
Abstract
Objective
To study the effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of glipizide.
Methods
Eighteen healthy male subjects were divided into three groups according to their genotypes: group I, CYP2C9*1/*1 and CYP2C19 extensive metabolizers (EMs); group II, CYP2C9*1/*1 and CYP2C19 poor metabolizers (PMs); and group III, CYP2C9*1/*3 and CYP2C19 EMs. After a single dose of a 5-mg glipizide tablet, plasma concentrations of glipizide for a 36-h period were determined. Meanwhile, plasma glucose levels and plasma insulin levels were determined from 0 to 4 h after dosing.
Results
The area under the plasma concentration-time curve (\( AU{C_{0 - \infty }} \)) was 2.0-fold higher and the oral clearance was 51.1% lower in group III than in group I. The change in fasting insulin level within 1 h (ΔAUECinsulin0–1h) in group III was 3.8-fold higher than that in group I. The glipizide parameters in group II exhibited similar tendencies to those in group III.
Conclusions
These results suggest that CYP2C9 polymorphism significantly influences the pharmacokinetics and pharmacodynamics of glipizide, which needs to be considered in clinical practice. CYP2C19 polymorphism exhibits a tendency to influence the effects of glipizide, to a certain extent similarly to CYP2C9 polymorphism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glipizide is a second-generation oral sulfonylurea hypoglycemic agent widely used in the treatment of type 2 diabetes mellitus. Glipizide is rapidly absorbed and extensively metabolized to two inactive metabolites through hydroxylation at the 3-cis position (15%) and 4-trans position (71%) of the cyclohexane ring. Any remaining glipizide and its metabolites are mainly excreted into the urine [1, 2].
CYP2C9 is believed to be responsible for the metabolism of glipizide based on a case report that a healthy Caucasian who carried homologous cytochrome P450 (CYP) 2C9*3 alleles had an obviously higher plasma glipizide concentration and lower blood glucose level than those who carried CYP2C9*1/*1 alleles [3]. Kim et al. recently reported that the area under the plasma concentration-time curve from time zero to infinity (\( AU{C_{0 - \infty }} \)) of glipizide was higher by 30% in CYP2C9*3 heterogeneous carriers compared with CYP2C9 wild-type carriers [4]. These findings imply that CYP2C9 polymorphism probably has clinical relevance with glipizide. However, more details are needed to clarify the results.
In our preliminary in vitro study, we found a novel major hydroxyl metabolite of glipizide that can be attributed to both CYP2C9 and CYP2C19. This finding indicated that CYP2C19 as well as CYP2C9 was involved in the metabolism of glipizide. To date, there have been no reports on the effect of CYP2C19 polymorphism on glipizide. In view of the relatively high prevalence of CYP2C19 PMs in the Asian population (12.6–23.1%) [5], it is valuable to investigate the effects of CYP2C19 polymorphisms on the metabolism and responses of glipizide.
Therefore, we simultaneously investigated the effects of CYP2C9 and CYP2C19 polymorphisms on glipizide in the study. The purpose of this study was to investigate the effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of glipizide in healthy Chinese subjects and evaluate the clinical impacts of the two types of genetic polymorphisms on glipizide.
Materials and methods
Subjects
Blood samples were obtained from 221 unrelated healthy male Chinese subjects. After genotyping, 18 subjects (age 21–27 years, weight 56.1–73.8 kg) with different genotypes of CYP2C9 and CYP2C19 were enrolled in the study. They were divided into three groups (n = 6 in each group) as follows: group I, CYP2C9*1/*1 and CYP2C19 extensive metabolizers (EM) (CYP2C19*1/*1) subjects; group II, CYP2C9*1/*1 and CYP2C19 poor metabolizers (PM) (CYP2C19*2/*2, CYP2C19*2/*3, or CYP2C19*3/*3) subjects; group III, CYP2C9*1/*3 and CYP2C19 EM (CYP2C19*1/*1 ) subjects. The study sample size (six persons in each group) would be able to provide 80% power for detecting a significant difference of 30%. The mean ages (± SD) of subjects in groups I, II, and III were 23.5 ± 2.3, 21.7 ± 1.2, and 22.2 ± 1.6 years, respectively. The mean body weights (± SD) were 65.7 ± 5.0, 63.8 ± 2.3, and 63.3 ± 5.6 kg, respectively. There were no significant differences among the three groups with respect to age or body weight.
All subjects were male nonsmokers and in good health, as judged by physical examination and routine clinical laboratory tests, including blood glucose levels and insulin levels. No subject had any family history of diabetes mellitus. Two weeks before and throughout the study, subjects were not allowed to take any drug, alcohol, foods containing caffeine, or grapefruits and juice. The study protocol was approved by the Clinical Research Ethics Committee of the Second Affiliated Hospital, Liaoning University of Traditional Chinese Medicine, Shenyang, China. Written informed consent was obtained from each subject before participation. The trial was registered in the U.S. National Institute of Health register (www.clinicaltrials.gov) as trial NCT00806013.
Study design
After an overnight fast, the 18 subjects received a single oral dose of a 5-mg glipizide tablet (Disha Pharmaceutical Group, Shandong, China) with 200 mL water followed by 75 g dextrose 1 h after dosing. Venous blood samples were collected immediately before and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 8, and 12 h after dosing. Blood samples, collected in EDTA tubes, were centrifuged (2,500 g) immediately for 10 min, and separated plasma samples were stored at −80°C until assay. To evaluate the pharmacodynamic effect, blood glucose levels were determined directly by use of a glucose meter (Accu-Chek, Roche, Germany) at 0, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, and 4 h after dosing. Blood insulin concentrations were also determined by use of an enzyme immunoassay method (AIA-Pack IRI; Tosoh, Tokyo, Japan) at an identical time course with that of blood glucose.
For safety, each subject who had hypoglycemic symptoms or a blood glucose concentration lower than 3 mmol/L (54 mg/dL) was immediately administered a 15-g oral portion of dextrose. No adverse effects occurred during the study.
Genotyping of CYP2C9 and CYP2C19
CYP2C9 and CYP2C19 genotypes were determined by the polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) method using genomic DNA isolated from venous blood with an extraction kit (TIANamp Blood DNA Kit, Tiangen BioTech, China).
The CYP2C9*1 and CYP2C9*3 alleles were identified by the absence or presence of the KpnI restriction site, using the method previously described [6]. The CYP2C19*1 allele and two mutant alleles (CYP2C19*2 and CYP2C19*3) were determined by the loss of BamHI and SmaI digestion sites in the PCR product, as described by de Morais et al. and Nasu et al. [7, 8]. The genotypes of all the subjects in our study had been confirmed by gene sequencing.
Determination of glipizide in plasma
Plasma concentrations of glipizide were determined using a validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method, as previously described with a slight modification [9]. For sample preparation, a methanol–water mixture (50:50, v/v), internal standard solution (gliclazide), and phosphate buffer (0.25 M) were added. The mixed samples were then extracted with hexane–dichloromethane (2:1, v/v), by shaking on a mechanical shaker for 15 min. After centrifugation at 2,000 g for 5 min, the upper organic layer was removed and evaporated to dryness under nitrogen gas. The residue was dissolved in 100 μL of the mobile phase (mobile phase A was water with 0.1% acetic acid and mobile phase B was acetonitrile with 0.1% acetic acid), and 20 μL was injected for analysis.
Chromatography was performed using a Zorbax SB-C8 column (150 mm × 4.6 mm ID, 5 μm, Agilent, USA) at 25°C. Quantification was performed using selected reaction monitoring (SRM) of the transitions m/z 446 → m/z 321 for glipizide and m/z 324 → m/z 127 for gliclazide. The lower limit of quantification of glipizide was 1.0 ng/mL. The coefficients of variation for the intra- and inter-run were 2.1–14.1 and 5.6–7.6%, respectively. The accuracy of the assay ranged from 102.3 to 106.0%.
Data analysis
The pharmacokinetic parameters of glipizide were estimated by a noncompartmental method, using the WinNonlin program (version 5.1; Pharsight, USA). The peak plasma concentration (Cmax) was obtained directly from the observed concentration-time data. The terminal elimination rate constant (λz) was estimated by linear regression of the terminal portion of the concentration-time curve, and the elimination half-time (t 1/2) was calculated as 0.693/λz. The area under the plasma concentration-time curve (AUC) was calculated by use of the linear trapezoidal rule and extrapolated to infinity (\( AU{C_{0 - \infty }} \)). The apparent oral clearance (CL/F) of glipizide was calculated as dose/\( AU{C_{0 - \infty }} \).
To compare the pharmacodynamic effects of glypizide in subjects with different genotypes, changes in blood glucose and serum insulin concentrations over time were calculated by subtracting their baseline values from the observed values after dosing [10]. The decremental area under the glucose concentration-time curve (ΔAUECglucose, presented as a positive value) and the incremental area under the insulin concentration-time curve (ΔAUECinsulin) were calculated by use of the linear trapezoidal rule, in a similar manner to that described earlier for AUC.
Statistical analysis
The data are presented as the mean ± SD. All calculated parameters were compared across the three genotype groups by one-way ANOVA. As appropriate, a post-hoc Dunnett’s test was used to assess the presence of statistical differences between the genotype groups when a statistically significant association was described by ANOVA. The relationship of glipizide pharmacokinetic parameters and pharmacodynamic parameters was evaluated by use of the Spearman rank correlation coefficient (r s). For all analyses, P < 0.05 was considered statistically significant. All analyses were performed with SPSS software (version 11.0; SPSS, USA).
Results
Pharmacokinetic differences between CYP2C9 and CYP2C19 polymorphisms
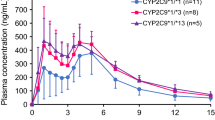
The glipizide plasma concentration-time curves for the three genotype groups are shown in Fig. 1a, and the calculated pharmacokinetic parameters are presented in Table 1. Glipizide parameters were significantly different between group I (CYP2C9*1/*1 and CYP2C19 EMs) and group III (CYP2C9*1/*3 and CYP2C19 EMs). In group III, the \( AU{C_{0 - \infty }} \) was significantly higher by 95.5% (P = 0.002) and the oral clearance (CL/F) was lower by 51.1% (P = 0.005) than in group I. Although the \( AU{C_{0 - \infty }} \) was higher by 52.6% (P = 0.072) and the CL/F was lower by 33.3% (P = 0.059) in group II (CYP2C9*1/*1 and CYP2C19 PMs) than in group I, there were no statistical differences between the two groups.

a Mean plasma concentration-time curves of glipizide in different genotype groups after a single dose of a 5-mg glipizide tablet. Data are expressed as mean values ± SEM. b Representative plasma concentration-time curve of glipizide in volunteer 9 after a single dose of a 5-mg glipizide tablet. The curve illustrates individual variation in Tmax
In addition, there was a second peak in the mean concentration-time curve among the three groups. However, the concentration-time curve for each individual showed one peak only (see Fig. 1b).
Pharmacodynamic differences between CYP2C9 and CYP2C19 polymorphisms
The glipizide pharmacodynamic responses (insulin and glucose responses) in the three genotype groups are shown in Figs. 2 and 3. The calculated pharmacodynamic parameters are presented in Table 2. The incremented area under the fasting insulin concentration-time curve (ΔAUECinsulin0–1h) was significantly higher by 278.4% (P = 0.012) in group III (CYP2C9*1/*1 and CYP2C19 PMs) compared with group I (CYP2C9*1/*1 and CYP2C19 EMs). However, the incremented area under the insulin concentration-time curve after oral dextrose (ΔAUECinsulin1–4h) in group III was 88.4% (P = 0.868) of that in group I, indicating that there was no increment. The ΔAUECinsulin0–1h and ΔAUECinsulin1–4h were higher by 76.8% (P = 0.520) and 16.7% (P = 0.751), respectively, in group II (CYP2C9*1/*1 and CYP2C19 PMs) than in group I, but none of these parameters were significantly different.

The mean changes in insulin level-time curves of glipizide in different genotype groups after a single dose of a 5-mg glipizide tablet. Data are expressed as mean values ± SEM of ΔAUECinsulin, the incremental area under the insulin concentration-time curve

The mean changes in glucose level-time curves of glipizide in different genotype groups after a single dose of a 5-mg glipizide tablet. Data are expressed as mean values ± SEM of ΔAUECglucose, the decremental area under the glucose concentration-time curve
The decremental area under the fasting glucose concentration-time curve (ΔAUECglucose0–1h) and the decremental area under the glucose concentration-time curve after oral dextrose (ΔAUECglucose1–4h) in group III were higher by 46.5% (P = 0.165) and 49.5% (P = 0.622), respectively, compared with those in group I. Similarly, ΔAUECglucose0–1h and ΔAUECglucose1–4h in group III were higher by 31.8% (P = 0.394) and 63.4% (P = 0.473), respectively. However, none of these parameters were significantly different.
A significant decrement in blood glucose concentration (<54 mg/dL) occurred in nine subjects at 1 h after dosing with glipizide tablet. Of these subjects, five belonged to group III, two belonged to group I, and two belonged to group II. One subject in group III even had hypoglycemic symptoms (i.e., faintness and sweating).
Association between glipizide pharmacokinetics and pharmacodynamics
In an examination of the relationship between glipizide pharmacokinetics and pharmacodynamics, glipizide exposure (AUC0–1h) in individual subjects was found to correlate well with the observed response from ΔAUEC insulin0–1h and ΔAUECglucose0–1h, with r s values of 0.652 (P = 0.006, Fig. 4a) and −0.646 (P = 0.004, Fig. 4b), respectively.

Correlation between individual pharmacokinetic parameters (a, b AUC0–1h and c, d AUC1–4h) of glipizide and its blood glucose-lowering effects, assessed by ΔAUECinsulin (during a 0–1 h and c 1–4 h), and ΔAUECglucose (during b 0–1 h and d 1–4 h). AUC Area under the curve, ΔAUEC insulin the incremental area under the insulin concentration-time curve, ΔAUEC glucose the decremental area under the glucose concentration-time curve
Discussion
In this study we found that CYP2C9 polymorphism significantly influenced the pharmacokinetics of glipizide. After a single dose of glipizide tablet, the \( AU{C_{0 - \infty }} \) and CL/F were 2.0-fold greater and 51.1% lower, respectively, in the CYP2C9*1/*3 subjects than those in the wild-type CYP2C9 subjects. Kidd et al. had reported the pharmacokinetic parameters for a healthy Caucasian who carried homozygous CYP2C9*3 alleles after a single dose of glipizide extended-release tablet (Glucotrol XL, Pfizer, USA) [3]. The \( AU{C_{0 - \infty }} \) and CL/F for this individual were 445.1 and 18%, respectively, compared with homozygous CYP2C9*1 alleles carriers. Consistent with the previous study, our study confirmed that CYP2C9 polymorphism had an important role in the pharmacokinetics of glipizide.
Furthermore, we found that CYP2C9 polymorphism had a significant influence on the pharmacodynamic response to glipizide. The ΔAUECinsulin0–1h in CYP2C9*1/*3 subjects was 3.8-fold greater than in the CYP2C9*1/*1 subjects. Meanwhile, a significantly higher rate (83.3% versus 33.3%) of hypoglycemic effect (blood glucose concentration <3 mmol/L) at 1 h after dosing with a glipizide tablet occurred in the CYP2C9*1/*3 subjects as compared with the CYP2C9*1/*1 subjects. ΔAUECglucose0–1h also tended to be greater in CYP2C9*1/*3 subjects, although the difference was not statistically significant (P = 0.225), probably because of the large variations in glucose response as well as the relatively small number of subjects included in each group. When compared with CYP2C9*1/*1 subjects, ΔAUECinsulin1–4h and ΔAUECglucose1–4h showed different changes in CYP2C9*1/*3 subjects: ΔAUEC glucose1–4h was higher whereas ΔAUECinsulin1–4h was lower. It may be that oral administration of dextrose at a high concentration results in a complex response to blood insulin and glucose that could obscure the influence of genetic polymorphism on glipizide.
In addition, this study revealed that there was a strong relationship between the pharmacokinetics (AUC0–1h) and pharmacodynamics (ΔAUECinsulin0–1h and ΔAUECglucose0–1h) of glipizide, supporting the idea that CYP2C9 polymorphism could have a significant clinical impact due to its influence on the pharmacokinetics of glipizide.
Taken together, our study strongly suggests that CYP2C9 polymorphism plays an important role in the pharmacokinetic and pharmacodynamic effects of glipizide and therefore a lower (i.e., CYP2C9*3 heterozygote) or nonfunctional (i.e., CYP2C9*3 homozygote) allele carrier should have an elevated plasma level of glipizide, which results in a greater hypoglycemic effect and an increased risk of hypoglycemia.
In this study we also found that CYP2C19 polymorphism had a tendency to influence the pharmacokinetics and pharmacodynamics of glipizide. Similarly to the effects of CYP2C9 polymorphism, there were detectable differences between CYP2C19 EMs and PMs in the pharmacokinetics and pharmacodynamics of glipizide, but none of these differences were statistically significant. However, the effects of CYP2C19 polymorphism on the pharmacokinetics and pharmacodynamics of glipizide could not be ignored, since the degree of changes for most parameter values exceeded 40% between CYP2C19 EMs and PMs. It is worth noting that the P values of \( AU{C_{0 - \infty }} \) and CL/F for CYP2C19 EMs and PMs were 0.072 and 0.059, respectively, close to the significance level (P < 0.05). Having a larger number of subjects in each group would be helpful to illuminate the effects of CYP2C19 polymorphism.
In contrast to our findings, a previous study showed insignificant effects of CYP2C9 polymorphism on the CL/F and pharmacodynamic parameters [4]. These differences are likely related to the differences in the study protocol, such as whether the effect of the CYP2C19 polymorphism was considered.
The distribution of the CYP2C9*3 allele is 1.1–3.3% among Asian population [11]. Therefore the frequency of homozygous CYP2C9*3 subjects is very low, less than 1/1,000 subjects, and our study did not include this genotype group. The CYP2C9*2 variant is another important variant in whites (8.0–19.1%) [11]. According to most in vitro data, the maximum rate of substrate metabolism (Vmax) in CYP2C9*2 is reduced to around 50% of that in CYP2C9*1, resulting in lower intrinsic clearance [12]. Therefore, the CYP2C9*2 carriers might also have enhanced exposure and similar clinical indications as CYP2C9*3 carriers [12]. However, we did not investigate this in this study for the same reason we did not study the homozygous CYP2C9*3 subjects [11]. Additionally, CYP2C19*17 is a novel variant allele that can increase the activity of CYP2C19 metabolism [13]. Due to its low frequency among the Chinese population (0.64%) [14], we did not investigate it in our study.
In our study, there was a second peak in the glipizide plasma concentration-time curves among three groups. It may be due to the interindividual variation in T max (see Table 1).
Given the small number of subjects in our study, the significance of the observations requires further confirmation in a larger population. In addition, our study was conducted in healthy subjects by use of a single-dose design. Further studies in patients with different CYP2C9 and CYP2C19 genotypes with multiple dosing will be needed to verify the conclusions and will be beneficial to the investigation of individual rational dosages and decreasing the risks of adverse effects [15-17].
In conclusion, CYP2C9 polymorphism significantly influences the pharmacokinetics and pharmacodynamics of glipizide. Dose adjustment based on CYP2C9 genotype may improve antidiabetic treatments and minimize the risk of adverse reactions. CYP2C19 polymorphism exhibits a tendency to influence the effects of glipizide, to a certain extent similar to CYP2C9 polymorphism. Further studies with multiple dosing in diabetic patients are needed to warrant these findings and will be beneficial for clinical practice.
References
Fuccella LM, Tamassia V, Valzelli G (1973) Metabolism and kinetics of the hypoglycemic agent glipizide in man — comparison with glibenclamide. J Clin Pharmacol New Drug 13:68–75
Balant L, Fabre J, Zahnd GR (1975) Comparison of the pharmacokinetics of glipizide and glibenclamide in man. Eur J Clin Pharmacol 8:63–69
Kidd RS, Straughn AB, Meyer MC, Blaisdell J, Goldstein JA, Dalton JT (1999) Pharmacokinetics of chlorpheniramine, phenytoin, glipizide and nifedipine in an individual homozygous for the CYP2C9*3 allele. Pharmacogenetics 9:71–80
Bae JW, Kim NT, Choi CI, Kim MJ, Jang CG, Lee SY (2007) Effects of CYP2C9 genetic polymorphism on the pharmacokinetics and pharmacodynamics of glipizide in healthy Korean subjects. FASEB J 21:lb362
Desta Z, Zhao X, Shin JG, Flockhart DA (2002) Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 41:913–958
Wang SL, Huang J, Lai MD, Tsai JJ (1995) Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics 5:37–42
De Morais SMF, Wilkinson GR, Blaisdell J, Nakamura K, Meyer UA, Goldstein JA (1994) The major genetic defect responsible for the polymorphism of S-mephenytoin metabolism in humans. J Biol Chem 269:15419–15422
De Morais SMF, Wilkinson GR, Blaisdell J, Meyer UA, Nakamura K, Goldstein JA (1994) Identification of a new genetic defect responsible for the polymorphism of (S)-mephenytoin metabolism in Japanese. Mol Pharmacol 46:594–598
Zhao XH, Song B, Zhong DF, Zhang SQ, Chen XY (2007) Simultaneous determination of metformin and glipizide in human plasma by liquid chromatography-tandem mass spectrometry. Acta Pharm Sin 42:1087–1091
Park JY, Kim KA, Park PW, Park CW, Shin JG (2003) Effect of rifampin on the pharmacokinetics and pharmacodynamics of gliclazide. Clin Pharmacol Ther 74:334–340
Xie HG, Prasad HC, Kim RB, Stein CM (2002) CYP2C9 allelic variants: ethnic distribution and functional significance. Adv Drug Deliv Rev 54:1257–1270
Lee CR, Goldstein JA, Pieper JA (2002) Cytochrome P450 2C9 polymorphisms: a comprehensive review of the in-vitro and human data. Pharmacogenetics 12:251–263
Sim SC, Risinger C, Dahl ML, Aklillu E, Christensen M, Bertilsson L et al (2006) A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther 79:103–113
Wang G, Lei HP, Li Z, Tan ZR, Guo D, Fan L et al (2009) The CYP2C19 ultra-rapid metabolizer genotype influences the pharmacokinetics of voriconazole in healthy male volunteers. Eur J Clin Pharmacol 65:281–285
Cefalu WT (2007) Pharmacotherapy for the treatment of patients with type 2 diabetes mellitus: rationale and specific agents. Clin Pharmacol Ther 81:636–649
Lindh JD, Holm L, Andersson ML, Rane A (2009) Influence of CYP2C9 genotype on warfarin dose requirements—a systematic review and meta-analysis. Eur J Clin Pharmacol 65:365–375
Becquemont L (2008) Evidence for a pharmacogenetic adapted dose of oral anticoagulant in routine medical practice. Eur J Clin Pharmacol 64:953–960
Acknowledgments
This study was supported by the Natural Science Foundation of China grant 30772621.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, B., Zhang, YF., Chen, XY. et al. The effects of CYP2C9 and CYP2C19 genetic polymorphisms on the pharmacokinetics and pharmacodynamics of glipizide in Chinese subjects. Eur J Clin Pharmacol 66, 145–151 (2010). https://doi.org/10.1007/s00228-009-0736-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-009-0736-2