for potentially-unnecessary and harmful interventions, such as chemo- and/or immunotherapy.
An important role for MRI in MGDA is in screening for various intracranial abnormalities that have been described in association with this condition. These wide-ranging abnormalities, often midline, include basal cephalocoeles, typically trans-sphenoidal [23,24,25], pituitary gland and/or stalk abnormalities [2, 15, 26, 27], corpus callosal agenesis/dysgenesis [21] and Chiari 1 deformities [28, 29]. The latter was observed in our cohort. We also report here, to the best of our knowledge, the first case of Chiari 2 malformation in the setting of MGDA; on balance, the Chiari 2 malformation and associated lumbar myelomeningocele are felt to be unrelated/incidental to the MGDA. Additionally, we report one complex congenital case whereby MGDA was seen alongside multiple cerebral, spinal and non-neuroaxis abnormalities. This raised suspicion for a systemic overgrowth syndrome, i.e., CLOVES (congenital lipomatous overgrowth, vascular malformations, epidermal nevi, skeletal and/or spinal anomalies), suggesting that MGDA can rarely occur as part of a syndrome. Other craniofacial (typically midline) abnormalities, i.e., cleft lip and palate [16] and facial hemangiomas/PHACES syndrome (posterior fossa abnormalities, hemangiomas, arterial anomalies, cardiac defects, eye abnormalities, sternal cleft and supraumbilical raphe) [17, 21, 30], which are associated with this condition may also be identified on MRI brain in this cohort.
Vascular abnormalities, most commonly Moya Moya disease (MMD) [27, 31] but also other internal carotid, anterior and middle cerebral artery vasculopathies, including stenoses [15, 32, 33] and agenesis [15, 17, 32] have been described in association with MGDA. Although these vascular abnormalities (apart from MMD) are typically static and likely represent congenital anomalies, reversible carotid artery narrowing has also been described in this context [34]. In our cohort, we encountered one case of terminal ICA stenosis, which did not progress during three years of follow-up.
Neurocristopathy, a disease of the neural crest cells, could explain the various associations of MGDA. Cephalic neural crest cells produce craniofacial mesenchyme, which forms the craniofacial skeleton (skull, adenohypophysis, eye tissues etc.) but also gives rise to smooth muscle cells in the cerebral arteries of the prosencephalon. Hence, cephalic neurocristopathy, a disease of the cephalic neural crest cells, could explain the co-existence of ocular anomalies, basal cephaloceles, pituitary gland/stalk abnormalities, corpus callosal agenesis/dysgenesis, craniofacial anomalies such as cleft lip and palate, PHACES syndrome and MMD [35].
There are several limitations to our study. Our cohort is small, owing to the rarity of this disease. Additionally, some patients with MGDA may not have been captured by our search strategy. Given the retrospective nature of our study and the long period over which data was acquired (almost 20 years), the quality of MRIs and the sequences performed varied between patients. More recently-performed studies are inherently of higher quality, having been acquired at 3 T field strength, with resultant increased signal-to-noise ratio, higher spatial resolution, potential for isotropic reconstruction and faster acquisition speeds, making images less likely to be motion-degraded [36]. Diagnostic accuracy may have been improved with higher quality studies. However, three findings—funnel-shaped morphology of the posterior optic disc, abnormal soft tissue associated with the retrobulbar optic nerve and effacement of adjacent subarachnoid spaces—were visible in 18 of 19 cases, regardless of study quality, thereby allowing the clinical diagnosis of MGDA to be confirmed.
Conclusions
While MGDA is typically diagnosed fundoscopically, MRI can aid in confirming the diagnosis and in detecting associated malformations—ocular, skull base/cerebral, vascular, and facial. MRI brain, with high-resolution, T2-weighted, fat-saturated imaging of the orbits, contrast-enhanced and angiographic sequences (at least on the index study), should be performed in cases of MGDA, both to assess the globe and to screen for associated abnormalities. Caliber abnormalities of the ipsilateral optic nerve and chiasm are a frequent finding in MGDA. Optic pathway enlargement should not be labeled “glioma”.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Abbreviations
- MGDA:
-
Morning glory disc anomaly
- MRI:
-
Magnetic resonance imaging
- T:
-
Tesla
- CT:
-
Computed tomography
- IAM:
-
Internal auditory meatus
- ICA:
-
Internal carotid artery
- CHARGE:
-
Coloboma, heart disease, choanal atresia, retardation, genital hypoplasia and ear anomalies
- OPG:
-
Optic pathway glioma
- OPE:
-
Optic pathway enlargement
- CLOVES :
-
Congenital lipomatous overgrowth, vascular malformations, epidermal nevi, skeletal and/or spinal anomalies
- PHACES :
-
Posterior fossa abnormalities, hemangiomas, arterial anomalies, cardiac defects, eye abnormalities, sternal cleft and supraumbilical raphe
- MMD:
-
Moya Moya disease
References
Nguyen D-T, Boddaert N, Bremond-Gignac D, Robert MP (2022) Optic nerve abnormalities in morning glory disc anomaly: an MRI study. J Neuroophthalmol 42:199–202. https://doi.org/10.1097/WNO.0000000000001412
de Pierre-Filho P, TP, Limeira-Soares PH, Marcondes AM, (2004) Morning glory syndrome associated with posterior pituitary ectopia and hypopituitarism. Acta Ophthalmol Scand 82:89–92. https://doi.org/10.1111/j.1395-3907.2004.00214.x
Kindler P (1970) Morning glory syndrome: unusual congenital optic disk anomaly. Am J Ophthalmol 69:376–384. https://doi.org/10.1016/0002-9394(70)92269-5
Ellika S, Robson CD, Heidary G, Paldino MJ (2013) Morning glory disc anomaly: characteristic MR imaging findings. AJNR Am J Neuroradiol 34:2010–2014. https://doi.org/10.3174/ajnr.A3542
Doneda C, Pinelli L, Scaramuzzi M et al (2017) Morning glory disc anomaly associated with ipsilateral optic nerve and chiasm thickening: three cases and review of the literature. Neuropediatrics 48:463–466. https://doi.org/10.1055/s-0037-1603642
Poillon G, Henry A, Bergès O et al (2021) Optic pathways enlargement on magnetic resonance imaging in patients with morning glory disc anomaly. Ophthalmology 128:172–174. https://doi.org/10.1016/j.ophtha.2020.06.060
Auber AE, O’Hara M (1999) Morning glory syndrome. MR imaging Clin Imaging 23:152–158. https://doi.org/10.1016/s0899-7071(99)00118-7
Massey LA, Micallef C, Paviour DC et al (2012) Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 27:1754–1762. https://doi.org/10.1002/mds.24968
Traboulsi EI, O’Neill JF (1988) The spectrum in the morphology of the so-called “morning glory disc anomaly.” J Pediatr Ophthalmol Strabismus 25:93–98. https://doi.org/10.3928/0191-3913-19880301-11
Koenig SB, Naidich TP, Lissner G (1982) The morning glory syndrome associated with sphenoidal encephalocele. Ophthalmology 89:1368–1373. https://doi.org/10.1016/s0161-6420(82)34623-0
Brodsky MC (1994) Morning glory disc anomaly or optic disc coloboma? Arch Ophthalmol 112:153. https://doi.org/10.1001/archopht.1994.01090140027002
Deb N, Das R, Roy IS (2003) Bilateral morning glory disc anomaly. Indian J Ophthalmol 51:182–183
Bhat R, Al-Samarraie M, Nada A et al (2021) Spotlight on the pediatric eye: a pictorial review of orbital anatomy and congenital orbital pathologies. Neuroradiol J 34:21–32. https://doi.org/10.1177/1971400920949232
Chang S, Gregory-Roberts E, Chen R (2012) Retinal detachment associated with optic disc colobomas and morning glory syndrome. Eye 26:494–500. https://doi.org/10.1038/eye.2011.354
Quah BL, Hamilton J, Blaser S et al (2005) Morning glory disc anomaly, midline cranial defects and abnormal carotid circulation: an association worth looking for. Pediatr Radiol 35:525–528. https://doi.org/10.1007/s00247-004-1345-y
Kumar J, Adenuga OO, Singh K et al (2021) Clinical characteristics of morning glory disc anomaly in South India. Taiwan J Ophthalmol 11:57–63. https://doi.org/10.4103/tjo.tjo_52_20
Puvanachandra N, Heran MK, Lyons CJ (2008) Morning glory disk anomaly with ipsilateral capillary hemangioma, agenesis of the internal carotid artery, and Horner syndrome: a variant of PHACES syndrome? J AAPOS 12:528–530. https://doi.org/10.1016/j.jaapos.2008.04.005
Kinori M, Smiley NP, Zeid JL (2018) Morning glory disc anomaly and ipsilateral sporadic optic pathway glioma. Am J Ophthalmol Case Rep 10:16–17. https://doi.org/10.1016/j.ajoc.2018.01.035
Bandopadhayay P, Dagi L, Robison N et al (2012) Morning glory disc anomaly in association with ipsilateral optic nerve glioma. Arch Ophthalmol 130:1082. https://doi.org/10.1001/archophthalmol.2012.412
Kornreich L, Blaser S, Schwarz M et al (2001) Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol 22:1963–1969
Ceynowa DJ, Wickström R, Olsson M et al (2015) Morning glory disc anomaly in childhood - a population-based study. Acta Ophthalmol 93:626–634. https://doi.org/10.1111/aos.12778
Farazdaghi MK, Katowitz WR, Avery RA (2019) Current treatment of optic nerve gliomas. Curr Opin Ophthalmol 30:356–363. https://doi.org/10.1097/ICU.0000000000000587
Koerner JC, Sweeney J, Rheeman C, Kenning TJ (2019) Delayed presentation of morning glory disc anomaly and transsphenoidal encephalocele: a management dilemma. Neuroophthalmology 43:95–101. https://doi.org/10.1080/01658107.2018.1479434
Chen CS, David D, Hanieh A (2004) Morning glory syndrome and basal encephalocele. Childs Nerv Syst 20:87–90. https://doi.org/10.1007/s00381-003-0869-z
Minotto I, Abdala N, Miachon AAS et al (2007) Basal encephalocele associated with morning glory syndrome: case report. Arq Neuropsiquiatr 65:988–991. https://doi.org/10.1590/s0004-282x2007000600013
Loddenkemper T, Friedman NR, Ruggieri PM et al (2008) Pituitary stalk duplication in association with moya moya disease and bilateral morning glory disc anomaly - broadening the clinical spectrum of midline defects. J Neurol 255:885–890. https://doi.org/10.1007/s00415-008-0799-5
Khodeiry MM, Chau VQ, Yasin A et al (2022) Morning glory disc anomaly associated with moyamoya disease and pituitary stalk duplication. Am J Ophthalmol Case Rep 27:101632. https://doi.org/10.1016/j.ajoc.2022.101632
Arlow T, Arepalli S, Flanders AE, Shields CL (2014) Morning glory disc anomaly with Chiari type I malformation. J Pediatr Ophthalmol Strabismus 51:e22-4. https://doi.org/10.3928/01913913-20140423-03
Razeghinejad MR, Masoumpour M (2006) Chiari type capital I, Ukrainian malformation associated with morning glory disc anomaly. J Neuroophthalmol 26:279–281. https://doi.org/10.1097/01.wno.0000249325.57604.34
Sathyan S, Chackochan M (2018) Morning glory disc anomaly and facial hemangiomas in a girl with moyamoya syndrome. Indian J Ophthalmol 66:1644–1646. https://doi.org/10.4103/ijo.IJO_538_18
Ponnatapura J (2018) Morning glory syndrome with Moyamoya disease: A rare association with role of imaging. Indian J Radiol Imaging 28:165–168. https://doi.org/10.4103/ijri.IJRI_219_17
Lenhart PD, Lambert SR, Newman NJ et al (2006) Intracranial vascular anomalies in patients with morning glory disk anomaly. Am J Ophthalmol 142:644–650. https://doi.org/10.1016/j.ajo.2006.05.040
Nezzar H, Mbekeani JN, Dalens H (2015) Morning glory syndrome with carotid and middle cerebral artery vasculopathy. Optom Vis Sci 92:e437–e441. https://doi.org/10.1097/OPX.0000000000000727
Murphy MA, Perlman EM, Rogg JM et al (2005) Reversible carotid artery narrowing in morning glory disc anomaly. J Neuroophthalmol 25:198–201. https://doi.org/10.1097/01.wno.0000177300.44845.a4
Ota T, Komiyama M (2021) Cephalic/cardiac neural crest cell and moyamoya disease. Neuroradiol J 34:529–533. https://doi.org/10.1177/19714009211021780
Graves MJ (2022) 3 T: the good, the bad and the ugly. Br J Radiol 95:20210708. https://doi.org/10.1259/bjr.20210708
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Declarations
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ní Leidhin, C., Erickson, J.P., Bynevelt, M. et al. (What’s the story) morning glory? MRI findings in morning glory disc anomaly. Neuroradiology 66, 1225–1233 (2024). https://doi.org/10.1007/s00234-024-03375-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-024-03375-2